DOI:
10.1039/C2RA20258A
(Paper)
RSC Adv., 2012,
2, 8145-8156
Ni supported high surface area CeO2–ZrO2 catalysts for hydrogen production from ethanol steam reforming
Received
14th February 2012
, Accepted 5th July 2012
First published on 3rd August 2012
Abstract
The catalytic activity of nano-sized x%Ni/Ce0.74Zr0.26O2 (x = 0, 2, 10 and 20wt%) catalysts have been investigated to develop highly active catalysts for ethanol steam reforming (ESR) into hydrogen. The structure and surface properties of the catalysts were tested by XRD, TPR, HRTEM and BET surface areas. The effect of reaction temperature from 200 °C to 600 °C was studied in a flow system at atmospheric pressure with an ethanol/water molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8. Selectivity was calculated for the catalytic products H2, CO, CO2 and CH4, as well as the intermediates C2H6, C2H4, C3H8, CH3CHO and CH3COCH3, at different reaction temperatures. It was found that complete conversion of ethanol with considerable amounts of H2 was obtained at 400 °C over all catalysts. H2 was produced at a very low temperature (200 °C) over 10% and 20% Ni loadings, while a maximum H2 selectivity (75%) is reached at 600 °C over the 2%Ni/Ce0.74Zr0.26O2 catalyst; this is most likely due to the small nickel particle size (2–4 nm) in 2%Ni, which results in enhancement of the metal–support interactions. Thermal decomposition of ethanol in an ethanol/water mixture under the same reaction conditions, but in the absence of catalyst, was also studied. HRTEM of the spent catalyst (8 h ESR) shows the deposition of carbon in the form of carbon nanotubes (CNTs).
:8. Selectivity was calculated for the catalytic products H2, CO, CO2 and CH4, as well as the intermediates C2H6, C2H4, C3H8, CH3CHO and CH3COCH3, at different reaction temperatures. It was found that complete conversion of ethanol with considerable amounts of H2 was obtained at 400 °C over all catalysts. H2 was produced at a very low temperature (200 °C) over 10% and 20% Ni loadings, while a maximum H2 selectivity (75%) is reached at 600 °C over the 2%Ni/Ce0.74Zr0.26O2 catalyst; this is most likely due to the small nickel particle size (2–4 nm) in 2%Ni, which results in enhancement of the metal–support interactions. Thermal decomposition of ethanol in an ethanol/water mixture under the same reaction conditions, but in the absence of catalyst, was also studied. HRTEM of the spent catalyst (8 h ESR) shows the deposition of carbon in the form of carbon nanotubes (CNTs).
1. Introduction
Hydrogen is a potentially very attractive source of energy since its combustion produces only water and energy. Molecular hydrogen is a clean burning fuel, can be stored as a liquid or gas, is distributed via pipelines, and has been described as a long term replacement for natural gas, so increased attention is focused on hydrogen production technologies.1 Several catalytic processes for hydrogen production from ethanol have been investigated, including steam reforming (SR), partial oxidation, and auto-thermal reforming.2
Ethanol is a promising future bio-fuel because of its relatively high hydrogen content (on a molar basis), non-toxicity, ease of storage and handling safety. Ethanol can be readily produced from renewable feedstocks such as sugar cane, municipal solid waste and agro-waste. Another advantage of using ethanol is that the CO2 produced during ethanol steam reforming (ESR) is equal to the CO2 required for biomass growth and thus provides a closed cycle for CO2 consumption. Therefore, no net pollutants are released into the environment.
Nickel-based catalysts are widely used in commercial reforming processes and are recognized as appropriate catalyst systems in SR for their low cost, relatively high activity towards C–C bond cleavage and hydrogenation/dehydrogenation reactions.3 The study by Kugai et al.4 has shown that ethanol is adsorbed on the surface of Rh and Ni metals as ethoxide species, which form an oxametallacycle intermediate and favors C–C bond rupture. Ni has a high activity for C–C bond and O–H bond breaking, and has high activity for hydrogenation, facilitating H atoms to form molecular H2.5
Supports also play important roles in the SR of ethanol, as supports help in the dispersion of the metal catalyst and may enhance the metal catalyst activity via metal–support interactions. Supports may promote migration of OH groups toward the metal catalyst in the presence of water at high temperature, facilitating SRR.6
In Ni-based catalysts, CeO2 has been used as either an effective promoter or support because of its characteristic oxygen storage capacity (OSC), which allows it to store and release oxygen, leading to the presence of highly active oxygen for the oxidation of CO and hydrocarbons. This makes the catalyst more active in many carbon formation-related reactions of hydrocarbons, such as SR, dry reforming and oxidation. The specific role of CeO2 in SR involves improving the dispersion of the active phase and enhances interaction with the supported metal.7
During the reforming process, in addition to the reactions on Ni surface, the gas–solid reactions between the gaseous components presented in the system (C2H6, C3H8, C2H4, CH4, CO2, CO, H2O, and H2) and the lattice oxygen (Ox) on the ceria surface also take place. The reactions of adsorbed surface hydrocarbons with the lattice oxygen (Ox) on the ceria surface (CnHm + Ox → nCO + m/2(H2) + COx−n) prevent the formation of carbon species on Ni surface from hydrocarbons decomposition reaction (CnHm ↔ nC + m/2H2). Moreover, the formation of carbon via the Boudouard reaction (2CO ↔ CO2 + C) is also reduced by the gas–solid reaction of carbon monoxide (produced from SR) with the lattice oxygen (CO + Ox ↔ CO2 + Ox−1).8
Ceria easily forms solid solutions with transition metals and other rare-earth elements. It has been established that introduction of zirconium into the ceria lattice greatly enhances the surface area, thermal stability and oxygen storage capacity, resulting in superior catalytic properties. Hence, ceria–zirconia solid solutions have been investigated with huge interest among other ceria-based mixed oxides.9,10 Seog et al.11 have been confirmed that Ni/Ce0.8Zr0.2O2 shows a higher BET surface area than Ni/CeO2 and better thermal resistance than Ni/ZrO2 during the reduction process at 600 °C. Electron paramagnetic resonance (EPR) spectroscopy has identified three types of paramagnetic defects in CeO2–ZrO2 oxides,12viz. electrons, quasi-free or trapped in anion vacancies, O− ions and O2− ions.13 These species arise by the following interactions:
The oxygen storage capacity of CeO2–ZrO2 catalysts was correlated to the concentration of the oxide ion species.13 Although it had been postulated that CeO2 promotes NiO reduction, no experimental evidence had been reported in the literature so far. The work of Srinivas et al.12 offers such a support, part of the Ni+ ions subsequently react with aerial oxygen and form Ni2+(O2·−) species, as described:
| | | Ce4+ − O2− − Zr4+ ↔ Ce3+ − O− − Zr4+ | (4) |
| | | Ce3+ + Ni2+ → Ce4+ + Ni+ | (5) |
| | | Ni+surf + O2 → Ni(O2·−) | (6) |
Three types of Ni species in NiO/CeO2–ZrO2 samples were observed by Srinivas et al.,12 (I) Ni substituted in the lattice forming Ni(O2·−) ions, (II) nanosize crystallites of NiO forming super paramagnetic metallic Ni and (III) larger particles of NiO forming ferromagnetic Ni. The sample containing 1% Ni contained species I and II, the sample containing 5% Ni contained all three species; while samples containing 40% Ni contained species I and III. The reducibility of these species decreased in the order: I > III > II.
Jones et al.14 suggested the elementary steps of ethanol decomposition in order to gain some insights into the reasons behind the product distribution. In the decomposition pathway of the ethoxy species Ni has a unique effect. It abstracts H from the CH3-group making a stable oxametallacycle intermediate (adsCH2–CH2–Oads). The stoichiometry of the reaction for maximum hydrogen production is given by:
| | | CH3CH2OH + 3H2O → 6H2 + 2CO2 | (7) |
Our study aimed at developing the amount of H2 produced from ESR over x%Ni/Ce0.74Zr0.26O2 catalysts. This goal was intended to firstly demonstrate the occurrence of ESR over the used catalysts by performing the reaction in the absence of the catalysts, i.e., thermal decomposition of ethanol. Secondly, to study the effect of the support and different Ni loadings on product distribution in the ESR reaction, and finally, to study the distribution in the gas and liquid phases to clarify the mechanism from intermediate distribution at different temperatures.
2. Experimental
2.1. Catalyst preparation
The Ce1−xZrxO2 mixed oxide supports were prepared by co-precipitation with ammonia using an aqueous solution of cerium nitrate (Ce(NO3)2·6H2O, 99% Fluka) and zirconium oxychloride (ZrOCl2·8H2O, 98% Aldrish). The weight ratio of CeO2/ZrO2 was 4 which corresponds to an x of 0.26. Aqueous ammonia solution (28–30%) was added dropwise to the aqueous solution containing (0.1 M) Ce and (0.1 M) Zr salts with constant stirring until the pH was 9–10. After precipitation, the obtained hydroxide was filtered, washed thoroughly with deionized water until chloride free (tested with silver nitrate Ag2NO3), and then dried overnight at 120 °C. The obtained solid was calcined in air at 600 °C for 5 h.
Ni loading was carried out by an incipient wetness impregnation method using an aqueous solution of (Ni(NO3)2·6H2O 99% Aldrich) dissolved in deionized water. Ni loadings were 0, 2, 10 and 20 wt%. After impregnation, the catalyst was dried overnight at 120 °C and then calcined at 600 °C for 6 h.
2.2. Characterization methods
Thermal gravimetric analysis (TGA) was carried out using SETARAM Labsys TG-DSC16 equipment in the temperature range of room temperature up to 1000 °C under a nitrogen flow, in order to follow the thermal stability of the prepared catalyst.
X-Ray diffraction analysis (XRD) was carried out by Shimadzu XD-1 diffractometer using Cu-target and Ni-filtered radiation, to trace the various changes in the crystalline structure and the different phases accompanied preparation method. Sample powders were packed in a glass holder, then measurements were taken of the diffraction intensity by step scanning in a 2θ range between 5° and 70°. The phase identification was made by comparison with the Joint Committee on Powder Diffraction Standards (JCPDS). The average crystallite size of the samples was determined from the XRD peaks using the Scherrer equation.
High resolution transmission electron microscopy (HRTEM) and energy dispersive X-ray (EDS) spectroscopy were conducted using a JEOL 2100F TEM at an accelerating voltage of 200 kV. To prepare the TEM samples, a dilute particle–ethanol colloidal mixture was ultrasonicated for 30 min and a drop of solution was placed on a carbon coated Cu TEM grid.
The textural properties were determined from the N2 adsorption–desorption isotherms measured at liquid nitrogen temperature (−196 °C) using a NOVA2000 gas sorption analyzer (Quantachrome Corporation) system. All samples were degassed at 200 °C for 17 h in a nitrogen atmosphere prior to adsorption to ensure a dry clean surface. The adsorption isotherm was constructed as the volume adsorbed (V cm3 g−1) versus the equilibrium relative pressure P/Po, where P is the equilibrium pressure and Po is the saturated vapor pressure of nitrogen.
Total amount of acidity was estimated from weight loss measurements of adsorbed pyridine using SETARAM Labsys TG-DSC16 equipment. First, a platinum crucible containing 50 mg of the sample was placed in a shallow porcelain plate and inserted into a glass reactor adapted to a tubular furnace. The sample was dehydrated in dried N2 (100 ml min−1) at 120 °C for 2 h, cooled to 70 °C, and then gaseous pyridine diluted in N2 was allowed to pass through the sample for 1.5 h. The temperature was held at 70 °C under N2 for 1 h to remove the physically adsorbed pyridine. After that, the samples were analysed by TGA.
Temperature programmed reduction (TPR) measurements were carried out to investigate the redox properties (the ease of reducibility of the metal oxide) over the resultant materials. The experiments were performed in automatic equipment (ChemBET 3000, Quantachrome). Typically, 100 mg of the pre-calcined sample was loaded into a quartz reactor and pretreated by heating under an inert atmosphere (20 ml min−1 nitrogen) at 200 °C for 3 h prior to running the TPR experiment, and was then cooled to room temperature in N2. Then the sample was submitted to a constant rate of heat treatment (10 °C min−1 up to 1000 °C) in a gas flow (80 ml min−1) of the hydrogen/nitrogen (5/95 vol%) mixture as a reducing gas. A thermal conductivity detector (TCD) was employed to monitor the amount of hydrogen consumption.
2.3. Catalytic activity
The experimental system used in this work is schematically shown in Fig. 1. Experiments were performed at atmospheric pressure in a continuous fixed-bed down flow vertical tubular reactor consisting of a quartz tube, 300 mm in length and 10 mm i.d. Another silica tube of 500 mm length and 14 mm i.d. attached to the first one served as the outlet to facilitate fast removal of the reactor effluent. The catalyst was placed on a quartz wool bed inside the reactor. 2 g of catalyst diluted with the same sized quartz particles was used for catalytic tests. Prior to a run, the catalyst was reduced in situ at 600 °C for 2 h under a hydrogen flow rate of 20 cm3 min−1, where the catalyst showed thermal stability up to 1000 °C (TGA, not shown). A water/ethanol mixture (EtOH/H2O = 1:8 mol/mol) premixed in a separate container was fed into the evaporator (at 150 °C) by a pump (Model RP-G6; FMI, USA) with a flow rate of 0.2 ml min−1. Nitrogen gas was fed into the evaporator with a flow rate 40 cm3 min−1 to uniformly carry the steam. This steam was then fed into the reactor. The reaction temperature was varied from 200 °C to 600 °C. The thermal decomposition of ethanol was studied under the same reaction conditions but in the absence of catalyst.
 |
| | Fig. 1 Schematic diagram of experimental setup: (1) feeding tank containing the water/ethanol mixture; (2) pump (Model RP-G6; FMI, USA); (3) evaporator; (4) catalyst packed reactor; (5) cooler; (6) gas collector; (7) liquid collector (8) hydrogen and nitrogen cylinder; and (9) heating and temperature controller. | |
The product stream was analyzed by the use of two gas chromatographs (GC) (Agilent 6890 plus HP, Varian Natural Gas Analyzer type C model CP-3800). The first one, equipped with a molecular sieve 5 Å capillary column (Molsive 5 Å, 15 m in length, 0.53 mm in internal diameter and 40 μm in film thickness) and TCD with (He) as the carrier gas, was used for the determination of H2. The first one was also used to analyze the composition of condensed liquid products on the capillary column (HP-30 50% phenyl/50% dimethyl polysiloxane, 30 meter in length, 0.53 mm in internal diameter and 0.32 μm in film thickness) using a flame ionization detector (FID) with N2 as the carrier gas and isopropanol was used as the internal standard. The second GC equipped with TCD and FID, three columns are used to accomplish the single channel analysis of the gas. Firstly, a molecular sieve column (4 ft in length, 1/8 inch in internal diameter and Molsieve 13X with a 45/60 mesh size) for oxygen, nitrogen, methane and carbon monoxide separation. Secondly, a Hayesep P column (6 ft in length, 1/8 inch in internal diameter and Hayesep P with an 80/100 mesh size) for separation of methane, carbon dioxide, ethane. The two packed columns, molecular sieve and Hayesep P, were attached with TCD. The third capillary column (CPSil 5, 60 meter in length, 0.25 mm in internal diameter and 1 μm in film thickness) used for separation of methane, ethane, ethylene, acetaldehyde, ethanol, acetone, was attached with FID. Helium gas was used as carrier gas with constant pressure 20 psi, 23.5 psi and 13.4 psi for the CPSil 5, Hayesep P and molecular sieve columns, respectively. The quantitative analysis of the gas mixture is based on an external standard, as the GC response is calibrated using a suite of certified reference external standard gas samples with known compositions and is used to calculate the response factor of each component in the products. Selectivity values were calculated as the molar percentage of products obtained, excluding water; Selectivity of product = (mol of product/total mol of H2 and carbon-containing products in the outlet) × 100.
3. Results and discussion
3.1. Catalyst characterization
3.1.1. X-Ray diffraction analysis.
The X-ray diffraction pattern of the CeO2–ZrO2 support, as well as the Ni/CeO2–ZrO2 catalysts calcined at 600 °C with various values of Ni loading (2–20%), are represented in Fig. 2.
 |
| | Fig. 2 XRD patterns of fresh CeO2–ZrO2 and x%Ni/CeO2–ZrO2 catalysts (ss: solid solution). | |
The XRD patterns of the CeO2–ZrO2 support (Fig. 2) showed a number of peaks corresponding to 2θ = 28.5, 33.3, 47.4 and 56.4, which are assigned to a typical cubic fluorite structure of Ce0.74Zr0.26O2 [ref. JCPDS card (00-028-0271)]. No separate phase due to CeO2 or ZrO2 was detected, which is ascribed to the insertion of zirconium ions of a smaller radius (0.084 nm) into the cubic lattice of CeO2 (0.097 nm) and the formation of a homogenous Ce1−xZxO2 solid solution (ss).15 This cubic phase CeO2–ZrO2 has the maximum degree of structural defects and oxygen storage capacity as reported by Kumar et al.16
The XRD patterns of the x%Ni/CeO2–ZrO2 catalysts impregnated with different Ni loadings all showed peaks corresponding to the cubic fluorite structure of the CeO2–ZrO2 solid solution. No separate NiO phases were found by XRD for 2%Ni/CeO2–ZrO2. The peak position of CeO2–ZrO2 was invariant with the Ni content, and the peak width changed only marginally. At higher Ni loadings (10 and 20% Ni), three new peaks at about 37°, 43° and 62° (2θ) [ref. JCPDS card (00-001-1239)] indicating a NiO phase were observed. The peak intensity was obviously stronger with increasing Ni loading.
Table 1 summarizes the crystallite sizes and lattice parameters of the prepared catalysts. The crystallite size of the support is in the range 11.5–9.4 nm, which confirms that a nano-crystallite size is achieved by the co-precipitation method. In addition, the crystallite size of NiO in the (2%Ni) catalyst is too small to measure, this indicates that the NiO species are finely dispersed on the support beyond the XRD detection limit (<3 nm), this is confirmed by the decrease in support lattice parameters, due to the incorporation of Ni2+ with a small ionic radius of (0.072 nm) into the support lattice.11,17 The dispersion and particle sizes were not identified by XRD, therefore HRTEM was utilized later (section 3.1.2) to discuss the presence and the size of NiO in the 2%Ni/CeO2–ZrO2 catalyst. The size of the NiO crystallites increased from 24.5 to 26.3 nm with increasing Ni content in the samples (Table 1).
Table 1 Textural properties for the prepared Ni/CeO2–ZrO2 catalyst
| Catalyst |
Acidity (mmol g−1) |
Crystal size (CeO2–ZrO2)a (nm) |
Crystal size (NiO)a (nm) |
Unit cell parameter (CeO2–ZrO2) (nm) |
BET surface areab (m2 g−1) |
Pore volumec (cm3 g−1) |
Pore radiusc (Å) |
|
According to XRD analysis using Scherrer equation.
BET surface area calculated from the linear portion of the BET plot in the relative pressure range of P/Po = 0.05–0.35.
Pore radius and total pore volume estimated using BJH method from the isothermal desorption data.
|
| CeO2–ZrO2 |
740.8 |
10.4 |
– |
0.542 |
133.5 |
0.112 |
18.2 |
| 2%NiO/CeO2–ZrO2 |
574.1 |
11.5 |
– |
0.538 |
82.9 |
0.155 |
40.3 |
| 10%NiO/CeO2–ZrO2 |
160.1 |
10.4 |
24.1 |
0.541 |
67.8 |
0.128 |
45.9 |
| 20%NiO/CeO2–ZrO2 |
63.3 |
9.4 |
26.1 |
0.542 |
41.9 |
0.1 |
31.9 |
3.1.2. High resolution transmission electron microscopy.
The HRTEM and STEM/EDS photographs obtained from the CeO2–ZrO2 and Ni/CeO2–ZrO2 catalysts are shown in Fig. 3. Analysis of the photographs for CeO2–ZrO2 (Fig. 3a) revealed particles with defined dimensions, having a grain size ranging from 10–19 nm. As per the literature,18 and taking into account the slight contraction of the crystal lattice, which is expected by partial substitution of Ce ions by smaller Zr ions, the cubic fluorite structure is quite obvious.19 Although no separate NiO phases were discernible by the XRD of 2%Ni/CeO2–ZrO2, STEM/EDS elemental mapping and HRTEM images (Fig. 3b) identified nickel clusters that were well dispersed within the CeO2–ZrO2 matrix. Therefore, at low Ni content, nickel oxide was present in CeO2–ZrO2, but, in nanoparticle form (2–4 nm).20 The 20%Ni/CeO2–ZrO2 (Fig. 3c) system is not homogeneous. In fact, two forms of crystals are observed, corresponding to the presence of the CeO2–ZrO2 fluorite structure and nickel oxide, as confirmed from XRD and STEM/EDS. The grains of NiO are readily recognizable by their opaque hexagonal shape, in contrast to the CeO2–ZrO2 particles, which are cubic and transparent. The mean size of the NiO aggregates is estimated to be in the range 57–95 nm and those of the CeO2–ZrO2 particles is in the range 8–12 nm.
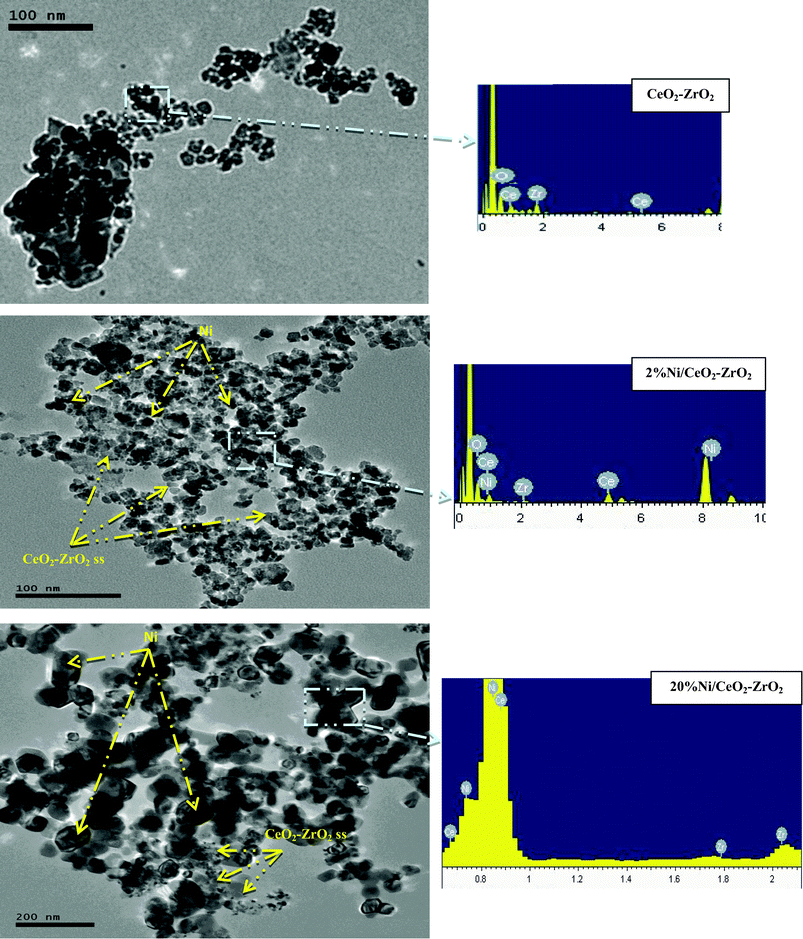 |
| | Fig. 3 HRTEM and STEM/EDS photographs obtained for the fresh CeO2–ZrO2 and x%Ni/CeO2–ZrO2. | |
3.1.3. Surface areas.
All samples displayed type IV isotherms, as illustrated in Fig. 4, according to Brunauer's classification and are characterized by well-developed mesoporous structures including micropores.21 The shape of the hysteresis loop is type H2, according to the IUPAC classification, which usually has non uniform sized or shaped pores.22
 |
| | Fig. 4 N2 adsorption–desorption isotherms for CeO2–ZrO2 and x%Ni/CeO2–ZrO2 catalysts. | |
Table 1 summarizes the BET surface area, pore volume and pore radius of the CeO2–ZrO2 and x%Ni/CeO2–ZrO2 catalysts, the surface area was found to be 133.5 m2 g−1 for the CeO2–ZrO2 support, which decreases with increases in Ni loading from 2–20 wt%. This might be a consequence of crystallite rearrangement during metal impregnation, as already reported by Alonso et al.,23 and/or due to the blockage of some pores by NiO particles.24 These surface areas are higher than those reported by other authors who have prepared ceria–zirconia supported nickel catalysts.25,26
The pore volume of the CeO2–ZrO2 support increases after impregnation with 2% Ni from 0.112 to 0.155 cm3 g−1, this may be attributed to low Ni loading, which is present in the form of nanoparticles, as illustrated from HRTEM (Fig. 3), so it may be incorporated into the support pore and increase the pore volume. On the other hand, as the Ni loading increases up to 20% the pore volume decreases, due to blocking of the pores by agglomerated NiO at the surface of the support.
3.1.4. Temperature programmed reduction (TPR).
TPR is a simple and quite useful method to obtain information on the reactivity of lattice oxygen towards hydrogen in CeO2–ZrO2 supports, as well as the Ni reducibility in x%Ni/CeO2–ZrO2 catalysts. As illustrated in Fig. 5, the CeO2–ZrO2 support shows two reduction peaks, at 450 and 600 °C. Since pure ZrO2 did not show any reduction peak in this wide temperature range, it is in agreement with the reported result.25 All the consumed hydrogen contributes to the reduction of Ce4+ to Ce3+. According to the reduction mechanism of CeO2 proposed by Pantu and Gavalas,27 the TPR profile of pure CeO2 shows a two-peak pattern at 503 °C and 752 °C, due to surface and bulk reduction. The outermost layers of the smaller crystalline CeO2 are first reduced at lower temperatures due to a lower enthalpy of reduction, and then bulk reduction occurrs at higher temperatures. In context, the TPR profiles of the mixed CeO2–ZrO2 oxides show a main broad reduction feature, which is in agreement with the promotion of the reduction of the mixed oxide, this means that the presence of Zr weakens the Ce–O bond in the solid solution of CeO2–ZrO2 and makes the CeO2 component more easily reduced.28 The low temperature peak at 450 °C is due to the reduction of the surface layer of CeO2 and the higher one at 600 °C is due to that of the material in the bulk.15
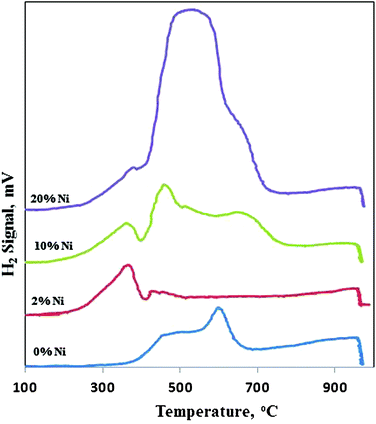 |
| | Fig. 5 TPR patterns of Ni/CeO2–ZrO2 catalysts with different Ni loading. | |
The TPR profile of the 2%Ni/CeO2–ZrO2 catalyst possesses two peaks: the first one is a peak at 370 °C, and the other peak is at 430 °C. The first peak is attributed to the reduction of NiO to Ni0 which is lower than the reduction at 490 °C in pure NiO.29 It is obvious that the reduction peak of NiO is shifted to a lower temperature when it is deposited on the high surface area CeO2–ZrO2 support; due to appropriate interaction between the metal and support. This interaction is an electronic interaction between CeO2 and Ni because Ce is rich in d-electrons and Ni has unfilled d-orbitals, where the unfilled d-orbitals of the Ni atoms accept d-electrons from Ce, resulting in an increase in d-electron density of the Ni atom.30 The second peak at 430 °C corresponds to the reduction of surface CeO2 in the CeO2–ZrO2 support.
With an increase in the metal loading from 2% to 10%, the intensity of the peaks at 370 °C and 430 °C increased and shifted towards higher temperatures (460 °C and 660 °C, respectively), which is significantly due to increasing NiO content. Also another small peak at about 360 °C was observed, which might be attributed to the reduction of free NiO particles 31 or the hydrogen spillover effect.32 Further increase in metal loading to 20 wt%, resulted in the peak at 460 °C to be shifted towards higher temperatures and merged with the second peak, which is attributed to agglomerated NiO.25
3.2. Catalytic Activity
The effect of reaction temperature (200–600 °C) and metal loading on ESR and product selectivity over the CeO2–ZrO2 support and x%Ni/CeO2–ZrO2 catalyst (x = 2, 10, 20) are studied. The catalytic products were H2, CO, CO2 and CH4. The presence of these products indicates that steam reforming (SR) and the water gas shift (WGS) reaction took place simultaneously in the reactor.33 Minor amounts of by-products, such as CH3CHO, C2H4, C2H6 and CH3COCH3, were also observed; their concentration exhibiting a strong dependence on reaction temperature. As expected, ethanol conversion increased with increasing temperature and with an increase in metal loading. Nearly complete conversion was obtained with nickel loaded catalysts at 400 °C. The high activity and variety of products is mainly attributed to the complex composition and multi-reactive sites, such as metal Ni, acidic/basic centers, and oxygen vacancies in the CeO2–ZrO2 solid solution.
3.2.1. Homogeneous contribution.
Reforming of ethanol under the same reaction conditions, but in the absence of catalyst, was studied to completely understand the role of the catalyst in the ESR reaction. The blank experiment shows that only 8.8–17.5% of ethanol was converted at temperatures in the range 400–600 °C (Table 2), in the absence of catalyst, which is probably due to ethanol thermal decomposition. The distribution of products cannot be attributed to the ESR reaction, where water is being produced and not consumed. The production of water can be explained by ethanol dehydration yielding ethylene, which is a coke precursor.
Table 2 Product distribution from ESR of ethanol in the absence of catalyst
| Reaction temperature (°C) |
Ethanol conversion (wt%) |
Selectivity (%) |
| H2 |
CH4 |
CO |
CO2 |
Ethane |
Ethylene |
| 400 |
8.8 |
0.00 |
42.59 |
2.51 |
12.64 |
35.56 |
6.70 |
| 500 |
10.6 |
7.18 |
25.39 |
17.71 |
3.14 |
17.54 |
29.04 |
| 600 |
17.5 |
10.78 |
29.21 |
32.13 |
0.00 |
24.84 |
3.04 |
On the basis of the obtained products, after we systemically investigated the variation of gaseous products, it was found that the amount of H2 and CO increased while CH4 and CO2 decreased as the reaction temperature rises. The following reactions may describe the changes in gaseous products with temperatures in our reaction system.
| | | CH3CH2OH → CO + H2 + CH4 | (8) |
The first reaction (eqn (8)) is the most endothermic one and it is thermodynamically favorable,34 so the high temperature favors the production of H2 and CO. Therefore, the higher the temperature, the larger the amount of H2 and CO obtained. The reaction in eqn (9) is also a thermodynamically favorable reaction, thus the synthesized CH4 in eqn (8) will partially decomposed to produce carbon and hydrogen. This is the reason that the amount of CH4 decreased when the reaction temperature increased. The most exothermic reaction is the Boudouard reaction (eqn (10)), which is favored at low temperatures, and leads to a decrease in the amount of CO2 when increasing the reaction temperature.
3.2.2. CeO2–ZrO2 support.
During catalytic ESR, ethanol decomposes mainly via two different routes:35 either by dehydration forming ethylene, or by dehydrogenation forming acetaldehyde. These two intermediate products further catalytically decomposed and are steam reformed to an equilibrated mixture of methane, carbon dioxide, carbon monoxide, hydrogen and water (Fig. 6).
 |
| | Fig. 6 Ethanol reaction pathways. | |
When CeO2–ZrO2 was used as a catalyst for ESR, the products distinguished were mainly H2, CO, CH4, C2H6, C3H8, C2H4, CH3CHO, and CH3COCH3, as shown in Table 3. At low temperatures (200–400 °C), ethylene, acetaldehyde and acetone were produced from side reactions, such as ethanol dehydrogenation and dehydration, and acetaldehyde decomposition. A higher selectivity toward ethylene reached 61%, resulting from ethanol dehydration according to the reaction in eqn (11). Generally, ethanol dehydration to ethylene is mainly promoted by acidic sites. Carbon deposition in the form of carbon nanotubes (CNTs) (as shown in HRTEM image (Fig. 10)), results when large amounts of ethylene are produced. This was ascribed to the high acidity (741 mmole g−1) of CeO2–ZrO2 catalysts as shown in Table 1, ethylene (C2H4) was ascribed as a severe carbon precursor resulting in rapid coke formation during the reforming process (eqn (12)).
Table 3 Product distribution from ESR
| — |
Reaction temperature (°C) |
Converted ethanol (%) |
Selectivity (%) |
| H2 |
CH4 |
CO |
CO2 |
C2H6 |
C2H4 |
C3H8 |
CH3CHO |
CH3COCH3 |
| CeO2–ZrO2 |
200 |
63 |
0.00 |
29.70 |
0.00 |
35.23 |
12.48 |
0.00 |
6.48 |
0.00 |
0.00 |
| 300 |
70 |
6.35 |
0.94 |
0.00 |
31.32 |
2.69 |
38.68 |
0.94 |
2.63 |
12.29 |
| 400 |
92 |
5.08 |
3.63 |
0.88 |
13.85 |
3.22 |
61.44 |
4.17 |
7.17 |
0.37 |
| 500 |
98 |
19.90 |
4.29 |
1.30 |
11.81 |
2.57 |
47.41 |
3.21 |
6.30 |
3.05 |
| 600 |
100 |
25.88 |
23.28 |
8.31 |
22.12 |
1.73 |
17.91 |
0.20 |
0.30 |
0.10 |
| 2%Ni/CeO2–ZrO2 |
200 |
82 |
0.00 |
36.94 |
0.00 |
4.01 |
28.19 |
0.66 |
16.22 |
10.47 |
0.00 |
| 300 |
96 |
50.72 |
14.96 |
17.23 |
14.17 |
0.05 |
0.13 |
0.03 |
1.74 |
0.84 |
| 400 |
100 |
54.80 |
16.87 |
1.54 |
26.76 |
0.00 |
0.00 |
0.00 |
0.02 |
0.02 |
| 500 |
100 |
72.49 |
4.05 |
4.36 |
19.10 |
0.00 |
0.00 |
0.00 |
0.01 |
0.01 |
| 600 |
100 |
75.34 |
0.39 |
8.00 |
16.27 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 10%Ni/CeO2–ZrO2 |
200 |
98 |
37.56 |
34.80 |
15.91 |
1.29 |
0.75 |
0.09 |
0.30 |
8.11 |
0.00 |
| 300 |
98 |
40.61 |
31.90 |
0.26 |
25.60 |
0.73 |
0.01 |
0.36 |
0.30 |
0.00 |
| 400 |
100 |
56.45 |
16.60 |
1.08 |
25.87 |
0.00 |
0.00 |
0.00 |
0.01 |
0.00 |
| 500 |
100 |
70.21 |
3.16 |
3.27 |
23.35 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 600 |
100 |
70.32 |
0.64 |
5.28 |
23.76 |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 20%Ni/CeO2–ZrO2 |
200 |
96 |
36.88 |
25.33 |
29.01 |
1.11 |
0.05 |
0.05 |
0.22 |
7.13 |
0.00 |
| 300 |
96 |
40.11 |
22.54 |
25.15 |
11.97 |
0.02 |
0.00 |
0.01 |
0.14 |
0.00 |
| 400 |
100 |
54.13 |
22.35 |
2.06 |
21.36 |
0.09 |
0.00 |
0.00 |
0.01 |
0.00 |
| 500 |
100 |
67.35 |
6.72 |
5.98 |
19.87 |
0.05 |
0.00 |
0.01 |
0.00 |
0.00 |
| 600 |
100 |
71.74 |
3.13 |
8.80 |
16.29 |
0.02 |
0.00 |
0.00 |
0.00 |
0.00 |
The formation of acetone (at 300 °C) proceeds through the dehydrogenation of ethanol to acetaldehyde, the aldol condensation to 3-hydroxybutanal, its oxidation to 3-hydroxybutyrate using the oxygen provided by the catalyst, and finally, the thermal decarboxylation and dehydrogenation of this product (generally by reaction shown in eqn (13)).28 C2H4 and CH3COCH3 are highly undesired products in the ESR reaction since they are precursors of coke, leading to catalyst deactivation.
| | | 2C2H5OH + H2O → CH3COCH3 + CO2 + 4H2 | (13) |
On the other hand, ethanol could be converted to acetaldehyde (eqn (14)),36 which is sequentially decomposed and converted into syngas (CO and H2) through dehydrogenation as the temperature is raised to 600 °C. The formation of hydrogen is based on the reactions of ESR and water-gas shift (eqn (15)).
Another important product of ESR over the CeO2–ZrO2 catalyst is C3H8, the reactivity of the surface towards higher hydrocarbons was related to the tendency to form multiple bonds between one carbon atom and the surface.37 CH4 was adsorbed on the surface, forming an intermediate surface hydrocarbon species with the lattice oxygen (OOx) at the CeO2 surface, as illustrated schematically below. Using the Kroger–Vink notation, Vo·· denotes as an oxygen vacancy with an effective charge 2*, and e− is an electron which either more or less localized on a cerium ion or delocalized in a conduction band.38
CH4 adsorption
Co-reactant (H2O and CO2) adsorption
Redox reactions of lattice oxygen (Oox) with C* and O*
| | | C* + OO* → CO* + Vo·· + 2e− | (23) |
| | | Vo·· + 2e− + O* ↔ Oox + * | (24) |
Inhibitory effects of CO and H2
| | | CO* + Oo* ↔ CO2* + Vo·· + 2e− | (27) |
As the temperature increased from 400 to 600 °C, the increase in selectivity of methane, carbon monoxide and carbon dioxide keeps increasing the selectivity of hydrogen, which indicates that the predominant reactions are the reforming reaction (eqn (7)) of ethanol and water.36 On the other hand, at high temperatures, ethane and propane undergo decomposition,39 or SR 40 to produce hydrogen, methane, ethylene, and carbon (eqn (28)–(34)):
| | | C2H6 + 2H2O → 5H2 + 2CO | (30) |
| | | C3H8 + 3H2O → 3CO + 7H2 | (33) |
| | | C3H8 → 2C + CH4 + 2H2 (dehydrogenation) | (34) |
3.2.3. Metal loading.
For all x%Ni/CeO2–ZrO2 catalysts, the main reaction products were H2, CO, CO2 and CH4. With increasing temperature, the selectivity to H2 and CO2 increased, where the selectivity to CH4 passed through a maxima and the selectivity to CO passed through a minima. On the other hand, selectivity to ethylene, acetaldehyde and acetone formation was negligible (Table 3).
At 200 °C; only the 10 and 20 wt% catalyst gave H2. These unexpected results clearly show evidence the there are higher amounts of reduced nickel available for reaction, as illustrated in the TPR analysis, and is the main factor in ESR for hydrogen production. On the other hand, Hernandez et al.,41 suggested that nickel is the phase which is mainly responsible for hydrogen production, although the ZrO2 support reduces CO formation. This data is a totally different picture compared to the results obtained in earlier studies on ESR, which stated that temperatures higher than 230 °C are necessary to carry out the ESR reaction. In a similar study, Duhamel, et al.42 investigated hydrogen production from ethanol in the presence of water (H2O/C2H5OH = 3) over cerium–nickel CeNixOy (0< x ≤ 5) mixed oxide catalysts. The influence of different reaction temperatures showed that a stable activity, i.e., ethanol conversion and H2 selectivity, was obtained at a very low temperature (200 °C) when the solid is previously in situ treated in H2, in a temperature range between 200 and 300 °C. After such a treatment, the solids studied are hydrogen reservoirs, called oxyhydrides, with the presence of a hydrogen species of hydride nature in the anionic vacancies of the solid.42 David et al.43 indicated that reduction experiments under SR conditions, in the presence of both H2 and hydrocarbons, have shown an increased reducibility of nickel ions in the presence of hydrocarbons and alcohol.
On the other hand, from the analysis of product distribution obtained at 200 °C, it can be observed that methane selectivity is equal to CO selectivity, suggesting that ethanol decomposition to CO, CH4 and H2 (eqn (8)) is the main reaction at 200 °C, while the lower selectivity to CO2 could be produced either by the WGS reaction (eqn (15)) or by ESR (eqn (7)). As the temperature increases from 200 to 300 °C, CH4 and CO2 selectivity increases while CO selectivity decreases. This means that at 300 °C the ESR (eqn (7)) instead ethanol decomposition (eqn (8)) becomes significant and low quantities of CO are produced by the reaction in eqn (35).
The selectivity to hydrogen obtained for the 10 and 20 wt% catalysts was comparable, as shown in Fig. 7, from this figure we can also observe that at lower reaction temperatures (< 400 °C) the 10%Ni/CeO2–ZrO2 is the most selective catalyst toward hydrogen production (H2 selectivity is 37% at 200 °C). Whereas, at higher reaction temperatures (> 400 °C), with an increase in metal loading, the selectivity of hydrogen substantially decreased. 2%Ni/CeO2–ZrO2 is the most selective catalyst towards hydrogen production (H2 selectivity is 75% at 600 °C), because it has a high specific surface area and large pore volumes (Table 1). Also, the small particle size of nickel in 2% Ni loading catalyst results in enhanced metal–support interactions, which accounts for the high activity of the catalyst.44
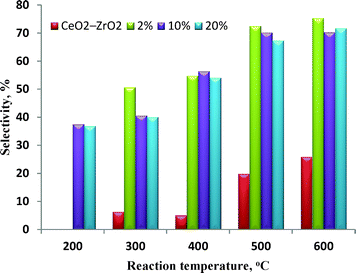 |
| | Fig. 7 Variation of H2 selectivity with temperature over x%Ni/CeO2–ZrO2 catalysts. | |
The presence of strong metal–support interactions (SMSIs) would prevent the nickel metal from sintering at moderate temperatures, thus increasing the dispersion of nickel and the length of the interfacial boundaries between Ni and the support. The presence of nickel with a small particle size guarantees high activity of the catalyst by its high J activity in C–H and C–C bond cleavages, while the abundant interfacial boundary between nickel and support facilitates oxidization of the carbon-containing intermediates to COx or other oxygenates by the active oxygen species on the surface of the support.44 This has also been demonstrated in our study. Whereas, the XRD pattern of the 2%Ni/CeO2–ZrO2 catalyst did not show a significant nickel peak; which encourages the strong metal–support interaction. HRTEM shows high nickel dispersion (Fig. 3). This was also established by the TPR data, where the reduction property of the 2%Ni/CeO2–ZrO2 catalyst seems to cause the peak corresponding to CeO2–ZrO2 to shift downward, and thus, overlaps the NiO reduction peak at about 460 °C. This indicates that a strong interaction between Ni and CeO2–ZrO2 or Ni incorporation into CeO2–ZrO2 makes ceria more reducible, which helps to produce mobile oxygen during the reforming reaction. The participation of the lattice oxygen would be subsequently supplemented with the oxygen from the water molecules and lead to high activity in SR reaction.45 As reported by Arias et al.,46 for ceria's ability to store and release oxygen is a crucial feature in its superior performance. Because oxygen desorption from ceria occurs at very high temperatures,47 oxygen-storage requires contact between ceria and a precious metal, with the reduction of ceria occurring through reaction with a reductant on the precious metal.48
For all catalysts, when the temperature is increased from 500 to 600 °C, methane selectivity decreases, carbon dioxide selectivity remains constant while hydrogen and carbon monoxide selectivity increase. CO produced from the reverse water gas shift reaction (WGSR) was expected (eqn (36)) to increase with increases in temperature.49 The decrease in CO2 and CH4 yields with increased temperature was ascribed to the RWGS, SR of CH4 (eqn (35)) and/or dry reforming, resulting in increase in the selectivity to H2 and CO2.50
| | | CH4 + CO2 → 2CO + 2H2 | (37) |
On the basis of intermediate distribution at lower and higher reaction temperatures (Table 3), we can conclude that ethanol dehydrogenation to acetaldehyde (eqn (14)) and ethanol decomposition to methane and carbon monoxide (eqn (8)) were the primary reactions at low temperatures. At higher temperatures, where all the ethanol and the intermediate compounds, like acetaldehyde and acetone (eqn (38) and (39)), were completely converted into hydrogen, carbon oxides and methane, SR of methane and WGS became the major reactions, which is in agreement with Zhang et al.51
| | | CH3CHO + H2O → 2CO + 3H2 | (39) |
A bi-functional mechanism for ESR was proposed by Aupretre et al.,52 as shown in Fig. 8. According to this mechanism, ethanol may be activated on the metal particle or on the support acidic and basic sites to produce the intermediate products (step 1). Acetaldehyde is produced on both the support (basic sites) and the metal. Ethylene is only produced on the support, with an essential role of acidic sites isn olefin formation. The water molecules are adsorbed on the support in the form of mobile hydroxyl groups (step 2). The support would promote the migration of these OH groups towards the metal particle (step 3). As a consequence, hydrophilic supports, with high oxygen mobility, are expected to act as promoters in SR reactions.53 Furthermore, the mobile oxygen species supplied by the support may participate in SR reactions and prevent the formation of coke on the metal particles.54 Ethanol dehydration to ethylene is mainly promoted by the acidic sites. Coke deposition results in deactivation of the catalysts when large amounts of ethylene are produced. On the other hand, ethanol could be converted to acetaldehyde, which is sequentially decomposed and converted into syngas through dehydrogenation. Two different mechanisms have been suggested for the dehydrogenation of alcohol: (i) the “carbonyl” mechanism, in which the hydrogen from the hydroxyl group and from the α-carbon atom are split, either simultaneously or in two steps; (ii) the “enolic” mechanism, which involves splitting of the β-hydrogen.55 While it is well documented that splitting of the α-hydrogen with formation of surface alkoxide is the rate determining step on oxide catalysts,55 little information is available on ethanol dehydrogenation on metal catalysts.
 |
| | Fig. 8 Mechanism of ethanol steam reforming.52 | |
3.3. Characterization of used catalyst
To gain some insight into the state of the catalyst after the ESR reaction, the XRD and HRTEM analysis of CeO2–ZrO2 and 10%Ni/CeO2–ZrO2 catalysts after 8 h of ESR reaction were carried out. It should be mentioned that the XRD and HRTEM of the fresh catalyst were taken after calcination, whereas for the used catalysts these analyses were done after in situ reduction followed by the reforming reaction. The CeO2–ZrO2 patterns (Fig. 9) reveal the presence of carbon nanotubes, as evidenced by reflections [(100) and (004)] at 43° and 53°, respectively [ref. JCPDS card (00-058-1638)]. Other reflections (002), at 26.2° is associated with carbon nanotube structures that are not clearly evidenced because they overlap with the reflection assigned to the CeO2–ZrO2 solid solution. The absence of low angle reflections indicates the formation of thick MWNTs.56 This matter will be pursued by HRTEM examination (Fig. 10). The HRTEM micrograph of this sample reveals that the deposited carbon appears either as large filaments with catalyst particles, or as a rather uniform carbon coating covering the catalyst particles.
 |
| | Fig. 9 XRD pattern of spent catalyst. | |
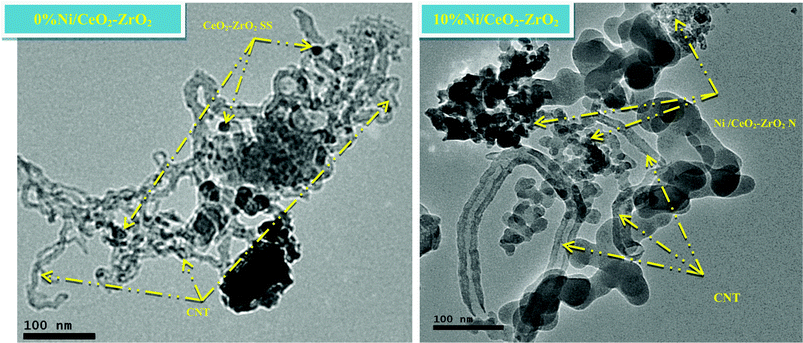 |
| | Fig. 10 HRTEM image of spent catalyst after 8 h of ESR reaction. | |
From the XRD of the 10%Ni/CeO2–ZrO2 catalyst (Fig. 9), we observed that all peaks corresponding to NiO completely disappeared and other reflections at 44.5° and 51.8° appeared, which were assigned to planes (111) and (200) respectively, of metallic Ni. Whereas the peaks corresponding to the CeO2–ZrO2 mixed oxide were almost unaffected. Although, no separate carbon nanotube phases were discernible by XRD of the used 10%Ni/CeO2–ZrO2 catalyst, the HRTEM image (Fig. 10) identified CNTs deposited within the catalyst matrix. The CNT reflections overlap with other reflections assigned to Ni-containing species also present in these samples. The center of the filaments in Fig. 10 seem hollow, which suggests that they are multiwalled nanotubes. Deactivation likely occurs because the active Ni nanoparticles become increasingly encapsulated by graphitic carbon.57 Suelves et al.56 suggested that the catalyst becomes deactivated when deposited carbon prevents reactant diffusion toward the Ni surface, and diffusion is prevented in higher extents by carbon-coating than by filaments.
4. Conclusion
The catalytic performance of a nickel supported ceria-zirconia catalyst on the ESR reaction was investigated at different temperatures and different nickel loadings to understand the role of the active metal and support oxides in the reaction pathways. The thermal decomposition of ethanol in the absence of catalyst produced low amounts of CO2, which reached zero at 600 °C, indicating the absence of the SR reaction. For all catalysts, the major products formed include H2, CH4, CO and CO2 at higher temperatures (400–600 °C) and intermediate compounds CH3CHO, C2H4, C2H6 and CH3COCH3 appear at lower temperatures (200–300 °C). Compared to nickel support catalysts, the ceria-zirconia support shows a significant amount of activity, and reaches 61% of ethylene, this is due to the high acidity of the support (740.8 mmole g−1). The main reaction route of dehydrogenation of ethanol, followed by decomposition of acetaldehyde, and subsequently the competition of SRM and WGSR, were proposed for ESR. Surprisingly, while the 10%Ni/CeO2–ZrO2 and 20%Ni/CeO2–ZrO2 catalysts showed a considerable amount of H2 at a very low temperature (200 °C), the 2%Ni/CeO2–ZrO2 catalyst showed high selectivity to H2 at 600 °C. XRD and HRTEM images indicated that the interaction between the nickel metal and the support is greatly strengthened by the small nickel particle size in the 2%Ni/CeO2–ZrO2 catalyst, which accounts for its high selectivity. Characterization of the spent catalyst shows that higher amounts of carbon deposition on the CeO2–ZrO2 support was observed, as shown by HRTEM. The deposited carbon appears as either large filaments with catalyst particles, or as a rather uniform carbon-coating that covers the catalyst particles.
References
- S. M. de Lima, R. C. Colman, G. Jacobs, B. H. Davis, K. R. Souza, A. F. F. de Lima, L. G. Appel, L. V. Mattos and F. B. Noronha, Catal. Today, 2009, 146, 110 CrossRef CAS.
- N. Kiratzis, P. Holtappels, C. E. Hatchwell, M. Mogensen and J. T. S. Irvine, Fuel Cells, 2001, 1, 211 CrossRef CAS.
- H. Muroyama, R. Nakase, T. Matsui and K. Eguchi, Int. J. Hydrogen Energy, 2010, 35, 1575 CrossRef CAS.
- J. Kugai, S. Velu and C. S. Song, Catal. Lett., 2005, 101, 255 CrossRef CAS.
- Ni. Meng, Y. C. L. Dennis and K. H. L. Michael, Int. J. Hydrogen Energy, 2007, 32, 3238 CrossRef.
- P. D. Vaidya and A. E. Rodrigues, Chem. Eng. J., 2006, 117, 39 CrossRef CAS.
- P. Biswas and D. Kunzru, Chem. Eng. J., 2008, 136, 41 CrossRef CAS.
- K. Otsuka and T. I. Y. Ushiyama, Chem. Lett., 1993, 15, 17 Search PubMed.
- R. Si, Y. W. Zhang, S. J. Li, B. X. Lin and C. H. Yan, J. Phys. Chem. B, 2004, 108, 12481 CrossRef CAS.
- R. D. Monte and J. Kaspar, J. Mater. Chem., 2005, 15, 633 RSC.
- H. Seog, I. H. Eum and D. W. Jeong, Renewable Energy, 2012, 42, 212–216 CrossRef.
- D. Srinivas, C. V. V. Satyanarayana, H. S. Potdar and P. Ratnasamy, Appl. Catal., A, 2003, 246, 323 CrossRef CAS.
- A. M. Arias, M. F. Garcia, L. N. Salamanca, R. X. Valenzuela, J. C. Conesa and J. Soria, J. Phys. Chem. B, 2000, 104, 4038 CrossRef.
- G. S. Jones, M. Mavrikakis, M. A. Barteau and J. M. Vohs, J. Am. Chem. Soc., 1998, 120, 3196 CrossRef CAS.
- A. Trovarelli, F. Zamar, J. Llorca, C. Leitenburg, G. Dolcetti and J. T. Kiss, J. Catal., 1997, 169, 490 CrossRef CAS.
- P. Kumar, S. Yanping and R. O. Idem, Energy Fuels, 2007, 21, 3113 CrossRef CAS.
- W. Shan, M. Luo, P. Ying, W. Shen and C. Li, Appl. Catal., A, 2003, 246, 1 CrossRef CAS.
- B. M. Reddy and A. Khan, Catal. Surv. Asia, 2005, 9, 155 CrossRef CAS.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 11475 CrossRef CAS.
- T. Zhu and M. F. Stephanopoulos, Appl. Catal., A, 2001, 208, 403 CrossRef CAS.
- F. Tomul and S. Balci, Appl. Clay Sci., 2009, 43, 13 CrossRef CAS.
- G. Leofanti, M. Padovan, G. Tozzola and B. Venturelli, Catal. Today, 1998, 41, 207 CrossRef CAS.
- C. G. Alonso, A. C. Furtado, M. P. Cantao, O. A. A. dos Santos and N. R. C. Fernandes-Machado, Int. J. Hydrogen Energy, 2009, 34, 3333 CrossRef CAS.
- S. C. Dantas, J. C. Escritori, R. R. Soares and C. E. Hori, Chem. Eng. J., 2010, 156, 380 CrossRef CAS.
- P. Biswas and D. Kunzru, Int. J. Hydrogen Energy, 2007, 32, 969 CrossRef CAS.
- N. Laosiripojana and S. Assabumrungrat, Appl. Catal., A, 2005, 290, 200 CrossRef CAS.
- P. K. K. Pantu and G. R. Gavalas, Appl. Catal., A, 2000, 193, 203 CrossRef CAS.
- C. Liang, Z. Ma, H. Lin, L. Ding, J. Qiu, W. Frandsen and D. Su, J. Mater. Chem., 2009, 19, 1417 RSC.
- P. Kumar, Y. Sun and R. O. Idem, Energy Fuels, 2008, 22, 3575 CrossRef CAS.
- Y. Yang, W. Li and H. Xu, React. Kinet. Catal. Lett., 2002, 77, 155 CrossRef CAS.
- H. Roh, K. Jun, W. Dong, J. Chang, S. Park and Y. Joe, J. Mol. Catal. A: Chem., 2002, 181, 13 CrossRef.
- T. Takeguchi, S. Furukawa and M. Inoue, J. Catal., 2001, 202, 14 CrossRef CAS.
- R. Padilla, M. Benito, L. Rodrıguez, A. Serrano, G. Munoz and L. Daza, Int. J. Hydrogen Energy, 2010, 35, 8921 CrossRef CAS.
- G. Wang, H. Wang, W. Lib and J. Bai, RSC Adv., 2011, 1, 1585 RSC.
- J. R. Hansen, C. H. Christensen, J. Sehested, S. Helveg, J. R. R. Nielsenb and S. Dahl, Green Chem., 2007, 9, 1016 RSC.
- J. Suna, X. P. Qiua, F. Wuc and W. T. Zhua, Int. J. Hydrogen Energy, 2005, 30, 437 CrossRef.
- E. H. Broekhoven and V. Ponec, Prog. Surf. Sci., 1985, 19, 351 CrossRef.
- N. Laosiripojana and S. Assabumrungrat, Appl. Catal., B, 2008, 82, 103 CrossRef CAS.
-
M.V. Twigg, Catalyst Handbook, Wolfe Publishing, London, 2nd edn, 1989, p. 253 Search PubMed.
- P. O. Graf, B. L. Mojet, J. G. Ommen and L. Lefferts, Appl. Catal., A, 2007, 332, 310 CrossRef CAS.
- R. P. Hernandez, A. G. Martınez, J. Palacios, M. V. Hernandez and V. R. Lugo, Int. J. Hydrogen Energy, 2011, 36, 6601 CrossRef.
- L. J. Duhamel, C. Pirez, M. Capron, F. Dumeignil and E. Payen, Catal. Today, 2010, 157, 456 CrossRef.
-
B. Zhou, S. Han, R. Raja and G. A. Somorjai, Nanotechnology in catalysis, Springer, Dordrecht, The Netherlands, 2007, vol. 3, p. 194 Search PubMed.
- H. V. Fajardo and L. F. D. Probst, Appl. Catal., A, 2006, 306, 134 CrossRef CAS.
- W. H. Dong, H. S. Roh, K. W. Jun, S. E. Park and Y. S. Oh, Appl. Catal., A, 2002, 226, 63 CrossRef CAS.
- A. M. Arias, M. F. Garcia, C. Belver, J. C. Conesa and J. Soria, Catal. Lett., 2000, 65, 197 CrossRef.
- M. Mavrikakis, D. J. Doren and M. A. Barteau, J. Phys. Chem. B, 1998, 102, 394 CrossRef CAS.
- L. J. Duhamel, Int. J. Hydrogen Energy, 2006, 31, 191 CrossRef.
- A. M. Karim, Y. Su, J. Sun, C. Yang, J. J. Strohm and D. L. King, Appl. Catal., B, 2010, 96, 441 CrossRef CAS.
- D. K. Liguras, D. I. Kondarides and X. E. Verykios, Appl. Catal., B, 2003, 43, 345 CrossRef CAS.
- B. Zhang, X. Tang, Y. Li, Y. Xu and W. Shen, Int. J. Hydrogen Energy, 2007, 32, 2367 CrossRef CAS.
- F. Aupretre, C. Descorme and D. Duprez, Ann. Chim. Sci. Mater., 2001, 26, 93 CrossRef CAS.
- L. S. F. Feio, C. E. Hori, S. Damyanova, F. B. Noronha, W. H. Cassinelli, C. M. P. Marques and J. M. C. Bueno, Appl. Catal., A, 2007, 316, 107 CrossRef CAS.
- Y. Wang, Y. H. Chin, R. T. Rozmiarek, B. R. Johnson, Y. Gao, J. Watson, A. Y. L. Tonkovich and D. P. Van der Wiel, Catal. Today, 2004, 98, 575 CrossRef CAS.
-
G. Ertl, H. Knozinger and J. Weitkamp, Handbook of Heterogeneous Catalysis, VCH, Weinheim-D, 1997, vol. 5, p. 2159 Search PubMed.
- I. Suelves, M. J. Lázaro, R. Moliner, B. M. Corbella and J. M. Palacios, Int. J. Hydrogen Energy, 2005, 30, 1555 CrossRef CAS.
- W. Shen, F. E. Huggins, N. Shah, G. Jacobs, Y. Wang, X. Shi and G. P. Huffman, Appl. Catal., A, 2008, 351, 102 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.