Recent progress in photofunctional lanthanide hybrid materials
Bing
Yan
*
Department of Chemistry, Tongji University, Shanghai 200092, China. E-mail: byan@tongji.edu.cn; Fax: +86-21-65982287; Tel: +86-21-65984663
First published on 18th July 2012
Abstract
This critical review mainly focuses on recent research progress in photofunctional lanthanide hybrid materials. The review covers hybrids with complexes of organically modified silanes as precursors for sol–gel processing, hybrids with lanthanide complex units grafted onto the interior of mesoporous hosts, hybrids with lanthanide complex units on polymer chains, and other non-silica or composite-based lanthanide hybrids. The photophysical properties of lanthanide hybrids are also discussed. Finally, future challenges and opportunities in this field are discussed.
 Bing Yan | Prof. Bing Yan received his BSc and MSc in Applied Chemistry from Harbin Institute of Technology in 1992 and 1995, respectively. Then he studied at Changchun Institute of Applied Chemistry, CAS under the direction of Prof. Jia-Zuan Ni and Prof. Hong-Jie Zhang, where he obtained his Ph.D. in Inorganic Chemistry in 1998. In 1998-2001, he came to City University of Hong Kong to be a research assistant and then became PostD fellow in Peking University and University of Sherebrook. At the end of 2001, he joined Tongji University as a professor. His current research interests are focused on rare earth chemistry and functional materials science, involving rare earth inorganic/organic/polymeric hybrid materials and functional rare earth compounds or rare earth submicro or nano solids. At present, he has published over 300 scientific papers as corresponding author in international journals. |
1. Introduction
Trivalent lanthanide ions have excellent photoactive properties such as sharp emission spectra for high color purity, broad emission bands covering the ultraviolet (UV)–visible–near infrared (NIR) region, a wide range of lifetimes from the microseconds to seconds level, and high luminescence quantum efficiencies. These properties have attracted much attention for a wide variety of applications in the fields of lighting devices (television and computer displays, optical fibers, optical amplifiers, lasers), and biomedical analysis (medical diagnosis and cell imaging).1,2 However, it is difficult to utilise lanthanide ions directly as luminescent materials because of their poor light absorption abilities. As is known, the f–f transitions of lanthanide ions are spin forbidden, so it is hard to generate efficient luminescence emission by direct excitation. The traditional doping of lanthanide ions into special matrices such as metallic oxides and oxy salts3 is an efficient method of obtaining excellent luminescence emission. On the other hand, one can design lanthanide complexes comprising of sensitizing ligands such as β-diketonates, aromatic carboxylates and heterocyclic ligands duo to their excellent coordination ability and appropriate energy matching. Unfortunately, the practical application of lanthanide complexes is still limited to a certain extent by their poor stability under high temperature, high pressure or moisture conditions, and their low mechanical strength.4Therefore, in order to improve the photostability of lanthanide complexes, the new concept of “organic–inorganic hybrid materials” provides photofunctional lanthanide materials that have the potential for new developments.5 The combination of organic and inorganic components at a molecular or nanometer scale integrates certain strong points of organic compounds (easy processing with conventional techniques, elasticity, and organic functionalities) with the advantages of inorganic components (hardness, thermal and chemical stabilities). According to the different interaction forces between the two phases, organic–inorganic hybrid materials are divided into two major classes. Physically doped hybrids have weak interactions (such as hydrogen bonding, van der Waals forces, or weak static effects) between the organic and inorganic phases,5 and these cannot overcome many problems such as clustering of emitters, inhomogeneous dispersion of the components, or leaching of the dopants. Chemically bonded hybrids have strong interactions such as covalent, ion–covalent, coordination, or Lewis acid/base bonds,6 and these can graft the lanthanide complexes onto sol–gel-derived hosts.7 The latter class of hybrids overcome the disadvantages of the former ones, since they are homogeneous and avoid the self-quenching of lanthanide ions, which results in increased incorporation concentrations. Luminescent lanthanide hybrid materials based on organically modified silanes (ORMOSILs) have therefore been widely developed. Carlos et al. have reviewed the topic8 based on their outstanding contribution to this field.9–13 In their review, they summarized four design strategies for lanthanide hybrid materials: conventional sol–gel technology (A), functionalized nano building-block assembly (B), template-directed assembly (C), and self-directed assembly (D). They discussed the spectroscopic principles and methods currently used to study the photophysical properties of these hybrids. Binnemans has also published an extensive review,14 first outlining the luminescence properties of lanthanide complexes, then giving a detailed introduction to typical hybrid systems such as sol–gel hybrids, porous hybrids, intercalation compounds, polyoxometalates, polymer hybrids, hybrid gels, and nanocomposites, and finally discussing the applications of photoluminescent lanthanide hybrids. Zhang's group and our research team have also done extensive work on covalently grafting ligands onto inorganic networks, in which the lanthanide complex luminescence centers are bonded to a siloxane matrix through Si–O linkages.16–30 More recently, Carlos recently reviewed the potential application of hybrid materials,31 giving an overview of major innovations in the use of lanthanide hybrids in the areas of phosphors, lighting, integrated optics and optical telecommunications, solar cells, and biomedicine.
In this review, we focus our attention on hybrids with ORMOSIL precursors for sol–gel processing, hybrids with lanthanide complex units grafted onto the interior of mesoporous materials or onto polymer chains, and other lanthanide hybrids based on non-silica or mixed matrices. We also discuss the photophysical properties of lanthanide hybrids. We hope that this review can objectively reflect the new and real progress in photofunctional lanthanide hybrid materials, especially the work undertaken since 2009.
2. Recent progress in research on lanthanide hybrid materials
2.1 Hybrids prepared using complexes with ligands featuring ORMOSILs as precursors for sol–gel processing
Previous research has shown that the key procedure in constructing these chemically bonded hybrid materials is designing the functional bridging molecule (ligand) by grafting; the bridging molecule behaves as a chemical linkage with the functions of coordinating or sensitizing lanthanide ions, and forming covalent Si–O networks.9–15,16,17,22–30 The photoactive groups of these bridging molecules are also important in photofunctional hybrids. Simple covalently linked bridging molecules are organic ligands with multifunctional groups such as nitrogen-heterocycle-conjugated carboxylates.32 More general bridging molecules, the ORMOSILs, need to be synthesized using modification reactions. The basic strategy is to graft a typical photoactive organic ligand for lanthanide ions onto a special functional silane crosslinking reagent. Many types of organic ligands have been functionalized and grafted onto silanes in order to achieve various molecular bridges (ORMOSILs); the modification paths can be summarized on the basis of different functional group reactions.16,22–30,33–39Some studies using amino group derivatives have been reported.16,17,22,23 Zhang's group initiated a study of linkages derived from bipyridine derivatives and developed a series of hybrid materials.16,17 Our group first focused on aminoaromatic carboxylic acid ligands22,23 and we are now trying to extend our work to other amino compounds33 such as aminoacridines, aminopyridines, 2,2′-bipyridylamine, thioacetamide, 2-amino-5-phenylthiazole, and aza-crown ethers. Here we give two examples to illustrate the detailed modification route.33d,33e The first example is 2-amino-5(4)-phenylthiazole, which has reactive hydrogen atoms and can be expected to realize hydrogen-transfer reactions with the silane crosslinking reagent 3-(triethoxysilyl)propyl isocyanate (TESPIC). The obtained silylated linkages (ORMOSILs) can be used as siloxane network precursors to construct lanthanide hybrids (Fig. 1).33d
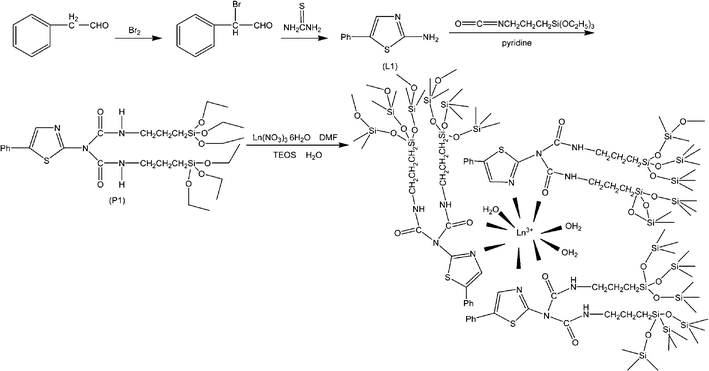 | ||
| Fig. 1 Scheme for synthesis of 2-amino-5-phenylthiazole and ORMOSILs, and the predicted composition of the corresponding lanthanide hybrid material. Reprinted with permission from ref. 33d. Copyright 2010, Wiley Publishing Company. | ||
Another example is the synthesis of three kinds of aza-crown ethers with rings containing 15, 16 and 18 atoms; these react with TESPIC to produce the functional molecular precursors (ORMOSILs) (Fig. 2).33e
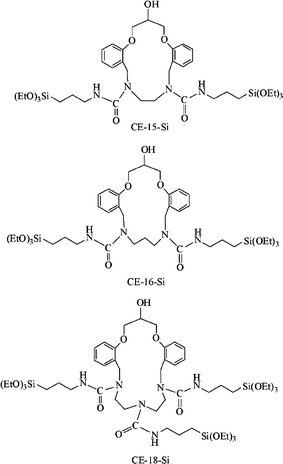 | ||
| Fig. 2 Chemical structures of silylated precursors containing aza-crown ether rings. Reprinted with permission from ref. 33e. Copyright 2011, Royal Society of Chemistry. | ||
There are many reports on the modification of the carboxylate groups of aromatic carboxylic acids, through two pathways; one pathway is to convert the carboxylate group to an acyl chloride and then to an amide derivative 24–27 through the amide reaction between an amino group crosslinking a silane and acyl chloride, as described in Liu's reports.16c In our work, we have found that the carboxylic groups of p-aminobenzoic acid (not the amino moiety) can be attacked by TESPIC and there is intense competition between the amino and carboxylic groups. The results reveal that the latter participate more actively than the former in the reaction and NMR and IR spectra confirm that the structure is totally different from that reported in ref 16c. It is therefore considered that the hydrogen atom of the p-aminobenzoic acid is more reactive than that of the amino group and that its acidity is a result of polarization by the carbonyl group and resonance stabilization of the resulting carboxylate.26a
With respect to modification of amino compounds with TESPIC, we predict that a similar hydrogen-transfer addition reaction could also occur between the hydroxyl group and TESPIC to achieve ORMOSILs.28,29,34 This method is suitable for all kinds of hydroxyl group compounds such as carboxylic acids,28 phenols29 and calixarene derivatives.34b,c For example, with regard to the attractive cavity in calixarene skeletons, the phenolic groups at the lower rim of the cup in calix[4]arene can be modified with TESPIC. We have found that the hydroxyl groups of the macrocyclic compound p-tert-butylcalix[4]arene can be converted into urethanesil (–NH(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O–)-grafted bridges, and we therefore attempted to modify the calixarene derivatives using TESPIC in molar ratios of 1
O)O–)-grafted bridges, and we therefore attempted to modify the calixarene derivatives using TESPIC in molar ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and 1:1;34b,c and found the reaction with 1:1 molar ratio is easiest to control (Fig. 3).34c
:3 and 1:1;34b,c and found the reaction with 1:1 molar ratio is easiest to control (Fig. 3).34c
![Typical preparation procedures of calix[4]arene-derived ORMOSILs and the corresponding lanthanide hybrids. Reprinted with permission from ref. 34c. Copyright 2008, American Chemical Society.](/image/article/2012/RA/c2ra20976d/c2ra20976d-f3.gif) | ||
| Fig. 3 Typical preparation procedures of calix[4]arene-derived ORMOSILs and the corresponding lanthanide hybrids. Reprinted with permission from ref. 34c. Copyright 2008, American Chemical Society. | ||
Our other work based on hydroxyl group modification deals with the synthesis of organic compounds such as 1,3-bis(2-formylphenoxy)-2-propanol (BFPP) containing two formylphenoxy groups, and then grafting to TESPIC to achieve a molecular precursor (ORMOSIL) BFPP-Si through an addition reaction between the hydroxyl group of BFPP and the isocyanate group of TESPIC (Fig. 4).34a
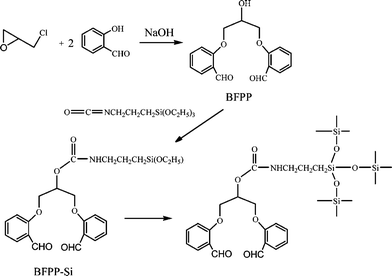 | ||
| Fig. 4 Scheme for synthesis of 1,3-bis(2-formylphenoxy)-2-propanol (BFPP) and ORMOSILs (BFPP-Si). Reprinted with permission from ref. 34a. Copyright 2009, American Chemical Society. | ||
As sulfur and oxygen are in the same group of the periodic table, hydrogen-transfer addition reactions can also occur between sulfide groups and the crosslinking silanes (3-chloropropyltrimethoxysilane (CPTMS), 3-aminopropyltrimethoxysilane (APS), 3-methacryloyloxypropyltrimethoxysilane, 3-glycidoxypropyltrimethoxysilane and TESPIC) (Fig. 5).37a
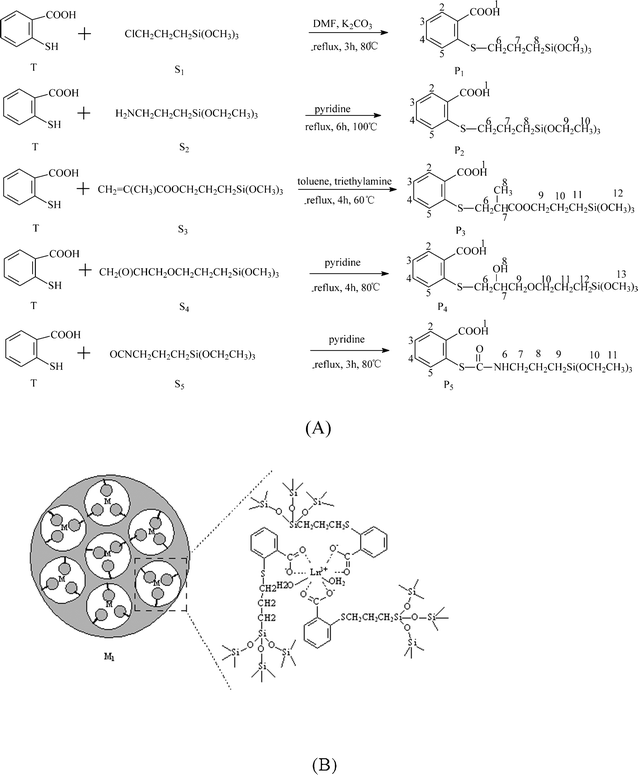 | ||
| Fig. 5 Scheme for synthesis of five ORMOSILs from the reactions of thiosalicylic acid with five silane crosslinking reagents (A) and the corresponding lanthanide hybrids (B). Reprinted with permission from ref. 37a. Copyright 2008, American Chemical Society. | ||
The above path can be extended to other sulfide compounds,37 thus we used a triazole heterocycle, 3-alkyl-4-amino-5-ylsulfanyl-1,2,4-triazole, as a precursor; this involves two reactions. The first step is to synthesize thiocarbohydrazide through the reaction of hydrazine hydrate and CS2. The second step is to prepare the linkage through the reaction between thiocarbohydrazide and acetic acid or propionic acid. The mercapto groups of the organic compounds also possess reactive hydrogen atoms and can realize hydrogen-transfer reactions with TESPIC to obtain silylated monomers (ORMOSILs) (Fig. 6).37e
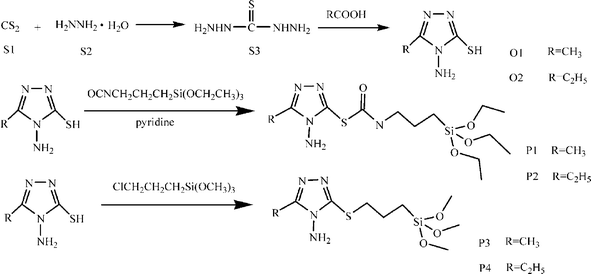 | ||
| Fig. 6 Scheme for synthesis of thiocarbohydrazide organic components and silylated precursors. Reprinted with permission from ref. 37e. Copyright 2008, American Chemical Society. | ||
Except for the aromatic ligand discussed above, β-diketonate ligands, especially thenoylmethyltrifluoroacetylacetonate (TTA), are the most important sensitizers for lanthanide ions, especially for Eu3+, through effective intramolecular energy transfer in their complexes. We first select a partly acidic β-diketonate and consider that its α-hydrogen (the hydrogen atom in the methylene) can be extracted by a base because of the polarization of C–H bonds by the adjacent carbonyl groups. Unlike our previous aminocarboxyl or hydroxyl moiety studies, we modify the active methylene group of the β-diketonate using TESPIC. Besides, we perform the single modification of one hydrogen atom in the methylene group, which is to decrease the steric hindrance effect of the large silylated groups and to maintain the unsymmetric structure to obtain effective photoactivity.38 This modification path can also be extended to lanthanide hybrids with sulfoxide-derived linkages (ORMOSILs), which have similar flexibilities and structures to those of β-diketonate linkages; these may provide good chelating groups for coordination, effectively sensitizing the luminescence of lanthanide ions. Three novel sulfoxide functional bridging molecules (ORMOSILs) have been prepared by modification of the methylene groups in sulfoxide ligands with TESPIC (Fig. 7).38d
 | ||
| Fig. 7 Schemes for synthesis of sulfoxide-derived ORMOSILs and the corresponding lanthanide hybrids. Reprinted with permission from ref. 38d. Copyright 2011, Royal Society of Chemistry. | ||
Sulfonamide linkages are selected for linking inorganic and organic parts, since these linkers are stable under acidic and basic conditions and can be converted to sulfonic chlorides. A novel sulfonamide linkage has been constructed, based on the modification of 5-sulfosalicylic acid (SSA) with 3-aminopropylmethyldiethoxylsilane or APS, because sulfosalicylic acid has a reactive functionalized sulfo group and an excellent photoactive unit (Fig. 8).36b The above modification path can be used for the modification of many kinds of typical ligands for coordination to lanthanide ions and further construction of the corresponding hybrid materials. Work is mainly focused on ORMOSIL-derived hybrids, possibly because versatile silane crosslinking reagents are easily modified with organic ligands.16,17,33–38. The hybridization of TEOS (tetraethylorthosilicate) and ORMOSILs involves a sol–gel reaction between the terminal silanol groups of ORMOSILs and the hydroxyl groups of hydrolyzed TEOS. At the beginning of the hybridization reaction, the individual hydrolyses of ORMOSILs and TEOS are predominant, but then the predominant process becomes polycondensation reactions between the hydroxyl groups of both ORMOSILs and TEOS.
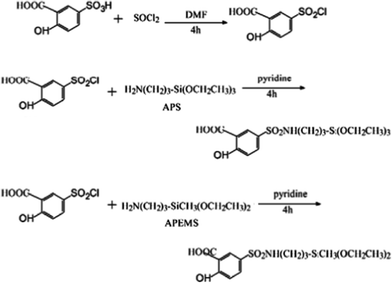 | ||
| Fig. 8 Schemes for synthesis of sulfonamide-derived ORMOSILs. Reprinted with permission from ref. 36b. Copyright 2010, Elsevier Ltd. | ||
Zhang's group has done a lot of work on hybrid materials containing ORMOSILs derived from pyridine derivatives (such as phen or bipy).16,17,39,40 The pyridine derivatives were modified using two paths. One is oxidation to the corresponding bipyridylcarboxylic acid derivatives, followed by modification of the carboxylic groups, for the construction of lanthanide hybrid materials through coordination between Ln3+ and the nitrogen atoms of the pyridine rings. The other path consists of modifying the amino-derived phen molecule with TESPIC through an addition reaction. The hybrid materials were assembled after coordination between the nitrogen atoms of the ORMOSILs and Ln3+ by a sol–gel process. The hybrid materials can be prepared as thin films by dip coating. In addition, because of their strong nucleophilic abilities, the two amino groups of 2,6-diaminopyridine were modified by TESPIC, and the two derived carbonyl groups and the pyridine nitrogen atom provided three coordination locations for lanthanide ions. Recently, Zhang's group reported a ternary europium monophase hybrid system with a bis-silylated bipy ligand derived from 4,4′-diamino-2,2′-bipyridine and three TTA ligands; this system has a much longer lifetime than that of the pure Eu complex of TTA. A transparent and luminescent ionogel monolith was prepared by hydrolysis and condensation of the silylated bipyridine; it can sensitize the luminescence of Eu3+ in the presence of a carboxyl-functionalized ionic liquid in which Eu3+ is coordinated to the oxygen atoms of carboxylate groups from the ionic liquid.40
In our research group, we used aromatic carboxylic acids with different functional groups as precursors in the design of ORMOSILs for assembling hybrids.22–29,33 Two paths were used for the direct modification of carboxylic groups, but neither of them can maintain coordination of the carboxylate group of the carboxylic acid molecule. In order to keep the carboxylate group as the Ln3+-coordinating group, we modified the substituent groups of carboxylate derivatives, such as amino, hydroxyl and mercapto groups.22–29,33,34 However, this produces another problem, which is that the coordination mode of the carboxylic group to Ln3+ is difficult to show clearly unless a single-crystal structure can be obtained. So, for these non-crystalline hybrid materials derived from the modification of aromatic carboxylic acids, it is difficult to interpret the coordination environment around Ln3+. Nevertheless, these types of hybrid materials still present some interesting results. The different ORMOSILs derived from aromatic carboxylic acids influence the microstructures of the final hybrids; the microstructure may be related to the coordination mode of the lanthanide complex with the carboxylic acid precursor. As is known, lanthanide complexes generally form polymeric microstructures, and scanning electron microscopy (SEM) shows that the corresponding hybrid systems with these kinds of carboxylic-acid-derived linkages often have a branch-like morphology (Fig. 9c, d).24e,d Hybrids with m-aminobenzoic acid (MBA)-functionalized linkages have a porous structure, which is related to the mononuclear structure of the terbium complex of MBA (Fig. 9a).22a Benzoic acid without a substituent group is hard to modify, but we have modified the carboxylic groups of benzoic acid to construct hybrids. It is interesting that the hybrids show a lotus-root-like morphology (Fig. 9b).24a
 | ||
| Fig. 9 Morphologies of different terbium hybrid systems of ORMOSILs with aromatic carboxylate groups (a: m-aminobenzoate, b: benzoate, c: picolinate and d: 2-chloronicotinate). Reprinted with permission from refs. 22b and 24a,e,f. Copyright 2004–2006, Royal Society of Chemistry and Elsevier Ltd. | ||
In our previous work, we also found that there were a variety of shapes (leaf-shaped, spherical) for lanthanide hybrids with different ORMOSILs, and this seemed to be related to the coordination abilities of different carboxylic acid ligands (see Fig. 10).23b,c,d It is worth pointing out that though self-assembly occurred in non-crystalline hybrid systems through chemical bonding this is rarely found. The SEM images of these hybrid materials confirm their texture.
 | ||
| Fig. 10 Morphologies of different terbium hybrid systems of ORMOSILs with derived ligands (a, b: benzimidazole-5-carboxylate, c: 2-aminopyridine and d: 2,2′-bipyridyl amine). Reprinted with permission from ref. 23b,c,d. Copyright 2008, Elsevier Ltd. | ||
We give two typical examples of lanthanide hybrids with silanes functionalized with macrocyclic compounds. The first is hybrids of ORMOSILs with calixarene derivatives, which show corresponding green, red and blue luminescence.34b A comparison of the SEM images of covalently bonded hybrids and doped hybrids shows that the former appear to be very homogenous and uniform. Moreover, quite a few spheroids (diameter approximately 2–3 μm) emerged at the fracture surface, and we suggest that both the coupling agent TESPIC and the cup-like calix[4]arene triggered formation of this attractive morphology. In contrast, calix[4]arene-doped silicate materials exhibited obvious phase separation because the organic moieties were simply dispersed in the surface of the host lattice, without strong chemical bonding linkages. This is in agreement with the luminescence lifetimes and quantum efficiencies for covalently bonded and doped hybrids. The second example is hybrids of crown ether linkages; we discuss the coordination mode with Ln3+:crown ether ratios of :2. In this case, each N-substituted chelating group (–CO) was unable to coordinate to the same Ln3+, located in the ring of the crown ether molecule, because of the limits of spatial geometry. The coordination interaction therefore occurs intermolecularly but not intramolecularly.33e
The lanthanide hybrids of functionalized β-diketonates are of particular interest to us. Novel sol–gel-derived hybrid materials covalently grafted with Ln(DBM-OH)3·2H2O (DBM-OH = o-hydroxydibenzoylmethane, Ln = Eu, Nd, Er, Yb, Sm) have been prepared using the primary β-diketonate ligand DBM-OH.41 Compared with Eu-DBM hybrids prepared by physically doping silicon dioxide with Eu(DBM-OH)3·2H2O, this Eu-DBM-Si hybrid material has a more efficient ligand-to-Eu3+ energy transfer and a significantly improved emission quantum yield. On excitation at the maximum absorption of the ligand, the resultant materials display excellent NIR luminescence. A series of dysprosium-complex-doped xerogels based on the same first ligand and different neutral ligands have been synthesized via an in situ sol–gel process, and they show characteristic NIR luminescence of the Dy3+ ion, showing that the neutral ligand triphenylphosphine oxide can increase the emission intensity of the dysprosium complex, whereas phen has a negative effect.41 Carlos et al. reported the unusual role played by a sol–gel-derived diurethane crosslinked poly(oxyethylene) (POE)/siloxane [diurethanesil, d-Ut(600)] hybrid matrix in the immobilization of the β-diketonate aqua complex Eu(BTFA)3(H2O)2 (BTFA = 4,4,4-trifluoro-1-phenyl-2,4-butanedionate) and in quasi-preservation in hybrids of the 5D0 quantum efficiency displayed by this complex in the isolated state (0.12 vs. 0.18).24 They demonstrated that the d-Ut(600) framework acted as an inert (although optically active) support for Eu(BTFA)3(H2O)2, enabling Eu3+ sensitization by energy transfer between the hybrid host excited states and the lanthanide intra-4f6 levels. They also provided evidence that the incorporation of the Eu(BTFA)3phen complex into the same hybrid matrix is disadvantageous in terms of the 5D0 quantum efficiency because of the severe steric hindrance between the POE chains of the host structure and the bulky phen molecules. This hindrance leads to the expulsion of these ligands from the Eu3+ first coordination sphere and to their replacement by two water molecules.42 In contrast, ionogels were obtained by dissolving lanthanide complexes in the ionic liquid 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, [C6mim][Tf2N], followed by confinement of the lanthanide-doped ionic liquid mixtures in the pores of a nanoporous silica network. [C6mim][Ln(TTA)4] (Ln = Nd, Sm, Eu, Ho, Er, Yb) and [choline]3[Tb(DPA)3] (DPA = pyridine-2,6-dicarboxylate) were chosen as the lanthanide complexes, and the ionogels are luminescent, ion-conductive inorganic–organic hybrid materials. The work presented highlights that the confinement did not disturb the first coordination sphere of the lanthanide ions and also showed the excellent luminescence performances of lanthanide tetrakis β-diketonate complexes.43
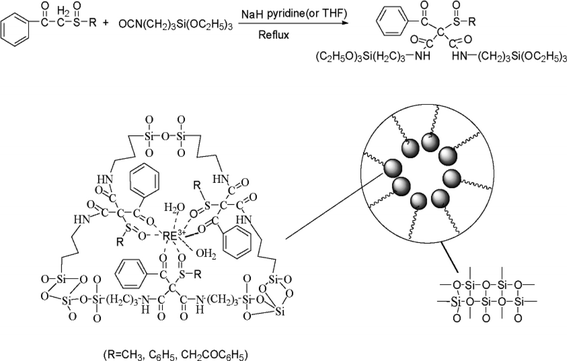
2.2 Hybrids prepared by grafting lanthanide complex units onto the interior of MCM-type mesoporous materials
On the basis of the above research on lanthanide sol–gel-derived hybrid materials with ORMOSILs, it is natural to attempt further assembly of lanthanide mesoporous silica hybrids by grafting lanthanide complex units with templates. As is known, ordered mesoporous materials are potential matrices because they possess a regular pore structure, and high photostability and thermal stability. Novel kinds of lanthanide organic–inorganic mesoporous hybrid materials, which combine the luminescence properties of lanthanide complexes and the particular properties of mesoporous materials, have therefore received considerable attention in recent years. The research has involved many kinds of typical mesoporous hosts such as MCM-41, SBA-15(16) and periodic mesoporous organosilicas (POMs). Functionalized microporous hosts such as zeolites have also been studied for the construction of hybrid materials. In particular, Li and Zhang have done significant work on the assembly of luminescent lanthanide species with zeolite microcrystals, and these have crystalline frameworks suitable for further practical applications.44 For example, zeolite L (ZL) loaded with lanthanide complexes was prepared by insertion of TTA into the nanochannels of Eu3+-exchanged zeolite crystals by gas diffusion. The novel luminescent core–shell material with a silica shell on the lanthanide-complex loaded ZL could be functionalized with silylated terbium complexes for use in the luminescence sensing of dipicolinic acid.44a Furthermore, the functionalized substrate can form monolayers of oriented ZL microcrystals bound to the hydroxyl groups on the quartz plate and coordinated to Ln3+. The functionalized plates can then be immersed in a suspension of ZL crystals. Organolanthanide complexes inside the nanochannels of ZL crystals have been synthesized using a ship-in-a-bottle procedure by inserting gas-phase bipy into the nanochannels of Ln3+-ZL crystals (see Fig. 11).44b Also, well-oriented and highly organized luminescent monolayers were realized by arranging ZL microcrystals on multifunctional-linker-modified quartz plates. Additional functionalization through coordination between Eu3+ ions and TTA moieties leads to strong luminescence with high quality ZL microcrystals. Alternatively, the luminescence color of the layers can be fine-tuned by arranging ZL microcrystals with Tb3+ ions and sensitizer loaded in the nanochannels on the luminescent quartz plates. Transparent, luminescent and uniformly oriented layers are obtained by simply coating poly(vinyl alcohol) on top of the ZL monolayers.44c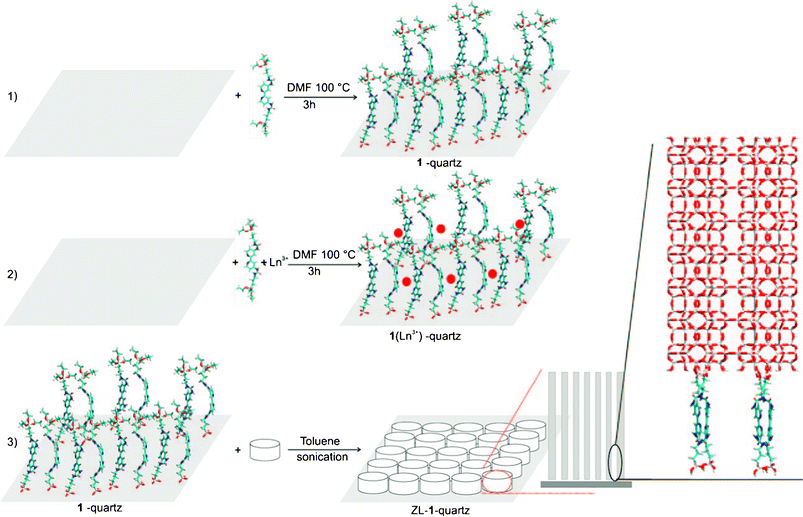 | ||
| Fig. 11 Scheme for the bonding of ORMOSILs to the hydroxyl groups of a quartz substrate and subsequent formation of oriented open-channel monolayers. Reprinted with permission from ref 44c. Copyright 2010, Wiley Publishing Company. | ||
Analogous nanomaterials with ordered arrays of uniform nanochannels are mesoporous materials, which are used as supports for lanthanide complexes.47–50 MCM-41, a member of the M41S family, possesses regular hexagonal arrays of mesopores, a variable pore diameter 1.5–30 nm, and tailorable interior surfaces.45 These properties, together with its thermal and mechanical stabilities, make it an ideal host for incorporation of active molecules, so some work has already been devoted to this area.45 SBA-15 has also become a very attractive host because of its high hydrothermal stability and the presence of hexagonally ordered large mesopores (P6mm symmetry group) interconnected by complementary micropores. The results reveal that the obtained materials possess good luminescence properties and photo and thermal stabilities. The mesoporous silica SBA-16 has also been considered as a good support on account of its three-dimensional structure, consisting of ordered interconnected spherical mesopores, which has a cubic cage-like structure with multidirectional and large pore systems allowing good access for both functionalization and adsorption.46,47 Periodic mesoporous organosilicas (PMOs) possess some obvious advantages over porous sol–gel-derived or grafted hybrid materials, such as highly ordered structures with very uniform pores, homogeneous distribution of functional groups throughout the whole framework, and high loadings.48
Functionalized mesoporous hybrid materials of photoactive lanthanide complexes have appeared in recent decades.18–21,49 The primary research originated from MCM-41 mesoporous hybrids fabricated with lanthanide complexes. Zink et al. took advantage of the different chemical and physical properties of regions in the sol–gel films and developed a new strategy for placing active molecules, e.g. lanthanide complex luminescent molecules, in desired inorganic silicate frameworks.49a There have been extensive studies of the encapsulation and assembly of guest molecules in mesoporous channels.49b,c Recently, Carlos et al.49d reported the syntheses of MCM-41 mesoporous materials covalently bonded with ternary europium complexes. It was shown that promising visible-luminescence properties can be obtained by linking ternary europium complexes to mesoporous materials. Zhang's group have studied MCM-41-based hybrids with pyridine derivative linkages in detail.18–21,50 They have tried several approaches to characterization of the mesoporous hybrids, but their emphasis has been on NIR luminescent mesoporous hybrids.50b For instance, new NIR luminescent mesoporous materials were prepared by linking ternary lanthanide (Er3+, Nd3+, Yb3+, Sm3+, Pr3+) complexes to MCM-41 through a functionalized phen group, 5-[N,N-bis-3-(triethoxysilyl)propyl]ureyl-1,10-phenanthroline. Upon excitation at the absorption wavelength of the organic ligands, all these materials show the characteristic NIR luminescence of the corresponding lanthanide ions.50a A new mesoporous hybrid material, Q-MCM-41, has been synthesized based on a bifunctional ligand, 8-hydroxyquinoline-functionalized organosilane (Q–Si). Furthermore, through ligand-exchange reactions, new NIR luminescent mesoporous LnQ3-MCM-41 (Ln = Er, Nd, Yb) materials have been prepared by linking lanthanide quinolinate complexes to Q-MCM-41.50b
Our group has also prepared some lanthanide mesoporous hybrids of MCM-41 hosts with modified β-diketonates and aromatic carboxylic acids.51 The lanthanide (Tb3+, Eu3+) complexes were covalently immobilized in MCM-41 by the modification of MBA with TESPIC, using a co-condensation method. The luminescence lifetimes and intensities of the two mesoporous materials both show that the triplet-state energy level of MBA-Si is more suitable for the central Tb3+ than for Eu3+. Similarly, other kinds of aromatic carboxylic acid ligands can be grafted onto an MCM-41 host.51e MCM-41 materials directly covalently bonded with modified β-diketonates have also been obtained. Zhou and Yan et al. reported MCM-41 mesoporous hybrids with dibenzoylmethane (DBM) and acetylacetone (ACAC) ORMOSILs.51a The hybrid materials with covalently bonded MCM-41 have higher intensities and longer lifetimes than those of pure complexes (Eu-DBM and Tb-ACAC). Moreover, the luminescence properties of mesoporous hybrids are comparable to those of sol–gel-derived hybrids with the same ORMOSILs.51c
Functionalized MCM-48 mesoporous hybrids with europium complexes were prepared using a simple wet impregnation method.52 The results reveal that Eu complexes were introduced into the pores of MCM-48 without disrupting the structure. Shifts in the absorption maxima were observed in the excitation spectra and discussed in relation to host–guest interactions between the organic complex and the silica matrix. Information on host–guest interactions was collected by analyzing the optical characteristics of the Eu3+ ions in different media. The role of the O2−–Eu3+ charge-transfer band and the impact of silylation on the luminescence properties at room and high temperatures were demonstrated.
There are many reports on photofunctional lanthanide SBA-15 mesoporous hybrids.19–21 Zhang's group concentrated on SBA-15-type hybrid materials with ORMOSILs derived from pyridine derivatives (bipy, phen, etc.) as the first ligand, and ancillary ligand such as β-diketonates [DBM, hfth (4,4,5,5,6,6,6-heptafluoro-1-(2-thienyl)hexane-1,3-dionate), tfnb (4,4,4-trifluoro-1-(2-naphthyl)-1,3-butanedionate]. They confirmed that these chemically bonded mesoporous hybrids show more favorable luminescence performances than those of the corresponding doped systems. Based on their work on visible-luminescent SBA-15 mesoporous hybrids, Zhang's research group has also obtained NIR-luminescent SBA-15 mesoporous hybrids with 8-hydroxyquinoline (Q-SBA-15), with a luminescence spectral region from 1300 to 1600 nm, which is of particular interest for telecommunications applications.53 We have also studied the functionalized SBA-15 mesoporous host with ORMOSILs derived from different organic precursors, including β-diketonates [1-(2-naphthoyl)-3,3,3-trifluoroacetonate (NTA), TTA, DBM, ACAC], calixarene derivatives (Calix, Calix-NH2, Calix-NO2), aromatic carboxylic acids, and sulfoxide ligands (oxobenzyldimethyl sulfoxide, benzylsulfinylacetylbenzene).54,55 They all have high surface areas and uniform mesostructures and crystallinities. The efficient intramolecular energy transfer in mesoporous materials mainly occurs between the modified ligand and the central Ln3+ ion. The ternary mesoporous material Eu(TTASi-SBA-15)3phen exhibits the characteristic emission of the Eu3+ ion with higher luminescence quantum efficiency intensity and a longer lifetime than those of the binary system, suggesting that the introduction of the phen ligand into the mesoporous matrix aids luminescence sensitization by replacing H2O groups that can quench the luminescence of Eu3+ ions.
We tried to prepare mesoporous SBA-16-type hybrids, TTA-S16 and DBM-S16, by co-condensation of a modified β-diketonate in the presence of Pluronic F127. Novel organic–inorganic mesoporous luminescent hybrids containing Ln3+ (Eu3+, Tb3+) complexes covalently attached to the functionalized ordered mesoporous SBA-16 (TTA-S16 and DBM-S16), denoted by bpy-Ln-TTA-S16 and bpy-RE-DBM-S16, were obtained by a sol–gel process. The luminescence properties of these resulting materials have been characterized in detail, and the results reveal that the mesoporous hybrid material bpy-Eu-TTA-S16 has a stronger luminescence intensity, longer lifetime, and higher luminescence quantum efficiency than the corresponding DBM-containing material bpy-Eu-DBM-S16, and bpy-Tb-DBM-S16 exhibits a stronger characteristic emission of Tb3+ and a longer lifetime than the corresponding TTA-containing material bpy-Tb-TTA-S16.56
Considering the high coordination number of lanthanide ions, a polymer can be introduced as a second ligand in assembling ternary lanthanide luminescent mesoporous [SBA-15(16)] hybrid materials (Fig. 12).57a All the resulting materials preserve their mesoscopically ordered structures and show highly uniform pore size distributions. Ternary polymer hybrid materials exhibit stronger luminescence intensities, longer lifetimes and higher quantum efficiencies than binary ones as a result of the introduction of an organic polymer, indicating that the introduction of an organic polymer chain is beneficial to the luminescence properties of the overall hybrid system. In addition, SBA-15 mesoporous hybrids show an overall increase in luminescence lifetime and quantum efficiency compared with those of SBA-16 mesoporous hybrids, indicating that SBA-15 is a better host material for lanthanide complexes than mesoporous silica SBA-16.57
![Scheme for synthesis and high-resolution transmission electron microscopy images of mesoporous hybrid material Eu(TTA-SBA-15)3PMMA recorded along the [100] (A) and [110] (B) zone axes. Reprinted with permission from ref. 57a. Copyright 2009, American Chemical Society.](/image/article/2012/RA/c2ra20976d/c2ra20976d-f12.gif) | ||
| Fig. 12 Scheme for synthesis and high-resolution transmission electron microscopy images of mesoporous hybrid material Eu(TTA-SBA-15)3PMMA recorded along the [100] (A) and [110] (B) zone axes. Reprinted with permission from ref. 57a. Copyright 2009, American Chemical Society. | ||
For mesoporous hybrids with PMO hosts, Zhang et al. have reported novel periodic mesoporous organosilica covalently grafted with phen-derived ORMOSILs (phen-PMO), synthesized via co-condensation of 1,2-bis(triethoxysilyl)ethane and phen-Si using a polyoxyethylene stearyl ether surfactant as a template (under acidic conditions). Accordingly, a series of PMO materials containing Eu(TTA)3phen were synthesized by impregnation of Eu(TTA)3·2H2O into phen-PMO through a ligand-exchange reaction. The results show that during the surfactant extraction process, the chelating organic ligand structure is preserved; this is confirmed by FTIR and 29Si CP-MAS NMR spectroscopies. Based on the emission spectra, the experimental intensity parameters for these europium hybrid systems were calculated according to the Judd–Ofelt theory. Compared with the pure complex, the resulting hybrid material exhibited better thermal stability and a similar emission quantum efficiency.58
In recent years, we have synthesized PMOs by linking lanthanide (Tb3+, Eu3+) complexes to mesoporous frameworks through modified 4-mercaptobenzoic acid, using a co-condensation method, in the presence of Pluronic P123 surfactant as a template.59a Recently, the ternary mesoporous hybrid material bpy-Ln-Calix-NH2-PMO (Ln = Eu, Tb) was prepared by linking ternary lanthanide (Eu3+, Tb3+) complexes to PMOs through functionalized Calix-NH2 ligands; this material shows stronger luminescence intensities and longer lifetimes than bpy-Eu-Calix-NH2-PMO hybrids. The luminescence properties show that the triplet-state energy level of Calix-NH2 is more suitable for the sensitization of Eu3+ than of Tb3+ and the luminescence intensity of the 5D0 → 7F2 transition and the 5D0 luminescence quantum efficiency of this material are higher than that of the pure bpy-Eu-Calix-NH2 complex, further confirming that the ternary complex bpy-Ln-Calix-NH2 is covalently bonded to the PMO silicon network (see Fig. 13).59b
![Scheme for synthesis and high-resolution transmission electron microscopy images of the ternary mesoporous hybrid bpy-Tb-Calix-NH2-PMO recorded along the [100] (A) and [110] (B) zone axes. Reprinted with permission from ref. 59b. Copyright 2011, Royal Society of Chemistry.](/image/article/2012/RA/c2ra20976d/c2ra20976d-f13.gif) | ||
| Fig. 13 Scheme for synthesis and high-resolution transmission electron microscopy images of the ternary mesoporous hybrid bpy-Tb-Calix-NH2-PMO recorded along the [100] (A) and [110] (B) zone axes. Reprinted with permission from ref. 59b. Copyright 2011, Royal Society of Chemistry. | ||
Based on the above work, it can be concluded that mesoporous hybrids of mesoporous hosts can be produced, and have luminescence performances comparable to those of silica-derived hybrids without mesostructures. Among these materials, the functionalized SBA-15 mesoporous hybrids have the optimum luminescence.
2.3 Hybrids prepared by attaching lanthanide complex units with ORMOSILs onto polymer chains
The introduction of lanthanide ions or their complexes into polymer hosts has been carried out for a long time.60 In general, there are four methods of preparing lanthanide-fabricated polymers:61 blending, polymerization and sol–gel methods and solution mixing; these can be more simply classified as chemical and physical incorporations. It has been shown that if a lanthanide ion is chemically bonded to the chains of a polymer, factors such as the content of lanthanide ion, the type of chemical interaction between the lanthanide ion and the polymer chain, and the distribution of lanthanide ions along the polymer chain will strongly influence the luminescence properties of the materials obtained.62 Compared with chemical incorporation, physical methods, in particular direct blending of lanthanide ions in organic complexes with polymers, have seldom been investigated, even though this method is convenient and inexpensive.62Carlos et al. have carried out detailed investigations of the aggregation, gelation and aging of urea-crosslinked siloxane-POE nanohybrids.63,64 In this work, the hybrid matrix was built by co-condensation of two diureasil frameworks [d-U(900) and d-U(600)], one for incorporating POE chains with about 15.5 oxyethylene repeating units and the other for about 8.5 oxyethylene repeating units. The co-condensed diureasil network was doped with a europium triflate complex; the effective interactions between the lanthanide ions and the host hybrid structure could account for the increased overall emission quantum yield (36%) and 5D0 quantum efficiency (57%) compared with those of the complex alone (29% and 27%, respectively).63a Three different types of photoluminescent hybrid materials containing trivalent lanthanide ions, chitosan and silica, with different structural features, have been prepared. The use of different silica sources led to hybrid materials with diverse microstructures, for example, a material with silica homogeneously dispersed in the chitosan, or formation of a core–shell morphology. The emission features of the core–shell materials are characterized by the distinct local environments of two Eu3+ ions, one associated with the chitosan core and the other with the silica shell.63b More recently, they prepared two structurally different but chemically similar families of alkyl/siloxane monoamidosil hosts doped with a wide range of concentrations of Eu(CF3SO3)3; changing the molar ratio of carbonyl groups per Eu3+ ion led to different microstructures. The hybrids are white-light emitters (maximum quantum yield: 0.08 ± 0.01), with a broad emission band in the blue/purplish-blue spectral region (ascribed to the hybrid host) superimposed on the 5D0 → 7F0–4 Eu3+ intra-4f6 transitions. Two Eu3+ local coordination sites have been identified in both systems.63c In addition, Er-doped organic–inorganic hybrids derived by solvolysis with different carboxylic acids (acetic, formic and valeric) have been prepared and characterized.64a The hybrid matrix is composed of a siliceous network, which is grafted onto both ends of a polymer chain with about 8.5 oxyethylene units. The host characteristic broad emission band in the visible region and the Er3+-specific emission at 1.5 μm were discerned. A bandwidth of 78 nm was observed for Er3+ emissions, which suggests the potential application of these hybrids in optical amplification devices.
We have summarized the three methods of constructing lanthanide hybrids with ORMOSILs and polymer units. In the first method, which is the most extensive, the polymer unit behaves as a ligand and coordinates to the central lanthanide ions.65–67 For example, a new polymer–inorganic material has been synthesized from terephthalic acid and lanthanide ion (Tb3+, Eu3+) complexes, in which the lanthanide ions are connected to the polymer by coordination bonds.65a The luminescent europium and terbium coordination polymers PET-Tb and PET-Eu [PET = poly(ethyl terephthalate)] are formed in situ through low-temperature solution polycondensation. SEM images show a homogeneous microstructure with typical polymeric aggregation, similar to synthetic tissue. The coordination numbers of lanthanide ions are generally 7–9; the coordinated groups of the PET polymer chains only provide two coordinated O atoms, to limit hindrance, thus there are two carbonyl groups of another PET polymer unit coordinating to Ln3+. In addition, the balancing ions (three NO3−) may also coordinate with Ln3+. In these polymer hybrid material systems, Ln3+ ions behave as bridges, linking the different adjacent PET polymer chain units though coordination. Finally, all the PET polymer chains can be assembled into an interpenetrating polymer network structure by Tb or Eu ions (Fig. 14).65a
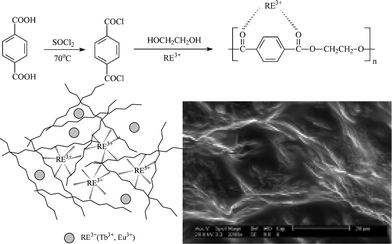 | ||
| Fig. 14 Scheme for synthesis and SEM images of lanthanide hybrid materials with PET. Reprinted with permission from ref. 65a. Copyright 2008, Elsevier Ltd. | ||
We have prepared a series of quaternary and ternary polymer hybrid materials from 3-methacryloxypropyltrimethoxysilane (MS), MAA (methacrylic acid), phen and lanthanide (Eu3+, Tb3+, Dy3+) precursors.65b MS and MSMAA (a co-polymer of MS and MAA) behave as structural functional components to form an inorganic polymer network or inorganic–organic polymer host through coordination to lanthanide ions, whereas phen acts as an energy sensitizer for the luminescence of Ln3+ ions. It is found that the introduction of organic polymer units influences the microstructure, and, in particular, the luminescence properties of hybrid materials. It is worth pointing out that quaternary hybrid materials phen–Eu3+(Tb3+, Dy3+)-MSMA with an organic polymer unit (MAA) have larger luminescence intensities and longer lifetimes than those of ternary materials without MAA (phen–Eu3+(Tb3+, Dy3+)-MS), suggesting that the introduction of an organic chain (MAA) is beneficial to the photophysical properties of the hybrids.65b A co-polymer of MMA (methyl methacrylate) and maleic anhydride (MAL) (MMA-co-MAL) was prepared by grafting onto APS, which behaves as the structural precursor for functional bridges, and then the covalently bonded systems were assembled through coordination between Ln3+ and the carboxylic groups of MAL;65c phen was used as the second functional ligand to sensitize the luminescence of Ln3+ by intramolecular energy transfer.
Polymer–inorganic hybrids (phen-Ln-MMA-co-MAL-Si) can be prepared through polyhydrolysis and condensation between the triethoxysilyl group of a modified co-polymer (MMA-co-MAL-APES) and TEOS, resulting in the characteristic red or green emissions of Eu or Tb ions. The introduction of co-polymer chains provides a template to induce the formation of a regular microstructure; however, there are few differences among the SEM images of these covalently bonded hybrid materials. The 2phen-Tb-MMA-co-MAL-Si hybrids have ordered particles of size 100–200 nm, which suggests that the terminal ligand of phen also affects the micro-morphology of the hybrid system. Atomic force microscopy images show that the covalent composites exhibit relatively regular column shapes (diameter range of around 200 nm) within rough upper surfaces, which may be caused by mutual influences originating from the polymer soft chains and silica coupling agent (Fig. 15).65c
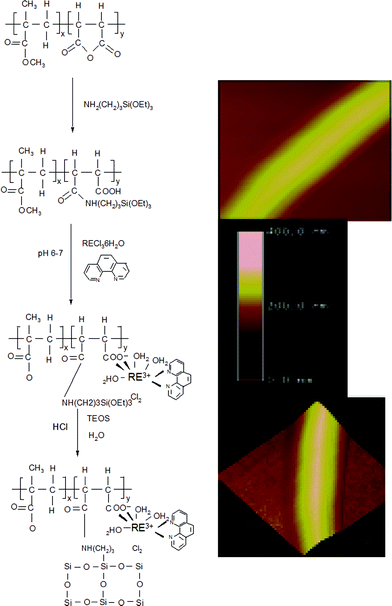 | ||
| Fig. 15 Scheme for synthesis and atomic force microscopy photographs of the surface of 2phen-Eu-MMA-co-MAL-Si. Reprinted with permission from ref. 65c. Copyright 2008, Elsevier Ltd. | ||
A series of chemically bonded lanthanide hybrids have been constructed using ORMOSILs (aromatic carboxylic acids, β-diketonates and calixarene derivatives) and polymer units.65d–f,66 All of them exhibit homogeneous, regular, ordered microstructures and morphologies, suggesting self-assembly of an inorganic network and organic chain. The ternary lanthanide/inorganic/organic polymer hybrids have better photoluminescence properties (stronger luminescence intensities, longer lifetimes and higher luminescence quantum efficiencies) than binary lanthanide/inorganic polymer hybrids, because the introduction of organic polymer chains is beneficial to the luminescence of the whole hybrid system. For hybrids with aromatic carboxylic acid linkages in particular, we can control the resulting different regularly ordered microstructures of lanthanide/inorganic/organic polymer hybrids through strong chemical bonds, to give structures such as dendritic stripes, sandwiches, planar circular disks, pin-holes and three-dimensional globes. Furthermore, the lanthanide ions and organic polymers are also important factors in the microstructure.65c,66d However, for lanthanide hybrids with β-diketonates or sulfoxide linkages and polymer units, similarly varied morphologies and microstructures cannot be obtained.65d–f,66a,b To explore the influence of the different polymer chains on the properties of lanthanide hybrids, the microstructures and photoluminescence properties of these hybrids have been studied in detail. It was found that four organic polymer chains with different structures not only coordinate to the lanthanide ions by their own carbonyl group, but also form a polymer matrix with the inorganic Si–O network. These results show that all the obtained hybrids have efficient intramolecular energy transfer and result in excellent characteristic emissions of lanthanide ions. Moreover, the different polymeric structures induce different microstructures and the different photoluminescence behaviors (lifetimes and quantum efficiencies) in this hybrid system (Fig. 16).65d
 | ||
| Fig. 16 Selected SEM images of lanthanide hybrid materials with 1,4-hydroxynicotinic-acid-derived ORMOSILs and polymers (polymethacrylic acid or co-polymer of methacrylic acid and acrylamide). Reprinted with permission from ref. 65d. Copyright 2009, American Chemical Society. | ||
For lanthanide polymer hybrids with calixarene-derived ORMOSILs, p-tert-butylcalix[4]arene derivatives with different functional groups may result in different thermal stabilities and microstructures because of their respective self-assembly mechanisms, and may also have an influence on the final luminescence properties.67d Different polymer chains also affect the luminescence properties of hybrid systems; Calix-Br-Si-Eu(Tb)-PMMA shows the longest luminescence decay time and largest emission quantum efficiency. It is speculated that the different central metal ions have different atomic radii, electron distributions, and coordination numbers, which could influence the coordination, co-polycondensation, and aging procedures. As well as the metal ions, the molecular precursors have important influences on the properties of the final hybrids (Fig. 17).
 | ||
| Fig. 17 Scheme for synthesis of hybrid polymer material Calix-Br-Si-Eu(Tb)-PMMA (a: Eu and b: Tb). Reprinted with permission from ref. 67d. Copyright 2011, Royal Society of Chemistry. | ||
Another path is to assemble lanthanide polymer hybrids with covalent bonds between ORMOSILs and polymers, in which no direct interactions (coordination bonds) exist between the lanthanide ions and the polymer units.68,69 In ref. 68b, TTA as the organic ligand and poly(ethylene glycol) (PEG400, with a molecular weight of 380–430), as the network precursor were each grafted onto the coupling agent TESPIC, in order to construct two precursors, namely TTA-Si and PEG-Si. The TTA-Si precursor and the terminal ligand phen were coordinated to the lanthanide ions by the carbonyl group or nitrogen atom to obtain binary or ternary hybrid polymer materials after hydrolysis and co-polycondensation of TEOS, water molecules, and the network precursor PEG-Si via a sol–gel process. The terminal ligand phen was used to investigate the differences between the photophysical and luminescence properties of binary and ternary hybrid materials, and the polymer network precursor PEG-Si was induced to show its own influence on the microstructure and thermal properties (Fig. 18).68b
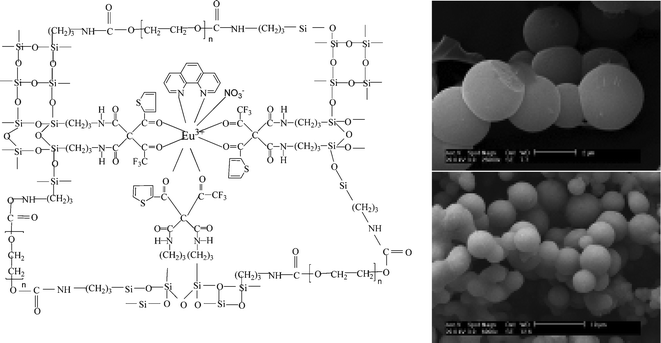 | ||
| Fig. 18 Scheme of synthesis of hybrid polymer materials and SEM images of TTA-Si-Eu-phen (top) and TTA-Si-Eu-phen-PEG (bottom). Reprinted with permission from ref. 68b. Copyright 2009, American Chemical Society. | ||
In ref. 69e, the organic chain unit is covalently bonded with inorganic silica through an Si–O network, but is not coordinated to lanthanide ions. The covalent Si–O bonds between the polymer and the lanthanide complex, through linkage, can have a direct influence on the sol–gel process for producing the ORMOSIL precursors, which is important to the microstructure formation. Generally, a polymer such as polyacrylamide (PAM) has no effective photoactive groups for energy transfer to lanthanide ions, so PAM mainly behaves as a structural host unit rather than a functional one. 5-Hydroxyisophthalic acid (HIPA) was modified by TESPIC through an addition reaction, so the two meta-carboxylate groups chelated with the lanthanide ions, and the remaining amide could react with the functional PAM, modified by CPTMS, through co-hydrolysis and co-polycondensation. Finally, the hybrids showed a relatively regular trunk or stripe. When PAMSi was introduced into the Eu-HIPA-Si hybrid material through a hydrolysis/polycondensation process, the emergence of circular particles was observed; the number of particles increased and the particle size decreased with increasing PAMSi content. This could be explained by the organic polymer chain unit PAMSi having two roles; one role is as a polymer chain, inducing orientation, and in the other role, the silanol group co-polymerizes with HIPASi. The dual control of orientation and co-polymerization affect the formation and growth of particles in the hybrid systems. The SEM images show that the particle sizes are on the nanometer scale and the microstructures tend to be regular and homogeneous (Fig. 19).
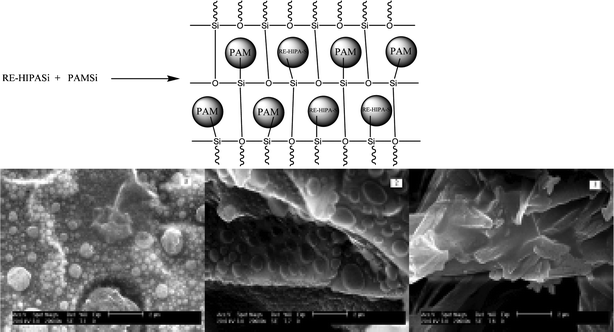 | ||
| Fig. 19 Scheme of the synthesis of the final hybrids RE-1HIPA-Si-1PAM and comparison of the SEM images of (1) Eu-HIPA-Si composite material, (2) Eu-2HIPA-Si-1PAM composite material with a PAM/HIPA molar ratio of 1:2, and (3) Eu-1HIPA-Si-1PAM composite material with a PAM/HIPA molar ratio of 1:1. Reprinted with permission from ref. 69e. Copyright 2011, Royal Society of Chemistry. | ||
The direct polymerization process can also be used to assemble lanthanide polymer hybrids.72 A new functional polysilsesquioxane linkage (VPBA-Si) has been achieved through a hydrogen-transfer addition reaction between 4-vinylphenylboronic acid and TESPIC. Three kinds of Eu3+- and Tb3+-centered hybrid materials were assembled with VPBA-Si and co-polymers through radical addition polymerization of VPBA-Si, trans-styrylacetic acid and N-vinylphthalimide, hydrolysis and co-polycondensation between the VPBA-Si unit and TEOS, and a coordination reaction between the ligand and rare-earth ions. At the same time, another kind of lanthanide (Eu3+, Tb3+) hybrid was constructed through addition polymerization of trans-styrylacetic acid and vinyltrimethoxysilane, without the VPBA-Si precursor. The photoluminescence properties were studied in depth, and the results show that the hybrids with trans-styrylacetic acid exhibit the most favorable characteristic luminescence properties (long lifetime and high quantum efficiency).70b
2.4 Other lanthanide hybrids which are non-silica based or composite based
In all the above work, the hybrids are mainly based on inorganic silica polymer networks. Recently, some studies have focused on the lanthanide hybrids of other inorganic frameworks such as M–O–M (M = B, Al, Ti).71b Li's group have prepared lanthanide hybrids of titania with simple organic pyridinecarboxylic acid linkages,32 and this system can be extended to a variety of lanthanide hybrids with silica-alumina and silica-titania composite hosts.71 In ref. 71a, we reported the synthesis of a series of novel Ln3+ mesoporous hybrid materials with different inorganic hosts. The differences in the profiles of the emission features, lifetime values and quantum efficiencies among the obtained materials confirm that the inorganic matrices in the hybrid materials influence the photoluminescence properties. The luminescent ternary europium mesoporous hybrid titania material Eu(Ti-MAB-S15)2(NTA)3 was successfully prepared by linking lanthanide Eu(NTA)3·2H2O complexes to the mesoporous hybrid titania material Ti-MAB-S15 via a ligand-exchange reaction. This method was used to assemble luminescent lanthanide molecular-based hybrid materials with both ordered mesoporous Si–O networks and amorphous Ti–O networks. A non-silica network can also be introduced into mesoporous silica systems. For example, investigation of the luminescence properties indicates that the europium mesoporous hybrid titania material Eu(Ti-MAB-S15)2(NTA)3 exhibits a larger 5D0 luminescence quantum efficiency and a longer lifetime than the pure Eu(NTA)3·2H2O complex and binary mesoporous hybrid titania material Eu(Ti-MAB-S15)4.71b More recently,71c an NTA-functionalized SBA-15 mesoporous hybrid material (NTA-S15) was synthesized by co-condensation of modified NTA (denoted by NTA-Si) and TEOS. Nicotinic acid (NA) was selected as the second ligand; The carboxylic acid group of NA can react with a metal alkoxide to moderate the reactivity towards hydrolysis and condensation, and the heterocyclic group can coordinate to lanthanide ions as well as sensitize their luminescence. The hybrid materials Ln(NA-Al)2(NTA-S15)3 show an overall increase in luminescence lifetime and quantum efficiency compared with mesoporous hybrid Ln(NA-Ti)2(NTA-S15)3, indicating that the Al–O network is a better host material for lanthanide complexes than the Ti–O network. Stable transparent titania thin films were fabricated at room temperature by combining TTA-modified titania precursors with the amphiphilic co-polymer P123. It has been reported that a facile strategy for tethering lanthanide complexes to organic–inorganic hybrid titania materials is to employ chemically modified titanium alkoxide as the precursor; the embedded organic ligand sensitizing the luminescence of lanthanide ions is bonded to the titania.71dPolysilsesquioxane bridges were obtained through the single modification of TESPIC by two β-diketonate ligands (TTA and trifluoroacetylacetone), which behave as linkages between europium ions and composite host Si–O–M (M = B or Ti) xerogels by controlling the hydrolysis rates of alkoxyl compounds (TEOS, titanium butoxide, and tributyl borate).72a The results reveal that these lanthanide complexes were covalently immobilized in the composite inorganic xerogels and the obtained hybrids have excellent light absorption abilities and luminescence emissions. The hybrids with composite Si–O–B xerogels have comparable photoluminescence properties (red emission intensities, lifetimes and quantum efficiencies) to those of hybrids of the pure silica oxygen network, and the hybrids with Si–O–B or Si–O–Si xerogels both show superior luminescence performances to those of the hybrids with Si–O–Ti xerogels. The single modification by TTA also gives excellent luminescence of the hybrid xerogels, and the luminescence quantum efficiencies of the hybrids with an Si–O–B host show the highest values among all the hosts. The Eu–TTASi′–O–Si–O–M hybrids have shorter lifetimes and lower quantum efficiencies than those of the corresponding Eu–TTASi–O–Si–O–M hybrids. This further verifies that the molecular linkage of a single modification is beneficial to the luminescence of the final hybrids. B and Si are diagonal elements in the periodic table, and they have similar features and chemical behaviors, as verified by the similar sol–gel processes of their alkoxyl compounds. However, the composite host of the Si–O–B systems shows little distinction from that of the pure Si–O host, and the results show that the hybrid gel with a composite Si–O–B host did not change the luminescence of the final hybrid. Ti is a transition metal, and has different chemical properties from Si. This work has been extended to other hybrid systems.72b–e
Some studies have achieved the introduction of various semiconductors, such as Si, ZnO, TiO2, ZnS, CdS, Ag2S, ZnSe, and GaN, into luminescent lanthanide hybrids.73 A color-tunable emitter comprising Eu-complex-capped ZnSe quantum dot (QD) organic–inorganic hybrid nanocrystals (NCs) was simply synthesized by a hot-injection method, with the addition of a Eu precursor. Hybrid NCs have the emissions of both Eu complexes and ZnSe QDs, resulting in a bluish-white light. The emission of the composite NC at Eu:Zn = 1.0 is 174% that of pristine ZnSe QDs as a result of the sensitization of the Eu complex as an antenna to transfer energy to the ZnSe QDs. The strong excitation band of the composite NCs in the near-UV region is expected to be particularly useful in white-light-emitting diodes. These new kinds of hybrid luminescent materials using the emissions of both QDs and lanthanide complexes could potentially serve as light sources in white displays.73a We have tried to modify ZnS QDs to obtain SiO2/ZnS nanocomposites, and then prepare lanthanide hybrid materials using TESPIC as an organic bridge. This organic bridge can coordinate to lanthanide ions (Eu3+, Tb3+, Sm3+, Dy3+) and form an inorganic Si–O–Si network with SiO2/ZnS nanocomposites to assemble the final material. The luminescence spectra of the hybrids show the dominant excitation of the TESPIC-MS-SiO2/ZnS unit and the unique emission of lanthanide ions, suggesting that the TESPIC-MS-SiO2/ZnS unit behaves as the main energy donor and effective energy transfer take place between this unit and lanthanide ions.73c Three kinds of semiconductor metal sulfide nanoparticles (CdS, ZnS and Ag2S) have been synthesized and then functionalized with MS to obtain organically modified MS–CdS(ZnS, Ag2S) composites. Through co-hydrolysis and co-polycondensation between the TTASi unit of phen(bipy)-Eu-TAASi and the MS unit of MS/CdS(ZnS, Ag2S), both the semiconductor unit and the lanthanide complex system were sol–gel assembled via covalently bonding Si–O to form the multicomponent inorganic–organic hybrids phen(bipy)-Eu-TAASi-SiO2-MS-CdS(ZnS, Ag2S). The luminescence properties of these hybrids have been studied in detail and show that the introduction of a semiconductor unit is favorable for the luminescence of europium ions (Fig. 20).73d
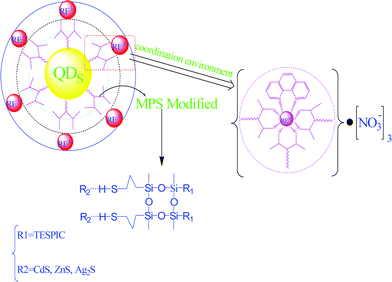 | ||
| Fig. 20 Scheme for synthesis of the hybrids phen-Eu-TAASi-SiO2-MS-CdS. Reprinted with permission from ref. 73d. Copyright 2010, Elsevier Ltd. | ||
Zhang et al. have prepared luminescent hybrid materials by physically doping (or covalently bonding) europium complexes into PMMA-co-Sn12Cluster, whose nanoscale space microstructure and soft skeleton are beneficial in grafting with europium complexes, and which has a high capacity for physical doping with europium complexes. PMMA-co-Sn12Cluster shows more extensive absorption spectra than the single PMMA matrix, revealing that it can prevent quenching of Eu3+ ions, thereby maintaining the luminescence lifetimes of the Eu3+ complexes.74a Novel NIR luminescent co-polymerized hybrid materials have been prepared by covalently grafting and physically doping Ln complexes (Ln = Er, Sm, Yb, Nd) into a co-polymer matrix built from nano building-blocks. Transparent films of these materials can be easily prepared through spin-coating on indium tin oxide glasses, taking advantage of their matrix nature (Fig. 21).74a The results show that these hybrid materials emit the characteristic NIR luminescence of the corresponding Ln3+ ions through intramolecular energy transfer from the ligands to Ln3+ ions.
![Proposed mechanisms for the formation of PMMA-co-Sn12Cluster/[Eu(TTA)3phen] and PMMA-co-Sn12Cluster-co-[EuAcAc(TTA)2phen] hybrid materials. Reprinted with permission from ref. 74a. Copyright 2010, Wiley Publishing Company.](/image/article/2012/RA/c2ra20976d/c2ra20976d-f21.gif) | ||
| Fig. 21 Proposed mechanisms for the formation of PMMA-co-Sn12Cluster/[Eu(TTA)3phen] and PMMA-co-Sn12Cluster-co-[EuAcAc(TTA)2phen] hybrid materials. Reprinted with permission from ref. 74a. Copyright 2010, Wiley Publishing Company. | ||
A new class of materials with a bifunctional architecture combining the useful functions of superparamagnetism and luminescence in one material have been developed. These bifunctional materials were prepared via two main steps, a modified Stöber method and a layer-by-layer assembly technique, and they exhibit superparamagnetic and photoluminescence behavior.74 Zhang et al. synthesized an NIR photoluminescent macromaterial and an NIR luminescent/magnetic bifunctional macromaterial using PS (polystyrene) and Fe3O4@PS, nanoparticles, respectively, as templates.76c Both of them show the characteristic emission of the Er3+ ion originating from an intra-4f shell transition (4I13/2 → 4I15/2), which is located at the third telecommunications window (centered at 1540 nm). This bifunctional macromaterial can be used to make NIR hybrids responding to magnetic fields.74d In addition, the advantages of mesoporous silica, magnetic Fe3O4 particles, and NIR luminescent lanthanide complexes have been combined to fabricate novel nanocomposites with magnetic separability and NIR luminescence properties.75b,c The Nd(DBM)3phen-MMS nanocomposite has potential applications in laser systems or optical amplifiers operating at 1.3 μm, and Yb(DBM)3-phen-MMS (MMS = magnetic mesoporous silica) nanocomposites have several advantages for potential applications in drug delivery or optical imaging.
Ionogels are solid-oxide host networks which contain mesoscale ionic liquids and retain their liquid nature. Ionogels are obtained by dissolving lanthanide complexes Ln(TTA)4 (Ln = Nd, Sm, Eu, Ho, Er, Yb) or Tb(DPA)3 in the ionic liquid [C6mim][Tf2N], followed by confinement of the lanthanide-doped ionic liquid mixtures in the pores of a nanoporous silica network, resulting in [C6mim][Ln(TTA)4] and [choline]3Tb(DPA)3, respectively. The luminescent, ion-conductive inorganic–organic hybrid ionogels can exhibit emissions in the visible or NIR regions of the electromagnetic spectrum, depending on the Ln3+ ion, and the confinement of ionogels does not disturb the first coordination sphere of Ln3+.76a Recently, work was reported on a Zn–Al layered double hydroxide pillared by 2,2′-bipyridine-5,5′-dicarboxylate anions, which behaves as a porous matrix and intercalates LnCl3 (Ln = Eu, Gd).76b The hybrid materials display both Eu3+ red emissions and free-ligand phosphorescent emissions at around 460 nm. The 5D0 quantum efficiency is estimated to be low (7.7%) as a result of a relatively high non-radiative transition, probably caused by water molecules in the first coordination shell. The number of water molecules is calculated to be around 3.6, suggesting that the incorporated europium ions are six-coordinated with four oxygen atoms from water molecules and two nitrogen atoms from a bidentate bipyridyl ligand.
Several experimental techniques have provided evidence for the encapsulation of suitably designed luminescent Eu complexes inside multiwalled carbon nanotubes (MWCNTs).77 Despite some quenching effects, the luminescence quantum yield of the selected luminophore remains reasonably high (7%). Appropriate organic functionalization of the external carbon wall and optimization of the synthesis procedure could lead to a new class of visible-light-emitting CNT-based hybrids, which would have potential applications in the biological and materials sciences, for example in biodiagnostics and non-toxic therapies (Fig. 22).77a Luminescent carbon-based materials have also been prepared by electrostatic self-assembly of negatively charged luminescent Eu complexes with imidazolium-functionalized MWCNTs.77b
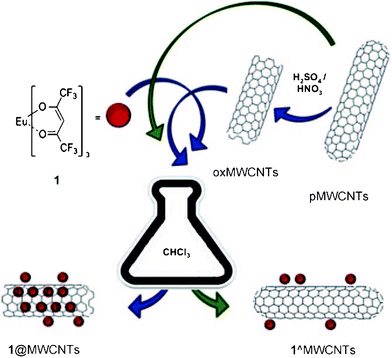 | ||
| Fig. 22 Schematic representation of the nano-extraction technique. The structure of complex 1 and the products of the functionalization of MWCNTs. Reprinted with permission from ref. 77a. Copyright 2011, Wiley Publishing Company. | ||
3. Photophysical properties of lanthanide hybrids
Photofunctional lanthanide hybrid materials mainly show the characteristic luminescence of the central lanthanide ions, and their photophysical properties have been discussed in detail by many research groups, especially Carlos' group.9–14 In hybrid systems, the ORMOSIL and other photoactive unit coordinated to the lanthanide ions function as the main energy donors to sensitize the luminescence of the lanthanide ions through effective energy transfer. The main photophysical behavior of the hybrid is therefore similar to that of the lanthanide complex, and the hybrid can be considered as a complex molecule-based system. For example, Lime and Carlos et al. have discussed the quantitative aspects of energy transfer in sol–gel-derived diureasil hybrids [Eu(BTFA)3(4,4′-bpy)(EtOH)] (BTFA = benzoyltrifluoroacetonate, 4,4′-bpy = 4,4′-bipyridine) or Eu(CF3SO3)3. They consider that the host-to-Eu3+ energy transfer occurs either via ligand singlet and triplet excited states or directly from the hybrid emitting centers through dipole–dipole, dipole–2λpole (λ = 2, 4 and 6) and exchange mechanisms. The ligand-to-Eu3+ energy transfer rate is typically one order of magnitude larger than the estimated value of direct hybrid-to-Eu3+ transfer, and the most efficient luminescence channel is concluded to be (S0)hybrid → (T)hybrid → (T)ligand → (5D1, 5D0) → 7F0–6.78a Recently, they studied the Eu3+-assisted short-range ordering of luminescent bridged silsesquioxanes, which were fabricated from the precursor (EtO)3Si(CH2)3NH(CO)NH(CH2)12NH(CO)NH(CH2)3Si(OEt)3 with multifunctional units of polymerizable silylated groups, urea functionalities, and alkyl chains,78b and via the supramolecular self-assembly mechanism by strong, ordered hydrogen-bonding interactions. Eu3+ plays totally different dual roles, as a structure-directing agent to arrange the assembly and as a structural probe to sense local morphological changes. The Eu3+ ion also shows other behavior, indicating the microstructure of the whole hybrid based on different concentrations in the different stages of the synthesis.
The inert lanthanide ions with a stable electronic configuration (the 4f shells are empty, half-filled or full), such as La3+, Y3+, Gd3+ and Lu3+, can enhance the luminescence of photoactive lanthanide ions (Eu3+, Tb3+). However, it is not easy to synthesize lanthanide complexes with mixed lanthanide ions in the same system. Chemically bonded hybrid materials can introduce different lanthanide ions into the same system (complex molecular network) with Si–O bonds; therefore, intramolecular energy transfer occurs between inert lanthanide ions and active lanthanide ions through the organically bonded Si–O group.79 In ref. 79a, synthesis of a new monomer, TBBA-APMES, by grafting 3-aminopropylmethyldiethoxylsiliane (APMES) on 4-tert-butylbenzoic acid (TBBA) was reported. Through co-hydrolysis and polycondensation, Tb3+ and La3+ can be introduced into the same molecular hybrids through the chemical Si–O network, and fluorescent enhancement of this hybrid is observed. Furthermore, the luminescence behavior has been studied with different ratios of Tb3+ to La3+, and the results suggest that the luminescence intensities of hybrids can be noticeably enhanced by the presence of La3+. A novel kind of organic–inorganic monomer, HIPA-TESPIC, has been achieved by modifying HIPA with TESPIC. The corresponding hybrid materials have been obtained, and they consist of an active Ln3+ (Tb3+, Eu3+)/inert Ln3+ (Y3+, La3+, Gd3+) complex covalently bonded to a silica-based network, and a luminescence enhancement effect is achieved.79b This study has been extended to lanthanide mesoporous or polymer hybrid systems. Luminescent hybrid mesoporous materials containing active lanthanide ion (Eu3+)/inert lanthanide ion (Y3+, La3+, Gd3+) complexes covalently bonded to mesoporous material networks have been obtained. The luminescence behaviors of the hybrid materials with different ratios of Eu3+ to Y3+, La3+ and Gd3+ have been studied, and the results proved that the presence of inert lanthanide ions can enhance the luminescence intensity of Eu3+. This may be a result of intramolecular energy transfer among Y3+, La3+, Gd3+ and Eu3+ through the covalently bonded mesoporous framework.79c The organic ligand SSA has been grafted by APS to achieve functionalized sulfonamide bridges (SSA-Si), which can both coordinate to Ln3+ to form luminescent centers and link with an inorganic Si–O network through hydrolysis and condensation reactions with TEOS. PMMA also acts as a precursor, and is prepared through the direct addition polymerization of MMA monomer in the presence of the initiator benzoyl peroxide. In these complicated hybrids, we have changed the ratios of Tb3+ to inert lanthanide ions (La3+, Gd3+, Y3+) and found that the introduction of the inert Ln3+ ions enhanced the luminescence intensity (Fig. 23).79d
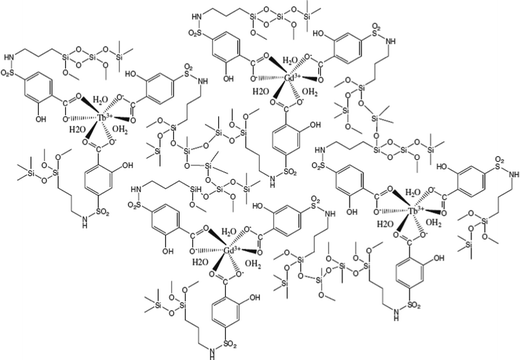 | ||
| Fig. 23 Schematic illustration of the cofabricated hybrids. Reprinted with permission from ref. 79d. Copyright 2011, Springer. | ||
Photofunctional lanthanide hybrids can be used as photophysical sensors for the recognition of various ions in solution.80 For example, luminescent porous europium materials have been prepared from the amide of DPA with APES in the presence or absence of the non-ionic surfactant F-127.80a The luminescence spectra of the hybrid system are used to identify and compare Eu3+ environments in amorphous matrices, as well as the accessibility of the material, using luminescence quenching by Cu(II). Solid-state devices have been built to test and compare their performances as potential Cu(II) sensors in terms of the values of their Stern–Volmer constants in quenching experiments. Materials templated with F127 show better response than non-templated materials under different synthesis conditions, permitting the in situ detection of Cu(II) in solution down to the 0.05 ppm level. Li's group has reported mesoporous hybrids based on a novel Eu complex Eu(DPIQ)(TTA)3 (DPIQ = 10H-dipyrido[f:h]indolo[3,2-b]quinoxaline) and explored their oxygen-sensing properties.80b The oxygen-sensing properties of hybrids with different loading levels of Eu(DPIQ)(TTA)3 were investigated. A sensitivity of 3.04, a short response time of 7 s, and good linearity were obtained for the hybrids with a loading level of 20 mg g−1. So far, these results are the best reported values for optical oxygen-sensing materials based on Eu3+.
Wang et al. have studied the application of photofunctional lanthanide hybrid systems in the field of sensors.81 A lanthanide complex, as a fluorescent receptor, was embedded in mesoporous silica through a sol–gel approach, and the nanoscale rod material exhibited excellent luminescence and thermal stability. The material showed significant changes in fluorescence upon hydrogen bonding to F− in DMSO–H2O (1:1), and the luminescence color varied from green to blue. Proton NMR titration studies also indicate that the organic ligand is participating in hydrogen-bonding interactions with fluoride guest anions. The detection limit for F− reached 1 μM, and reusability cycles were performed more than 10 times.81c To design anion sensors, a terbium complex of 2-methylimidazole-4,5-dicarboxylic acid was encapsulated in TEOS as the inorganic host;81b the strong green emission could still be observed when the material was dispersed in pure water. It was found that the luminescence of the hybrid material was selectively and rapidly turned off by hydrogen sulfate, whereas the addition of anions such as F−, Cl−, Br− and I− did not change the luminescence. A lanthanide TTA complex based on 2-(3,5-difluorophenyl)imidazo[4,5-f]-1,10-phenanthroline, with red luminescence, acts as an effective receptor for F− and AcO− anions.81c Spectroscopic studies showed that the sensor exhibits striking emission changes after the addition of F− (green) and AcO− anions (yellow–red to green) as a result of hydrogen-bonding interactions. More importantly, transparent hybrid thick films composed of a europium complex and PMMA matrix have been successfully prepared. The results show that all the films exhibit intense red emissions of Eu3+ and the optimum concentration of the luminescent species is 0.5 g/5 mL of MMA. The derived films give rise to luminescence changes in F− DMSO solution (Fig. 24).81a
![Synthesis of the hybrid material and photograph of hybrid material solution (10 μg/mL in water) and upon addition of 10−4 M [Bu4N]HSO4 (excited by UV lamp at 254 nm). Reprinted with permission from ref. 81a. Copyright 2010, Wiley Publishing Company.](/image/article/2012/RA/c2ra20976d/c2ra20976d-f24.gif) | ||
| Fig. 24 Synthesis of the hybrid material and photograph of hybrid material solution (10 μg/mL in water) and upon addition of 10−4 M [Bu4N]HSO4 (excited by UV lamp at 254 nm). Reprinted with permission from ref. 81a. Copyright 2010, Wiley Publishing Company. | ||
4. Summary and outlook
In conclusion, recent research progress on the four most active topics of photofunctional lanthanide hybrid materials have been summarized: chemically bonded hybrids with ORMOSILs as precursors for sol–gel processing, hybrids with lanthanide complex units grafted onto the interior channels of mesoporous materials, hybrids with lanthanide complex units on polymer chains, and lanthanide hybrids with non-silica or mixed matrices. The photophysical properties of these lanthanide hybrids are also introduced for the subject of continuing research. However, some challenges still exist in the field of lanthanide hybrid materials. The first problem is that the non-crystalline nature of the hybrid materials makes it difficult to determine their exact structures. Therefore the focus has shifted to the introduction of crystalline building blocks into hybrid systems to combine inorganic–organic hybrids and nanocomposites, giving materials that show the luminescence behaviors of both inorganic phosphors and molecular materials. The controlled preparation and relevant formation mechanism of the microstructures is a second problem for these lanthanide hybrid materials. In the syntheses, some important mechanisms should be emphasized, such as the sol–gel and assembly processes, chemical bonding and weak interactions including the template effects of the polymer unit. The third area for improvement is luminescence enhancement and functional integration of the lanthanide hybrids. Here it is worth pointing out that it is important to develop visible-excitation lanthanide hybrid systems and to obtain white luminescence through multicomponent assembly of lanthanide hybrids and inorganic nanocomposites. The fourth challenge is applications of the photofunctional lanthanide hybrid materials, which strongly depends on the solution of the other three aspects. So far, two important applications have been developed: one is as sensors or in labeling applications in biomedicine, which depend on controlling the scale and structure of the hybrids; the other is in luminescent devices, which depend on the preparation technology.Acknowledgements
This work was supported by the National Natural Science Foundation of China (20301013, 20671072, 20971100, 91122003). The author also thanks Dr Y.J. Li, Dr X.F. Qiao, Dr L. Guo, Dr J.L. Liu, Dr Y. Li, Dr H.F. Lu, Dr Q.M. Wang, and Dr Q.P. Li for their research work.References
- (a) A. de Bettencourt-Dias, Curr. Org. Chem., 2007, 11, 1460 CrossRef CAS; (b) J. C. G. Bunzli and C. Piguet, Chem. Rev., 2002, 102, 1897 CrossRef; (c) D. Parker, Chem. Soc. Rev., 2004, 33, 156 RSC; (d) D. Parker, R. S. Dickins, H. Puschmann, C. Crossl and J. A. K. Howard, Chem. Rev., 2002, 102, 1977 CrossRef CAS; (e) S. V. Eliseeva and J. C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189 RSC.
- (a) J. C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC; (b) J. C. G. Bunzli, Acc. Chem. Res., 2006, 39, 53 CrossRef; (c) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS; (d) K. L. Haas and J. Katherine, Chem. Rev., 2009, 109, 4921 CrossRef CAS; (e) J. C. G. Bunzli, Chem. Rev., 2010, 110, 2729 CrossRef.
- (a) R. C. Ropp, Luminescence and the Solid State, 2nd edn, Elsevier Science, Amsterdam, 2004 Search PubMed; (b) A. Kitai, Luminescent Materials and Applications, Wiley, New York, 2008 Search PubMed.
- (a) N. Sabbatini, M. Guardigli and J. M. Lehn, Coord. Chem. Rev., 1993, 123, 201 CrossRef CAS; (b) A. J. Wilkinson, D. Maffeo, A. Beeby A, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2007, 46, 9438 CrossRef CAS; (c) S. V. Eliseeva and J. C. G. Bunzli, Chem. Soc. Rev., 2010, 39, 189 RSC; (d) J. Kido and Y. Okamoto, Chem. Rev., 2002, 102, 2357 CrossRef CAS.
- C. Sanchez and F. Ribot, New J. Chem., 1994, 18, 1007 CAS.
- (a) L. R. Matthews and E. T. Knobbe, Chem. Mater., 1993, 5, 1697 CrossRef CAS; (b) A. M. Klonkowski, S. Lis, M. Pietraszkiewicz, Z. Hnatejko, K. Czarnobaj and M. Elbanowski, Chem. Mater., 2003, 15, 656 CrossRef CAS.
- (a) C. Sanchez, G. J. D. A. Sloer-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061 CrossRef CAS; (b) A. C. Franville, D. Zambon and R. Mahiou, Chem. Mater., 2000, 12, 428 CrossRef CAS; (c) P. Escribano, B. Julián-López, J. Planelles-Aragó, E. Cordoncillo, B. Viana and C. Sanchez, J. Mater. Chem., 2008, 18, 23 RSC; (d) B. H. Tong, S. J. Wang, Y. Z. Meng and B. Wang, Photochem. Photobiol. Sci., 2007, 6, 519 RSC; (e) Y. F. Ma, H. P. Wang, W. S. Liu, Q. Wang, J. Xu and Y. Tang, J. Phys. Chem. B, 2009, 113, 14139 CrossRef CAS; (f) T. H. Tran, M. M. Lezhnina and U. Kynast, J. Mater. Chem., 2011, 21, 12819 RSC.
- (a) L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509 CrossRef CAS; (b) K. Binnemans, Chem. Rev., 2009, 109, 4283 CrossRef CAS.
- (a) D. F. Parra, H. F. Brito, J. D. R. Matos and L. D. Carlos, J. Appl. Polym. Sci., 2002, 83, 2716 CrossRef CAS; (b) K. Dahmouche, L. D. Carlos, C. V. Santilli, V. D. Bermudez and A. F. Craievich, J. Phys. Chem. B, 2002, 106, 4377 CrossRef CAS; (c) V. D. Bermudez, D. Ostrovskii, M. C. Goncüalves, S. Lavoryk, L. D. Carlos and R. A. S. Ferreira, J. Phys. Chem. B, 2005, 109, 7110 CrossRef; (d) M. C. Goncalves, N. J. O. Silva, V. D. Bermudez, R. A. S. Ferreira, L. D. Carlos, K. Dahmouche, C. V. Santilli, D. Ostrovskii, I. C. C. Vilela and A. F. Craievich, J. Phys. Chem. B, 2005, 109, 20093 CrossRef CAS.
- (a) L. S. Fu, R. A. S. Ferreira, S. S. Nobre, L. D. Carlos and J. Rocha, J. Lumin., 2007, 122–123, 265 CrossRef CAS; (b) C. Molina, R. A. S. Ferreira, G. Poirier, L. S. Fu, S. J. L. Ribeiro, Y. Messsaddeq and L. D. Carlos, J. Phys. Chem. C, 2008, 112, 19346 CrossRef CAS.
- (a) L. D. Carlos, R. A. S. Ferreira, J. P. Rainho and V. D. Bermudez, Adv. Funct. Mater., 2002, 12, 819 CrossRef CAS; (b) Lianshe Fu, R. A. S. Ferreira, N. J. O. Silva, L. D. Carlos, V. de Zea Bermudez and J. Rocha, Chem. Mater., 2004, 16, 1507 CrossRef CAS; (c) L. S. Fu, R. A. S. Ferreira, N. J. O. Silva, J. A. Fernandes, P. Ribeiro-Claro, I. S. Goncalves, V. D. Bermudez and L. D. Carlos, J. Mater. Chem., 2005, 15, 3117 RSC.
- (a) S. C. Nunes, V. D. Bermudez, J. Cybinska, R. A. S. Ferreira, J. Legendziewicz, L. D. Carlos, M. M. Silva, M. J. Smith, D. Ostrovskii and J. Rocha, J. Mater. Chem., 2005, 15, 3876 RSC; (b) C. Molina, P. J. Moreira, R. R. Goncalves, R. A. S. Ferreira, Y. Messaddeq, S. J. L. Ribeiro, O. Soppera, A. P. Leite, P. V. S. Marques, V. D. Bermudez and L. D. Carlos, J. Mater. Chem., 2005, 15, 3937 RSC; (c) P. P. Lima, R. A. S Ferreira, R. O. Freire, F. A. A. Paz, L. S. Fu, S. Alves Jr., L. D. Carlos and O. L. Malta, ChemPhysChem, 2006, 7, 735 CrossRef CAS.
- (a) M. Fernandes, V. D. Bermudez, R. A. S. Ferreira, L. D. Carlos, M. M. Silva and M. J. Smith, Chem. Mater., 2007, 19, 3892 CrossRef CAS; (b) F. Y. Liu, L. D. Carlos, R. A. S. Ferreira, J. Rocha, M. C. Gaudino, M. Robitzer and F. Quignard, Biomolecules, 2008, 9, 1945 CAS; (c) S. S. Nobre, C. D. S. Brites, R. A. S. Ferreira, V. D. Bermudez, C. Carcel, J. I. E. Moreau, J. Rocha, M. W. C. Man and L. D. Carlos, J. Mater. Chem., 2008, 18, 4172–4182 RSC.
- (a) S. Gago, J. A. Fernandes, J. P. Rainho, R. A. S. Ferreira, M. Pillinger, A. A. Valente, T. M. Santos, L. D. Carlos, P. J. A. Ribeiro-Claro and I. S. Goncüalves, Chem. Mater., 2005, 17, 5077 CrossRef CAS; (b) S. M. Bruno, A. C. Coelho, R. A. S. Ferreira, L. D. Carlos, M. Pillinger, A. A. Valente, P. Ribeiro-Claro and I. S. Gonçalves, Eur. J. Inorg. Chem., 2008, 3786 CrossRef CAS; (c) S. M. Bruno, R. A. S. Ferreira, L. D. Carlos, M. Pillinger, P. Ribeiro-Claro and I. S. Goncalves, Microporous Mesoporous Mater., 2008, 113, 453 CrossRef CAS.
- (a) K. Binnemans, P. Lenaerts, K. Driesen and C. Gorller-Walrand, J. Mater. Chem., 2004, 14, 191 RSC; (b) P. Lenaerts, A. Storms, J. Mullens, J. D'Haen, C. Gorller-Walrand, K. Binnemans and K. Driesen, Chem. Mater., 2005, 17, 5194 CrossRef CAS; (c) K. Lunstroot, K. Driesen, P. Nockemann, C. Gorller-Walrand, K. Binnemans, S. Bellayer, J. Le Bideau and A. Vioux, Chem. Mater., 2006, 18, 5711 CrossRef CAS.
- (a) H. R. Li, J. Lin, H. J. Zhang, H. C. Li, L. S. Fu and Q. G. Meng, Chem. Commun., 2001, 1212 RSC; (b) H. R. Li, J. Lin, H. J. Zhang, L. S. Fu, Q. G. Meng and S. B. Wang, Chem. Mater., 2002, 14, 3651 CrossRef CAS; (c) F. Y. Liu, L. S. Fu, J. Wang, Q. G. Meng, H. R. Li, J. F. Guo and H. J. Zhang, New J. Chem., 2003, 27, 233 RSC.
- (a) H. R. Li, J. B. Yu, F. Y. Liu, H. J. Zhang, L. S. Fu, Q. G. Meng, C. Y. Peng and J. Lin, New J. Chem., 2004, 28, 1137 RSC; (b) L. H. Lu, F. Y. Liu, G. Y. Sun, H. J. Zhang, S. Q. Xi and H. S. Wang, J. Mater. Chem., 2004, 14, 2760 RSC; (c) L. N. Sun, H. J. Zhang, L. S. Fu, F. Y. Liu, Q. G. Meng, C. Y. Peng and J. B. Liu, Adv. Funct. Mater., 2005, 15, 1041 CrossRef CAS; (d) L. N. Sun, H. J. Zhang, Q. G. Meng, F. Y. Liu, L. S. Fu, C. Y. Peng, J. B. Yu, G. L. Zheng and S. B. Wang, J. Phys. Chem. B, 2005, 109, 6174 CrossRef CAS.
- (a) H. R. Li, J. Lin, L. S. Fu, J. F. Guo, Q. G. Meng, F. Y. Liu and H. J. Zhang, Microporous Mesoporous Mater., 2002, 55, 103 CrossRef CAS; (b) H. R. Li, L. S. Fu, F. Y. Liu, S. B. Wang and H. J. Zhang, New J. Chem., 2002, 26, 674 RSC.
- (a) X. M. Guo, L. S. Fu, H. J. Zhang, L. D. Carlos, C. Y. Peng, J. F. Guo, J. B. Yu, R. P. Deng and L. N. Sun, New J. Chem., 2005, 29, 1351 RSC; (b) L. N. Sun, J. B. Yu, H. J. Zhang, Q. G. Meng, E. Ma, C. Y. Peng and K.Y. Yang, Microporous Mesoporous Mater., 2007, 98, 156 CrossRef CAS; (c) L. N. Sun, Y. Zhang, B. Yu, S. Y. Yu, S. Dang, C. Y. Peng and H. J. Zhang, Microporous Mesoporous Mater., 2008, 115, 535 CrossRef CAS.
- (a) C. Y. Peng, H. J. Zhang, J. B. Yu, Q. G. Meng, L. S. Fu, H. R. Li, L. N. Sun and X. M. Guo, J. Phys. Chem. B, 2005, 109, 15278 CrossRef CAS; (b) L. N. Sun, H. J. Zhang, C. Y. Peng, J. B. Yu, Q. G. Meng, L. S. Fu, F. Y. Liu and X. M. Guo, J. Phys. Chem. B, 2006, 110, 7249 CrossRef CAS.
- (a) S. Dang, L. N. Sun, H. J. Zhang, X. M. Guo, Z. F. Li, J. Feng, H. D. Guo and Z. Y. Guo, J. Phys. Chem. C, 2008, 112, 13240 CrossRef CAS; (b) L. N. Sun, H. J. Zhang, J. B. Yu, S. Y. Yu, C. Y. Peng, S. Dang, X. M. Guo and J. Feng, Langmuir, 2008, 24, 5500 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, Inorg. Chem. Commun., 2004, 7, 747 CrossRef CAS; (b) Q. M. Wang and B. Yan, J. Mater. Chem., 2004, 14, 2450 RSC; (c) Q. M. Wang and B. Yan, J. Photochem. Photobiol., A, 2006, 178, 70 CrossRef CAS.
- (a) B. Yan, B. Zhou and Q. M. Wang, J. Lumin., 2007, 126, 556 CrossRef CAS; (b) H. F. Lu and B. Yan, J. Photochem. Photobiol., A, 2008, 194, 136 CrossRef CAS; (c) H. F. Lu and B. Yan, J. Photochem. Photobiol., A, 2008, 197, 351 CrossRef CAS; (d) H. F. Lu and B. Yan, J. Fluoresc., 2008, 18, 763 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, Cryst. Growth Des., 2005, 5, 497 CrossRef CAS; (b) B. Yan and L. M. Zhao, Mater. Lett., 2005, 59, 795 CrossRef CAS; (c) Q. M. Wang and B. Yan, J. Mater. Res., 2011, 20, 592 CrossRef; (d) Q. M. Wang and B. Yan, Opt. Mater., 2007, 29, 510 CrossRef CAS; (e) B. Yan and Y. L. Sui, Opt. Mater., 2006, 28, 1216 CrossRef CAS; (f) Q. M. Wang and B. Yan, J. Photochem. Photobiol., A, 2005, 175, 159 CrossRef CAS.
- (a) L. M. Zhao and B. Yan, Mater. Res. Bull., 2006, 41, 1 CrossRef CAS; (b) L. M. Zhao and B. Yan, Colloids Surf., A, 2006, 275, 64 CrossRef CAS; (c) Y. L. Sui and B. Yan, Appl. Surf. Sci., 2006, 252, 4306 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, Mater. Lett., 2006, 60, 3420 CrossRef CAS; (b) B. Yan, B. Zhou and Q. M. Wang, Appl. Organomet. Chem., 2006, 20, 835 CrossRef CAS; (c) B. Yan and Y. L. Sui, Mater. Lett., 2007, 61, 3715 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, J. Photochem. Photobiol., A, 2006, 177, 1 CrossRef CAS; (b) B. Yan, R. F. Yao and Q. M. Wang, Mater. Lett., 2006, 60, 3063 CrossRef CAS; (c) B. Yan, R. F. Yao and L. M. Zhao, J. Non-Cryst. Solids, 2006, 352, 2292 CrossRef CAS; (d) B. Yan and Q. M. Wang, Opt. Mater., 2007, 30, 617 CrossRef CAS.
- (a) L. M. Zhao and B. Yan, Appl. Organomet. Chem., 2005, 19, 1060 CrossRef CAS; (b) Q. M. Wang and B. Yan, J. Organomet. Chem., 2006, 691, 540 CrossRef; (c) Q. M. Wang and B. Yan, J. Organomet. Chem., 2006, 691, 3567 CrossRef CAS; (d) Y. L. Sui and B. Yan, J. Photochem. Photobiol., A, 2006, 182, 1 CrossRef CAS.
- (a) B. Yan and D. J. Ma, J. Solid State Chem., 2006, 179, 2059 CrossRef CAS; (b) B. Yan and X. F. Qiao, Photochem. Photobiol., 2007, 83, 971 CrossRef CAS.
- (a) Q. M. Wang and B. Yan, Inorg. Chem. Commun., 2004, 7, 1124 CrossRef CAS; (b) Q. M. Wang and B. Yan, Appl. Organomet. Chem., 2005, 19, 952 CrossRef CAS; (c) B. Yan and F. F. Wang, J. Organomet. Chem., 2007, 692, 2395 CrossRef CAS; (d) B. Yan, S. Xu and H. F. Lu, J. Fluoresc., 2007, 17, 155 CrossRef CAS; (e) B. Yan, K. Qian and H. F. Lu, Photochem. Photobiol., 2007, 83, 1481 CrossRef CAS.
- L. D. Carlos, R. A. S. Ferreira, V. D. Bermudez, B. Julian-Lopez and P. Escribano, Chem. Soc. Rev., 2011, 40, 536 RSC.
- (a) Y. G. Wang, L. Wang, H. R. Li, P. Liu, D. S. Qin, B.Y. Liu, W. J. Zhang, R.P. Deng and H. J. Zhang, J. Solid State Chem., 2008, 181, 562 CrossRef CAS; (b) P. Liu, H. R. Li, Y. G. Wang, B. Y. Liu, W. J. Zhang, Y. J. Wang, W. D. Yan, H. J. Zhang and U. Schubert, J. Mater. Chem., 2008, 18, 735 RSC; (c) H. R. Li, P. Liu, Y. G. Wang, L. Zhang, J. B. Yu, H. J. Zhang, B. Y. Liu and U. Schubert, J. Phys. Chem. C, 2009, 113, 3945 CrossRef CAS.
- (a) B. Yan and H. F. Lu, J. Organomet. Chem., 2009, 694, 2597 CrossRef CAS; (b) B. Yan and H. F. Lu, J. Photochem. Photobiol., A, 2009, 205, 122 CrossRef CAS; (c) J. L. Liu and B. Yan, J. Organomet. Chem., 2010, 695, 580–587 CrossRef CAS; (d) L. Guo and B. Yan, Eur. J. Inorg. Chem., 2010, 1267 CrossRef CAS; (e) J. L. Liu and B. Yan, Dalton Trans., 2011, 40, 1961 RSC; (f) B. Yan, Y. L. Sui and J. L. Liu, J. Alloys Compd., 2009, 476, 826 CrossRef CAS.
- (a) J. L. Liu and B. Yan, J. Phys. Chem. B, 2008, 112, 10898 CrossRef CAS; (b) B. Yan, Q. M. Wang and D. J. Ma, Inorg. Chem., 2009, 48, 36 CrossRef CAS; (c) H. F. Lu, B. Yan and J. L. Liu, Inorg. Chem., 2009, 48, 3966 CrossRef CAS.
- (a) B. Yan and K. Qian, J. Photochem. Photobiol., A, 2009, 207, 217 CrossRef CAS; (b) B. Yan and K. Qian, Photochem. Photobiol., 2009, 85, 1278 CrossRef CAS; (c) J. L. Liu, B. Yan and L. Guo, Eur. J. Inorg. Chem., 2010, 2290 CrossRef; (d) Y. Chen, Q. Chen, L. Song, H. P. Li and F. Z. Hou, J. Alloys Compd., 2010, 490, 264 CrossRef CAS.
- (a) H. F. Lu and B. Yan, J. Non-Cryst. Solids, 2006, 352, 5331 CrossRef CAS; (b) K. Qian and B. Yan, Polyhedron, 2010, 29, 226 CrossRef CAS.
- (a) B. Yan and H. F. Lu, Inorg. Chem., 2008, 47, 5601 CrossRef CAS; (b) J. L. Liu and B. Yan, J. Photochem. Photobiol., A, 2009, 206, 32 CrossRef CAS; (c) B. Yan, K. Qian and H. F. Lu, J. Organomet. Chem., 2009, 694, 3160 CrossRef CAS; (d) B. Yan, X. L. Wang, K. Qian and H. F. Lu, J. Photochem. Photobiol., A, 2010, 212, 75 CrossRef CAS; (e) J. L. Liu and B. Yan, J. Phys. Chem. C, 2008, 112, 14168 CrossRef CAS.
- (a) B. Yan and Q. M. Wang, Cryst. Growth Des., 2008, 8, 1484 CrossRef CAS; (b) B. Yan, L. L. Kong and B. Zhou, J. Non-Cryst. Solids, 2009, 355, 1281 CrossRef CAS; (c) L. Guo and B. Yan, Inorg. Chem. Commun., 2010, 13, 358 CrossRef CAS; (d) L. Guo, B. Yan and J. L. Liu, Dalton Trans., 2011, 40, 4933 RSC; (e) Y. Y. Li, B. Yan and L. Guo, Inorg. Chem. Commun., 2011, 47, 910 Search PubMed.
- (a) N. N. Lin, H. R. Li, Y. G. Wang, Y. Feng, D. S. Qin, Q. Y. Gan and S. D. Chen, Eur. J. Inorg. Chem., 2008, 4781 CrossRef CAS; (b) J. Feng, J. B. Yu, S. Y. Song, L. N. Sun, W. Q. Fan, X. M. Guo, S. Dang and H. J. Zhang, Dalton Trans., 2009, 2406 RSC.
- (a) H. R. Li, N. N. Lin, Y. G. Wang, Y. Feng, Q. Y. Gan, H. J. Zhang, Q. L. Dong and Y. Chen, Eur. J. Inorg. Chem., 2009, 519 CrossRef CAS; (b) Y. Feng, H. R. Li, Q. Y. Gan, Y. G. Wang, B. Y. Liu and H. J. Zhang, J. Mater. Chem., 2010, 20, 972 RSC; (c) H. S. He, M. Dubey, A. G. Sykes and P. S. May, Dalton Trans., 2010, 39, 6466 RSC.
- (a) X. M. Guo, H. D. Guo, L. S. Fu, H. Zhang, L. D. Carlos, R. P. Deng and J. B. Yu, J. Photochem. Photobiol., A, 2008, 200, 318 CrossRef CAS; (b) X. M. Guo, H. D. Guo, L. S. Fu, L. D. Carlos, R. A. S. Ferreira, L. N. Sun, R. P. Deng and H. J. Zhang, J. Phys. Chem. C, 2009, 113, 12538 CrossRef CAS; (c) J. Feng, L. Zhou, S. Y. Song, Z. F. Li, W. Q. Fan, L. N. Sun, Y. N. Yu and H. J. Zhang, Dalton Trans., 2009, 6593 RSC; (d) Y. H. Wang, B. Li, L. M. Zhang, Q. H. Zuo, L. N. Liu and P. Li, J. Colloid Interface Sci., 2010, 349, 505 CrossRef CAS.
- M. Fernandes, S. S. Nobre, M. C. Goncalves, A. Charas, J. Morgado, R. A. S. Ferreira, L. D. Carlos and V. D. Bermudez, J. Mater. Chem., 2009, 19, 733 RSC.
- K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Goerller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298 RSC.
- (a) Y. G. Wang, H. R. Li, J. J. Gu, Q. Y. Gan, Y. N. Li and G. Calzaferri, Microporous Mesoporous Mater., 2009, 121, 1 CrossRef CAS; (b) H. R. Li, W. J. Cheng, Y. Wang, B. Y. Liu, W. J. Zhang and H. J. Zhang, Chem.–Eur. J., 2010, 16, 2125 CrossRef CAS; (c) Y. Wang, H. R. Li, Y. Feng, H. J. Zhang, G. Calzaferri and T. Z. Ren, Angew. Chem., Int. Ed., 2010, 49, 1434 CrossRef CAS; (d) P. P. Cao, Y. G. Wang, H. R. Li and X. Y. Yu, J. Mater. Chem., 2011, 21, 2709 RSC; (e) Y. Wang, Y. G. Wang, P. P. Wang, Y. N. Li and H. R. Li, CrystEngComm, 2011, 13, 177 RSC.
- (a) C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS; (b) J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS; (c) B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 1001, 3140 Search PubMed; (d) M. H. Lim and A. Stein, Chem. Mater., 1999, 11, 3285 CrossRef CAS.
- (a) D. Y. Zhao, J. P. Feng, Q. S. Huo, N. Melosh, C. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 348 Search PubMed; (b) D. Y. Zhao, J. Y. Sun, Q. Z. Li and G. D. Stucky, Chem. Mater., 2000, 12, 275 CrossRef CAS; (c) B. L. Newalkar, S. Komarneni and H. Katsuki, Chem. Commun., 2000, 2389 RSC; (d) S. Madhugiri, A. Dalton, J. Gutierrez, J. P. Ferraris and K. J. Balkus, J. Am. Chem. Soc., 2003, 125, 14531 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- (a) B. J. Melde, B. T. Holland, C. F. Blanford and A. Stein, Chem. Mater., 1999, 11, 3302 CrossRef CAS; (b) S. Inagaki, S. Guan, Y. Fukushima, T. Ohsuna and O. Terasaki, J. Am. Chem. Soc., 1999, 121, 9611 CrossRef CAS; (c) T. Asefa, M. J. MacLachlan, N. Coombs and G. A. Ozin, Nature, 1999, 402, 867 CAS; (d) A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403 CrossRef CAS.
- (a) P. N. Minoofar, R. Hernandez, S. Chia, B. Dunn, J. I. Zink and A. C. Franville, J. Am. Chem. Soc., 2002, 124, 14388 CrossRef CAS; (b) J. T. Mitchell-Koch and A. S. Borovith, Chem. Mater., 2003, 15, 3490 CrossRef CAS; (c) Q. H. Xu, L. S. Li, B. Li, J. H. Yu and R. R. Xu, Microporous Mesoporous Mater., 2000, 38, 351 CrossRef CAS; (d) S. Gago, J. A. Fernandes, J. P. Rainho, R. A. S. Ferreira, M. Pillinger, A. A. Valente, T. M. Santos, L. D. Carlos, P. J. A. Ribeiro-Claro and I. S. Goncalves, Chem. Mater., 2005, 17, 5077 CrossRef CAS.
- (a) J. Feng, S. Y. Song, Y. Xing, H. J. Zhang, Z. F. Li, L. N. Sun, X. M. Guo and W. Q. Fan, J. Solid State Chem., 2009, 182, 435 CrossRef CAS; (b) J. Feng, S. Y. Song, W. Q. Fan, L. N. Sun, X. M. Guo, C. Y. Peng, J. B. Y. N. Yu and H. J. Zhang, Microporous Mesoporous Mater., 2009, 117, 278 CrossRef CAS.
- (a) B. Yan and B. Zhou, J. Photochem. Photobiol., A, 2008, 195, 314 CrossRef CAS; (b) Y. Li and B. Yan, J. Solid State Chem., 2008, 181, 1032 CrossRef CAS; (c) B. Yan, B. Zhou and Y. Li, Microporous Mesoporous Mater., 2009, 120, 317 CrossRef CAS; (d) Y. Li and B. Yan, J. Fluoresc., 2008, 19, 191–201 CrossRef; (e) Y. Li and B. Yan, Microporous Mesoporous Mater., 2010, 128, 62 CrossRef CAS; (f) Y. J. Li, B. Yan and L. Wang, J. Solid State Chem., 2011, 184, 2571 CrossRef CAS.
- (a) Q. G. Meng, P. Boutinaud, A. C. Franville, H. J. Zhang and R. Mahiou, Microporous Mesoporous Mater., 2003, 65, 127 CrossRef CAS; (b) Q. G. Meng, P. Boutinaud, H. J. Zhang and R. Mahiou, J. Lumin., 2007, 124, 15 CrossRef CAS.
- L. N. Sun, S. Dang, J. B. Yu, J. Feng, L. Y. Shi and H. J. Zhang, J. Phys. Chem. B, 2010, 114, 16393 CrossRef CAS.
- (a) Y. Li, B. Yan and H. Yang, J. Phys. Chem. C, 2008, 112, 3959 CrossRef CAS; (b) B. Yan and Y. Li, Dalton Trans., 2010, 39, 1480 RSC; (c) Y. J. Li, B. Yan and Y. Li, Microporous Mesoporous Mater., 2010, 131, 82 CrossRef CAS; (d) Y. Y. Li, B. Yan, L. Guo and Y. J. Li, Microporous Mesoporous Mater., 2012, 48, 73 Search PubMed.
- (a) L. L. Kong, B. Yan, Y. J. Li and Y. Li, Microporous Mesoporous Mater., 2010, 135, 45 CrossRef CAS; (b) L. L. Kong, B. Yan and Y. Li, J. Alloys Compd., 2009, 481, 549 CrossRef CAS; (c) L. L. Kong, B. Yan and Y. Li, J. Solid State Chem., 2009, 182, 1631 CrossRef; (d) Y. J. Li, B. Yan and L. Wang, Dalton Trans., 2011, 40, 6722 RSC.
- Y. J. Li, B. Yan and Y. Li, J. Solid State Chem., 2010, 183, 871–877 CrossRef CAS.
- (a) Y. J. Li and B. Yan, Inorg. Chem., 2009, 48, 8276 CrossRef CAS; (b) Y. J. Li and B. Yan, Dalton Trans., 2010, 39, 2554 RSC; (c) Y. J. Li, B. Yan and Y. Li, Chem.–Asian J., 2010, 5, 1642 CrossRef CAS.
- (a) X. M. Guo, X. M. Wang, H. J. Zhang, L. S. Fu, H. D. Guo, J. B. Yu, L.D. Carlos and K. Y. Yang, Microporous Mesoporous Mater., 2008, 116, 28 CrossRef CAS; (b) X. M. Guo, H. D. Guo, L. S. Fu, H. J. Zhang, R. P. Deng, L. N. Sun, J. Feng and S. Dang, Microporous Mesoporous Mater., 2009, 119, 252 CrossRef CAS; (c) X. M. Guo, H. D. Guo, L. S. Fu, R. P. Deng, W. Chen, J. Feng, S. Dang and H. J. Zhang, J. Phys. Chem. C, 2009, 113, 2603 CrossRef CAS.
- (a) Y. Li, B. Yan and Y. J. Li, Microporous Mesoporous Mater., 2010, 132, 87 CrossRef CAS; (b) Y. J. Li, L. Wang and B. Yan, J. Mater. Chem., 2011, 21, 1130 RSC.
- (a) Y. Ueba, E. Banks and Y. Okmoto, J. Appl. Polym. Sci., 1980, 25, 2007 CrossRef CAS; (b) Y. Okmoto, Y. Ueba, N. F. D. Zhanibekov and E. Banks, Macromolecules, 1981, 14, 17 CrossRef.
- (a) J. M. Huang, V. Bekeari, P. Lianos and S. Carios, J. Lumin., 1999, 81, 285 CrossRef CAS; (b) R. Moleski, E. Stathatos, V. Bekeari and P. Lianos, Thin Solid Films, 2002, 416, 279 CrossRef CAS; (c) A. C. Franville, D. Zambon, R. Mahiou and Y. Troin, Chem. Mater., 2000, 12, 428 CrossRef CAS; (d) V. Bekeari, P. Lianos, U. L. Stangar, B. Orel and P. Judeinstein, Chem. Mater., 2000, 12, 3095 CrossRef; (e) R. Shunmugam and G. N. Tew, Macromol. Rapid Commun., 2008, 29, 1355 CrossRef CAS; (f) Z. Q. Huang, H. H. Zhang, J. X. Guo, J. L. Wang and Y. Q. Chen, Polym. Eng. Sci., 2009, 49, 1273 CrossRef CAS.
- (a) C. Guizard and P. Lacan, New J. Chem., 1994, 18, 1097 CAS; (b) H. Y. Chen and R. D. Archer, Macromolecules, 1996, 29, 1957 CrossRef CAS; (c) V. Bekiari, G. Pistolis and P. Lianos, Chem. Mater., 1999, 11, 3189 CrossRef CAS; (d) L. H. Wang, W. Wang, W. G. Zhang, E. T. Kang and W. Huang, Chem. Mater., 2000, 12, 2212 CrossRef CAS; (e) X. G. Huang, Q. Wang, X. H. Yan, J. Xu, W. S. Liu, Q. Wang and Y. Tang, J. Phys. Chem. C, 2011, 115, 2332 CrossRef CAS.
- (a) M. Fernandes, V. D. Bermudez, R. A. S. Ferreira, L. D. Carlos and N. V. Martins, J. Lumin., 2008, 128, 205 CrossRef CAS; (b) F. Y. Liu, L. D. Carlos, R. A. S. Ferreira, J. Rocha, M. C. Ferro, A. Tourrette, F. Quignard and M. Robitzer, J. Phys. Chem. B, 2010, 114, 77 CrossRef CAS; (c) S. C. Nunes, J. Planelles-Arago, R. A. S. Ferreira, L. D. Carlos and V. D. Bermudez, Eur. J. Inorg. Chem., 2010, 2688 CrossRef CAS; (d) C. Molina, R. A. S. Ferreira, G. Poirier, L. S. Fu, S. J. L. Ribeiro, Y. Messsaddeq and L. D. Carlos, J. Phys. Chem. C, 2008, 112, 19346 CrossRef CAS.
- (a) P. P. Lima, R. A. S. Ferreira, S. A. Júnior, O. L. Malta and L. D. Carlos, J. Photochem. Photobiol., A, 2009, 201, 214 CrossRef CAS; (b) M. E. Mesquita, S. S. Nobre, M. Fernandes, R. A. S. Ferreira, S. C. G. Santos, M. O. Rodrigues, L. D. Carlos and V. D. Bermudezc, J. Photochem. Photobiol., A, 2009, 205, 156 CrossRef CAS.
- (a) L. M. Zhao and B. Yan, J. Non-Cryst. Solids, 2007, 353, 4654 CrossRef CAS; (b) B. Yan, L. M. Zhao and J. L. Liu, J. Photochem. Photobiol., A, 2008, 199, 50 CrossRef CAS; (c) B. Yan and Q. M. Wang, J. Photochem. Photobiol., A, 2008, 197, 213 CrossRef CAS; (d) B. Yan and X. F. Qiao, J. Phys. Chem. B, 2007, 111, 12362 CrossRef CAS; (e) X. F. Qiao and B. Yan, J. Photochem. Photobiol., A, 2008, 199, 188 CrossRef CAS; (f) X. F. Qiao and B. Yan, J. Phys. Chem. B, 2008, 112, 14742 CrossRef CAS.
- (a) X. F. Qiao and B. Yan, Inorg. Chem., 2009, 48, 4714 CrossRef CAS; (b) X. F. Qiao and B. Yan, Eur. Polym. J., 2009, 45, 2002 CrossRef CAS; (c) K. Sheng and B. Yan, J. Photochem. Photobiol., A, 2009, 206, 140 CrossRef CAS; (d) X. F. Qiao and B. Yan, Dalton Trans., 2009, 8509 RSC; (e) K. Sheng, B. Yan, X. F. Qiao and L. Guo, J. Photochem. Photobiol., A, 2010, 210, 36 CrossRef CAS.
- (a) L. Guo, B. Yan and Y. Li, Photochem. Photobiol., 2010, 86, 813 CrossRef CAS; (b) K. Sheng, B. Yan, H. F. Lu and L. Guo, Eur. J. Inorg. Chem., 2010, 3498 CrossRef CAS; (c) B. Yan, K. Qian and X. L. Wang, Inorg. Chim. Acta, 2011, 376, 302 CrossRef CAS; (d) X. F. Qiao, H. Y. Zhang and B. Yan, Dalton Trans., 2010, 39, 8882 RSC; (e) L. Guo, B. Yan and K. Sheng, Dalton Trans., 2011, 40, 632 RSC; (f) L. Guo and B. Yan, Photochem. Photobiol., 2010, 86, 1185 CrossRef CAS.
- (a) X. F. Qiao and B. Yan, J. Organomet. Chem., 2009, 694, 3232 CrossRef CAS; (b) X. F. Qiao and B. Yan, J. Phys. Chem. B, 2009, 113, 11865 CrossRef CAS; (c) X. L. Wang, B. Yan and J. L. Liu, Colloid Polym. Sci., 2010, 288, 1139 CrossRef CAS; (d) M. Guo, B. Yan, L. Guo and X. F. Qiao, Colloids Surf., A, 2011, 380, 53 CrossRef CAS.
- (a) X. L. Wang and B. Yan, Colloid Polym. Sci., 2011, 289, 423 CrossRef CAS; (b) X. L. Wang, B. Yan and J. L. Liu, Photochem. Photobiol. Sci., 2011, 10, 580 RSC; (c) B. Yan, X. L. Wang and J. L. Liu, Photochem. Photobiol., 2011, 87, 602 CrossRef CAS; (d) B. Yan, M. Guo and X. F. Qiao, Photochem. Photobiol., 2011, 87, 786 CrossRef CAS; (e) B. Yan, L. M. Zhao, X. L. Wang and Y. Zhao, RSC Adv., 2011, 1, 1064–1071 RSC.
- (a) D. M. Wang, J. H. Zhang, Q. Lin, L. S. Fu, H. J. Zhang and B. Yang, J. Mater. Chem., 2003, 13, 2279 RSC; (b) X. F. Qiao and B. Yan, New J. Chem., 2011, 35, 568 RSC.
- (a) L. Guo and B. Yan, J. Photochem. Photobiol., A, 2011, 224, 141 CrossRef CAS; (b) Y. J. Li and B. Yan, J. Mater. Chem., 2011, 21, 8129 RSC; (c) B. Yan and Y. J. Li, J. Mater. Chem., 2011, 21, 18464 Search PubMed; (d) K. Lunstroot, K. Driesen, P. Nockemann, K. Van Hecke, L. Van Meervelt, C. Goerller-Walrand, K. Binnemans, S. Bellayer, L. Viau, J. Le Bideau and A. Vioux, Dalton Trans., 2009, 298 RSC.
- (a) C. Wang, B. Yan, J. L. Liu and L. Guo, Eur. J. Inorg. Chem., 2011, 879 CrossRef; (b) B. Yan, C. Wang, L. Guo and J. L. Liu, Photochem. Photobiol., 2010, 86, 499 CrossRef CAS; (c) C. Wang and B. Yan, J. Fluoresc., 2011, 21, 1239 CrossRef CAS; (d) B. Yan and C. Wang, Inorg. Chem. Commun., 2011, 14, 1494 CrossRef CAS; (e) B. Yan and C. Wang, Mater. Res. Bull., 2011, 46, 2515 CrossRef.
- (a) B. H. Kwon, H. S. Jang, H. S. Yoo, S. W. Kim, D. S. Kang, S. Maeng, D. S. Jang, H. Kimand and D. Y. Jeon, J. Mater. Chem., 2011, 21, 12812 RSC; (b) Y. J. Li and B. Yan, Photochem. Photobiol., 2010, 86, 1008 CrossRef CAS; (c) B. Yan, Y. Zhao and Y. J. Li, Photochem. Photobiol., 2011, 87, 757 CrossRef CAS; (d) B. Yan, Y. Zhao and Q. P. Li, J. Photochem. Photobiol., A, 2011, 222, 351 CrossRef CAS; (e) Y. Zhao and B. Yan, Photochem. Photobiol., 2012, 88, 21 CrossRef CAS.
- (a) W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, G. L. Zheng and H. J. Zhang, Chem.–Eur. J., 2010, 16, 1903 CrossRef CAS; (b) W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, L. Zhou, G. L. Zheng, S. Dang, S. Wang and H. J. Zhang, Nanoscale, 2010, 2, 2096 RSC; (c) W. Q. Fan, J. Feng, S. Y. Song, Y. Q. Lei, Y. Xing, R. P. Deng, S. Dang and H. J. Zhang, Eur. J. Inorg. Chem., 2008, 5513 CrossRef CAS.
- (a) S. Y. Yu, H. J. Zhang, J. B. Yu, C. Wang, L. N. Sun and W. D. Shi, Langmuir, 2007, 23, 7836 CrossRef CAS; (b) J. Feng, S. Y. Song, R. P. Deng, W. Q. Fan and H. J. Zhang, Langmuir, 2010, 26, 3596 CrossRef CAS; (c) J. Feng, W. Q. Fan, S. Y. Song, Y. N. Yu, R. P. Deng and H. J. Zhang, Dalton Trans., 2010, 39, 5166 RSC.
- (a) S. Gago, M. Pillinger, R. A. S. Ferreira, L. D. Carlos, T. M. Santos and I. S. Goncüalves, Chem. Mater., 2005, 17, 5803 CrossRef CAS; (b) E. H. de Faria, E. J. Nassar, K. J. Ciuffi, M. A. Vicente, R. Trujillano, V. Rives and P. S. Calefi, ACS Appl. Mater. Interfaces, 2011, 3, 1311 CrossRef CAS.
- (a) L. Maggini, J. Mohanraj, H. Traboulsi, A. Parisini, G. Accorsi, N. Armaroli and D. Bonifazi, Chem.–Eur. J., 2011, 17, 8533 CrossRef CAS; (b) L. Maggini, H. Traboulsi, K. Yoosaf, J. Mohanraj, J. Wouters, O. Pietraszkiewicz, M. Pietraszkiewicz, N. Armaroli and D. Bonifazi, Chem. Commun., 2011, 47, 1625 RSC.
- (a) P. P. Lima, S. S. Nobre, R. O. Freire, S. A. Junior, R. A. S. Ferreira, U. Pischel, O. L. Malta and L. D. Carlos, J. Phys. Chem. C, 2007, 111, 17627 CrossRef CAS; (b) S. S. Nobre, X. Cattoen, R. A. S. Ferreira, C. Carcel, V. D. Bermudez, M. W. C. Man and L. D. Carlos, Chem. Mater., 2010, 22, 3599 CrossRef CAS.
- (a) L. M. Zhao and B. Yan, J. Lumin., 2006, 118, 317 CrossRef CAS; (b) F. F. Wang and B. Yan, J. Photochem. Photobiol., A, 2008, 194, 238 CrossRef CAS; (c) B. Yan and L. Kong, Nanoscale Res. Lett., 2010, 5, 1195 CrossRef CAS; (d) K. Sheng, B. Yan and X. F. Qiao, J. Fluoresc., 2010, 21, 653 CrossRef.
- (a) B. Carmen Barja and P. F. Aramendia, Photochem. Photobiol. Sci., 2008, 7, 1391 RSC; (b) Q. H. Zuo, B. Li, L. M. Zhang, Y. H. Wang, Y. H. Liu, J. Zhang, Y. Chen and L. F. Guo, J. Solid State Chem., 2010, 183, 1715 CrossRef CAS.
- (a) C. L. Tan, Q. M. Wang and L. J. Ma, Photochem. Photobiol., 2010, 86, 1191 CrossRef CAS; (b) Q. M. Wang, C. L. Tan, H. Y. Chen and H. Tamiaki, J. Phys. Chem. C, 2010, 114, 13879 CAS; (c) J. T. Lin, Q. M. Wang, C. L. Tan and H. Y. Chen, Synth. Met., 2010, 160, 1780 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |