Synthesis of graphene-based nanomaterials and their application in energy-related and environmental-related areas
Guixia
Zhao
,
Tao
Wen
,
Changlun
Chen
and
Xiangke
Wang
*
Key Laboratory of Novel Thin Film Solar Cells, Institute of Plasma Physics, Chinese Academy of Sciences, Hefei, 230031, China. E-mail: xkwang@ipp.ac.cn; Fax: +86-0551-5591310; Tel: +86-0551-5592788
First published on 26th July 2012
Abstract
As a fascinating two-dimensional carbon allotrope, graphene has triggered a ‘gold rush’ all over scientific research areas especially since the Nobel Prize for Physics in 2010. To exploit the prominent properties of graphene-based nanomaterials, two important problems are focused in this review: one is the synthesis of these graphene-based nanomaterials with different kinds of well-defined structures, and the other is the effective application of them as active nanomaterials in functional devices or processes. In this critical review, from the viewpoint of chemistry and materials, we give a brief overview of the recent significant advances in the synthesis of graphene-based nanomaterials and their applications in energy-related areas and environmental pollution remediation areas, including supercapacitors, lithium ion batteries, solar cells, adsorption, and degradation of organic/inorganic pollutants from large volumes of aqueous solutions in environmental pollution cleanup. The main challenges and perspectives of the materials for future research are also discussed.
1. Introduction
Owing to its extraordinary structure and properties, graphene, a honeycomb network of sp2 carbon atoms, with single-atom thickness, has attained a central position as a “miraculous” material in the beginning of the 21st century. Graphene exhibits excellent electronic, optical, mechanical and thermal properties, such as the high values of its mobility of charge carriers (200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm2 V−1 s−1),1 fracture strength (125 GPa),2 Young's modulus (∼1100 GPa),2 thermal conductivity (∼5000 W m−1 K−1),3 specific surface area (theoretical value of 2630 m2 g−1),4 and high optical transmittance.4 These attractive properties have triggered huge interest from different research fields concerned with energy conversion/storage and environmental pollution remediation, both of which are the most pressing and hottest issues in modern society. However, for those practical applications in supercapacitors, lithium ion batteries, solar cells, adsorption and degradation of different kinds of pollutants from large volumes of aqueous solutions, graphene needs to be available and processable in large quantity and good quality. To fully utilise the superior properties of graphene, fabrication of graphene composites with other functional materials is another important academic and technological endeavor, for which extraordinary performances have been witnessed in many scientific research areas. For instance, in energy-related areas, modified graphene materials have been used in solar cells5 while metallic/metal oxides combined with graphene have been used in lithium ion batteries,6 supercapacitors7 and in fuel cells as catalysts.8 In the environmental pollution remediation area, many graphene and magnetic graphene nanomaterials have been used as adsorbents for heavy metal ions and organic pollutants9 while several transition-metal oxide graphene hybrids have been used for the degradation of toxic organic pollutants.10 Some graphene-based materials have been fabricated as sensor devices for pollutant analysis.11 These treatments toward graphene not only improve its pristine properties, but also introduce some specific functional groups or other functional nanomaterials on the surfaces of graphene layers. Thus, these studies have extended the research and application of graphene-based nanomaterials in multidisciplinary areas. Although some researchers have studied the toxicity of graphene nanosheets,12 we believe that their intriguing properties outweigh possible biohazards and it is hoped that any possible toxicity can be reduced or avoided in the near future.
000 cm2 V−1 s−1),1 fracture strength (125 GPa),2 Young's modulus (∼1100 GPa),2 thermal conductivity (∼5000 W m−1 K−1),3 specific surface area (theoretical value of 2630 m2 g−1),4 and high optical transmittance.4 These attractive properties have triggered huge interest from different research fields concerned with energy conversion/storage and environmental pollution remediation, both of which are the most pressing and hottest issues in modern society. However, for those practical applications in supercapacitors, lithium ion batteries, solar cells, adsorption and degradation of different kinds of pollutants from large volumes of aqueous solutions, graphene needs to be available and processable in large quantity and good quality. To fully utilise the superior properties of graphene, fabrication of graphene composites with other functional materials is another important academic and technological endeavor, for which extraordinary performances have been witnessed in many scientific research areas. For instance, in energy-related areas, modified graphene materials have been used in solar cells5 while metallic/metal oxides combined with graphene have been used in lithium ion batteries,6 supercapacitors7 and in fuel cells as catalysts.8 In the environmental pollution remediation area, many graphene and magnetic graphene nanomaterials have been used as adsorbents for heavy metal ions and organic pollutants9 while several transition-metal oxide graphene hybrids have been used for the degradation of toxic organic pollutants.10 Some graphene-based materials have been fabricated as sensor devices for pollutant analysis.11 These treatments toward graphene not only improve its pristine properties, but also introduce some specific functional groups or other functional nanomaterials on the surfaces of graphene layers. Thus, these studies have extended the research and application of graphene-based nanomaterials in multidisciplinary areas. Although some researchers have studied the toxicity of graphene nanosheets,12 we believe that their intriguing properties outweigh possible biohazards and it is hoped that any possible toxicity can be reduced or avoided in the near future.
Herein, the recent significant advance in the synthesis of graphene-based nanomaterials and their applications in energy-related areas and environmental-related areas, including supercapacitors, lithium ion batteries, solar cells and organic/inorganic pollutant management, are focused and reviewed. We believe that this comprehensive review will provide a general understanding to the present research about graphene-based nanomaterials and their perspective in future nanomaterials research, although clearly it can not include all of the published work, due to the large activity of this field.
2. Synthesis of graphene-based nanomaterials
Since the micromechanical exfoliation of graphene in 1999, a wide variety of studies have been focused on its fabrication. To realize their various applications, graphene-based nanomaterials are firstly needed to be synthesized with controlled size, thickness and morphology. Therefore, the development of effective approaches for their synthesis is of crucial importance. Up to now, there are two main strategies for the synthesis of graphene originated from different carbon sources, the ‘top-down’ and ‘bottom-up’ approaches.2.1. The top-down approach
The graphitic materials, such as 0D fullerenes, 1D nanotubes and 3D graphite, can be chosen as carbon source for the synthesis of graphene. The most popular top-down method is via the oxidative exfoliation of graphite through the Staudemaier, Brodie and Hummers methods.13 The prepared graphite oxides are further expanded into one- or few-layered graphenes by fast heating-treatment or exfoliated into individual graphene oxide nanosheets by ultrasonic treatment in aqueous solution. In Chen's group, a lot of research on the controlled synthesis of graphene oxides has been carried out.14Fig. 1 shows the basic process of the oxidative exfoliation. The graphene oxides are then reduced into graphene by means of thermal annealing,15 solvothermal reduction,16 electrochemical reduction,17 hydrogen plasma treatment,18 or radiation-induced reduction,19etc. Among these methods, the most frequently used method is chemical reduction using reductants such as hydrazine, sodium borohydride, metal Fe, Vitamin C, dimethylhydrazine, alcohols and hydroquinone.20 By using sulfur-containing compounds such as NaHSO3, Na2SO3, Na2S2O3, Na2S, Chen et al.21 found that the reducing ability of NaHSO3 is comparable to that of hydrazine, with the advantages of low toxicity and non-volatility. However, these reductions are processed at high reduction temperature. Recently, great efforts have been devoted to develop mild reduction processes. For example, Kaminska et al.22 proposed an easy and environmental friendly method for the reduction and simultaneously noncovalent functionalization of graphene oxide by using dopamine at room temperature. Under a similar condition, Tung et al.23 presented a solution-based approach to synthesize large-scale and single-layer chemically converted graphene by directly dispersing graphene oxide paper in pure hydrazine. At a lower temperature (i.e., subzero temperature), a new reducing agent system, hydriodic acid with trifluoroacetic acid can chemically reduce graphene oxide into graphene according to Cui et al.24 The above mentioned methods are processed in solution, while Liang et al.25 put forward a facile green approach for flexible graphene film by the reduction of graphene oxide in the gas phase, in which a solid graphene oxide film can be efficiently reduced by hydrogen at room temperature with a small amount of Pd as catalyst. Thermal expansion of graphite oxide is a useful method for the synthesis of functionalized single-layered graphene.15a,26 McAllister and co-workers26 provided a detailed analysis on the thermal expansion mechanism. It is considered that the exfoliation takes place when the decomposition rate of the epoxy and hydroxyl sites of graphite oxide exceeds the diffusion rate of the evolved gases.26 Peng et al.17 introduced an electrochemical method to reduce graphene oxide under constant potential. By control of the electrical current, applied voltage, reduction time and the amount of precursor (graphene oxide), high quality electrochemically reduced graphene oxide film with controllable size and thickness was obtained. More conveniently, Kumar et al.19 reported the radiation-induced reduction of graphene oxide by using sunlight, UV light and KrF excimer laser. It has been found that after prolonged irradiation under sunlight or ultraviolet light, graphene oxide can be well reduced. Within a short time under laser irradiation, graphene with negligible oxygen functionalities can be obtained.19 | ||
| Fig. 1 (A) Outlined oxidization/intercalation process for the preparation of FGO and GO; (B) XRD patterns of FGO and GO.14b | ||
It is obvious that the exfoliation of the graphite oxide and the subsequent chemical reduction are convenient for the mass production, functionalization and solution processing of graphene. However, this method inevitably introduces large amounts of defects into the graphene framework because of the formation of oxygen-containing groups, the ultrasonic treatment and the incomplete reduction. These defects lead to deterioration of the electronic properties of the reduced graphene oxide. Several groups reported that pristine graphite could be exfoliated to defect-free graphene monolayers in different solvents such as N,N-dimethylformamide (DMF), ortho-dichlorobenzene, N-methylpyrrolidone (NMP), benzylamine, ionic liquid, or in some surfactant–water solutions. For example, as shown in Fig. 2, Hernandez et al.27 demonstrated that through dispersion and exfoliation of graphite in organic solvents such as N-methylpyrrolidone, whose surface energy matches that of graphene, the graphite layers can be exfoliated because the required energy for the exfoliation is balanced by the solvent–graphene interaction. One of the problems for this method is that the effective solvents often have high boiling points, which makes it difficult to remove the solvents. According to O'Neill et al.,28 the exfoliation of graphene at higher concentration in low-boiling point solvents such as chloroform and isopropanol can be achieved, with a thickness of less than 10 layers. Apart from these exfoliation methods, high-yield graphene with good quality was also prepared via the exfoliation–reinteraction–expansion of graphite, where graphite was treated with oleum without excessive chemical functionalization.29 Through fluorination of graphite, thermal expansion process and the followed ultrasonic treatment, the obtained graphene can be dispersed well in organic solvents or in aqueous solutions containing sodium dodecylbenzene sulfonate as surfactant according to Lee et al.30
 | ||
| Fig. 2 Electron microscopy of graphite and graphene: (a) SEM image of sieved, pristine graphite (scale bar: 500 μm). (b) SEM image of sediment after centrifugation (scale bar: 25 μm). (c–e): Bright-field TEM images of monolayer graphene flakes deposited from γ-butyrolactone (GBL) (c), 1,3-dimethyl-2-imidazolidinone (DMEU) (d) and NMP (e), respectively (scale bar: 500 nm). (f), (g), Bright-field TEM images of a folded graphene sheet and multilayer graphene, both deposited from NMP (scale bar: 500 nm). (h), Histogram of the number of visual observations of flakes as a function of the number of monolayers per flake for NMP dispersions.27 | ||
There is also a more facile method for the exfoliation of graphite oxide by using laser excitation other than using any chemical reducing agent.31 Abdelsayed et al.31a reported a solution processable synthesis of individual graphene nanosheets in water without reductants. According to their report, the authors confirmed the high performance of graphite oxide and laser converted graphene in the conversion of the laser radiation into usable heat. More specifically, Sokolov et al.31b found that graphene features could be produced via continuous-wave (532 nm) or pulsed (532 and 355 nm) laser excitation of graphite oxide. According to the Sokolov et al.,31b initial excitation results in electron–hole plasmas within the material. Then the strain associated with the oxygen-containing groups leads to the trapping of excitons and holes. The phonon coupling results in efficient heating and material removal, which lead to an expanding plasma plume. Finally, the formation and growth of graphene nanoparticles/sheets in the gas phase and on the surface are realized.31b
Another interesting carbon source for top-down synthesis of graphene is carbon nanotubes (CNTs), especially for the synthesis of graphene nanoribbons (GNRs). The chemical unzipping of CNTs resulted in elongated graphene strips by using acid reactions,32 plasma treatment,33 liquid NH3 and Li intercalation–exfoliation34 and catalytic approaches.35 According to Kosynkin and co-workers,32 a high-yielding procedure for the fabrication of single- and few-layered GNRs through oxidative longitudinal unzipping of multiwalled carbon nanotubes (MWCNTs) in sulfuric acid was realized. However, due to the excessive oxidation, those narrow ribbons derived from the inner tubes are readily destroyed while the contiguous regions of the basal plane from the wider ribbons are often disrupted, resulting in holes of various shapes and sizes; as a consequence the electronic performance is detrimentally affected.32 To overcome this disadvantage an improved method was further proposed and changes in the reaction conditions such as acid ratio, time and temperature were investigated (see Fig. 3). It was found that sufficiently concentrated H2SO4 (∼90 vol%) and the elevation of the reaction temperature to 60 °C were important for the formation and exfoliation of GONRs, and the addition of 10 vol% of a second acid (H3PO4 or trifluoroacetic acid) can greatly enhance the quality of the GONRs.36 In Dai's group,37 the atomic structures, Raman spectroscopic and the electrical transport properties of individual graphene nanoribbons obtained by sonochemical unzipping of MWCNTs were clearly investigated, and they found that a large fraction of GNRs were bent with smooth edges, and most of them were two-layered.
 | ||
| Fig. 3
a
Previously published conditions and results.32bWeight equivalents of KMnO4 relative to MWCNT weight in the starting material. cWeight gain (%) upon isolation of the product relative to the starting weight of MWCNTs. dAll scale bars are 100 nm. eWeight percent remaining at 250 °C by TGA at 10 °C min−1 under Ar. fWeight percent remaining at 950 °C. gRecorded in water. hThe darker portion in the TEM micrograph is part of the lacey carbon grid. iPreferred conditions for optimized GONRs.36 | ||
To better control the quality of graphene layers along with the high-field preparation, Shinde et al.38 used electrochemical oxidation to accurately control the degree and sites of oxidation under ambient conditions with the unique advantage of thickness and orientation control. In their method, an interfacial electric field is able to orient the CNTs and longitudinal unzipping is more favorable instead of a random breakdown in chemical methods because possible C–C cleavage initiated at topological defects has sufficient strain which makes the C–C cleavage is easy to realize.
In 2011, Perdigão and co-workers39 reported graphene formation by the thermal induced decomposition of C60 in combination with a Ni thin film. The formation of graphene was verified by releasing the resulting layers by etching the metal substrate. The main processes are illustrated in Fig. 4. They also found that the carbon present at a buried metal/SiO2 layer can diffuse and segregate at the surface, and graphene can grow from the adsorbed carbon.39 However, from the high D/G intensity ratio in the Raman spectra, it can be concluded that graphene produced by this method appears to be highly defective.
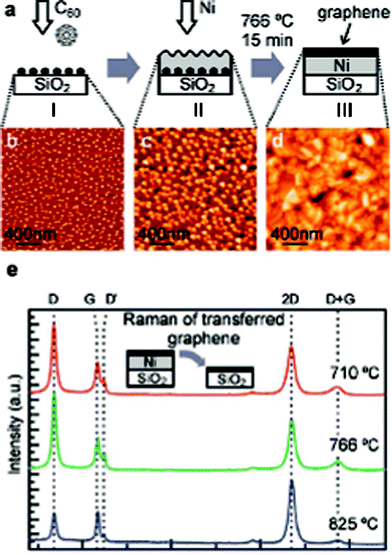 | ||
| Fig. 4 Graphene synthesized from C60 buried under a nickel film on a SiO2 surface. (a) The preparation procedure performed in vacuum: (I) ∼1.6 nm of C60 was deposited on a predegassed SiO2 surface, (II) nickel film was grown by evaporation, and then (III) the whole assembly was annealed at a chosen temperature for 15 min. (b–d) tapping mode 2 × 2 μm AFM images of samples taken out after stages I–III, respectively. (e) Raman spectra of transferred graphene prepared at different annealing temperatures, with the peak assignments D, G, D′, 2D and D + G shown by dashed lines.39 | ||
There are some new methods for graphene synthesis.40 Theoretically, Miyamato et al.40a proposed to use ultrashort laser pulses to detach graphene monolayers from graphite, one at a time. After calculations by an ab initio study, the authors inferred that photo-exfoliation should be able to produce intact graphene monolayers free of contaminants and defects at a high rate. Laser-induced melting of graphite under different pressure conditions has been investigated by Garcia and Jeschke.40b Their results showed that two steps were involved in the laser-induced melting process: the destruction of the graphite sheets via bond breaking and merging of the melted layers. Under the external pressure (10 GPa), the separation of the two steps is more evident for graphite, although it is also present in graphite films at normal pressure. Experimentally, Carey et al.40c investigated the interaction between the laser and graphite from various laser and graphite parameters, which supports well the formal theoretical researches. Recently, Zhao et al.40d attempted to synthesize graphene by etching of graphite using a low-temperature plasma technique. The firstly introduced H2O2 plasma technique is an easy and environmental friendly method although the yield, as yet, is not high enough. The oxidation and etching of reduced graphene oxide by thermal oxidation in air, ultraviolet-generated ozone, microwave oxygen plasma and scanning tunneling microscopy lithography have been investigated by Solís-Fernández et al.41 It was found that reduced graphene oxide exhibited a higher reactivity toward oxidation than the pristine graphite. Another new wet chemical bulk functionalization route using pristine graphite was achieved by coupling reductive graphite activation with oxidative arylation by organic aryldiazonium salts in a single-step procedure, resulting in single-layered graphene without reaggregation and substrate-induced doping.42
Graphene obtained through the top-down approach has been used to blend with other components to synthesize functional composites because of the intriguing properties derived from the synergistic effects of the components and graphene. For example, Chidembo et al.43 employed an efficient and versatile in situ spray pyrolysis method to synthesize globular metal oxide–graphene composites (rGO-Co3O4 or rGO-NiO) with highly porous morphologies. Zhao et al.44 mixed a graphene oxide solution with a Ni(NO3)2 solution, then added NH4HCO3 to produce basic nickel carbonate depositing on both sides of graphene layers. The precursor was heated at 400 °C to obtain graphene/NiO nanocomposites. Liang et al.45 reported well-distributed TiO2 nanocrystals grown on graphene as shown in Fig. 5. This synthesis process has been applied to obtain other transition-metal oxides/graphene composites, such as MnO2/GNS,46 Fe3O4/GNS,47 TiO2/GNS,48 SnO2/GNS,49 Co3O4/GNS.50
 | ||
| Fig. 5 (a) SEM image, (b) low magnification and (c) high magnification TEM images of TiO2 nanocrystals grown on GO sheets. The scale bar is 400 nm for the SEM image in (a) and 20 nm for the TEM image in (b). (d) An XRD pattern of the graphene/TiO2 nanocrystals hybrid.45 | ||
Apart from complexation with inorganic compounds, graphene also has been utilized in the synthesis of polymer–graphene composites. For instance, in situ reduction of graphite oxide in polymer powder has been carried out using focused solar electromagnetic radiation, where graphene oxide is completely reduced in polyvinylidene fluoride (PVDF) through photo-chemical reduction, resulting in highly conducting graphene–PVDF composites.51 Tang et al.52 reported a simple casting of a polymer solution which contains dispersed graphene oxide, followed by thermal reduction, for producing nanocomposites containing well-isolated monolayer reduced-graphene oxide nanosheets. Potts et al.53 firstly reported polymer composites using microwave-exfoliated graphite oxide (MEGO) to produce MEGO/polycarbonate composites at various loadings, which resulted in improvements of multifunctional properties as compared with neat polycarbonate. In these composites, graphene and polymer are noncovalently mixed together. By chemically grafting an organosilane, 3-aminopropyl triethoxysilane, onto the graphene skeleton, a covalent functionalization of graphene nanosheets (f-GNS) was realized by Wang and co-workers.54
The top-down approach is widely used for graphene synthesis in large quantity, and the resulting graphene nanosheets can be conveniently complexed with other functional components. However, this method has several disadvantages such as: (i) use of hazardous and toxic reagents (i.e., concentrated sulfuric acid, organic solvents) in the synthesis processes; and (ii) the as-prepared graphene frameworks contain a large amounts of defects, which limit the application of synthesized graphene in many research areas.
2.2. The bottom-up approach
Different from the top-down chemical approach, which starts from graphitic materials and provides a feasible way for the mass production of graphene, the bottom-up approach starting from small organic molecules is another important method for precise control over the morphology and structure of graphene. It has been well documented that the decomposition of hydrocarbons into graphitic materials can be catalyzed by metal surfaces through chemical vapor deposition (CVD).55 Hydrocarbon gases such as methane were chosen as carbon source and large domains of single-crystalline graphene were successfully grown via different CVD processes on various metal substrates. One of the best templates for the deposition is the Ni(111) surface due to the small lattice mismatch of this surface with the graphene surface. It has been reported that graphene films with resistances of 280 Ω sq−1 (80% transparent) and 770 Ω sq−1 (90% transparent) were realized through the growth of graphene on Ni film.56 Furthermore, the control in graphene scale makes CVD the most attractive method for devices fabrication. However, precise controlling the edge structure and topology of graphene is still a great challenge for the CVD method. Xu et al.57 produced extended single-layer and centimeter-scale graphene on a nickel surface deposited on a highly oriented pyrolytic graphite substrate by the diffusion of carbon atoms through the nickel template. Their results demonstrate the optimization of the relevant parameters for graphene growth (annealing time and temperature) to yield fine control of thickness and structure of the graphene layers. Fig. 6 shows the synthetic process for patterned graphene film growth on the thin nickel layers.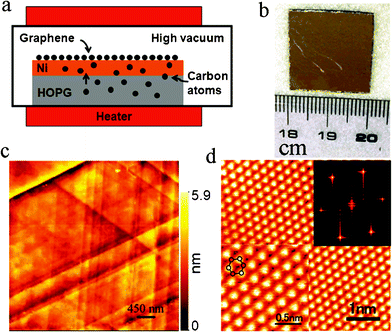 | ||
| Fig. 6 Novel Ni(111)/HOPG(0001) system for production of high-quality single-atomic graphene layers: (a) schematic diagram of graphene growth system and formation mechanism; (b) graphene on Ni(111)/HOPG (0001) (size: 2 cm × 2 cm); (c) AFM image of our graphene sheet; (d) constant current STM image of the as-prepared graphene sheet. The top-right and bottom-left insets show a two-dimensional fast Fourier transform taken from the STM image and STM image superimposed by the honeycomb lattice of graphene, respectively.57 | ||
In Ruoff's group and Kaner's group, more researches were focused on CVD growth of graphene on Cu substrates.55,58 Bhaviripudi et al.59 presented the role of kinetic factors in CVD synthesis of uniform large area graphene using Cu catalyst. It was found out that the growth of graphene varied from a monolayer at low methane concentration (ppm) to multilayers at higher methane concentration (5–10% by volume) under atmospheric pressure. One difficulty in the CVD synthesis on Cu substrates is that the domain size of the resulting graphene is not large enough.55a Li et al.55c studied the effect of growth parameters and developed a two-step CVD process to synthesize graphene films with an area of hundreds of square micrometers. In Ahn and Hong's group,60 a new route to large-scale synthesis of high-quality graphene films was demonstrated by using centimeter-scale Cu substrates. This cost- and time-effective roll-to-roll production method resulted in 30-inch graphene films and simultaneously provided an efficient synthesis of graphene with large scale and good quality for practical application.60 From the work in Kong's group61 and Ruoff's group,62 the transfer of these deposited graphene onto arbitrary substrates was also been launched.
Another common method for graphene synthesis is the epitaxial growth of graphene on SiC wafer surfaces through the decomposition of SiC followed by the desorption of Si from the surface.63 By using in situ low-temperature scanning tunneling microscopy to study the epitaxial growth, Huang et al.63d inferred the bottom-up growth mechanism. According to Heer et al.,63a the quantum Hall effect was suppressed due to the absence of localized states in the bulk of the as-prepared material. The advantages of the epitaxial growth can be summarized by: (i) the resulting material does not need to be transferred from the metal to another dielectric substrate; (ii) there are no trapped impurities under the graphene; and (iii) the growth can be directed by proper tailoring of the substrate.63c
Interestingly, an organic synthesis method for graphene has been put forward, in which precursors are those discotic aromatic hydrocarbons with precise chemical structures and functional groups.64 In cyclodehydrogenation of well-defined polyphenylene precursors, dendritic polyphenylene is easier to process in solution due to its good solubility, which decreases π–π stacking significantly.65 By designing the structure of the precursor monomers, a wide range of nanographenes with various topologies and widths can be produced.65b,66 In the organic synthesis field, there are some researches on nanographene synthesis, where the crucial problem is the practical processability of large polycyclic aromatic hydrocarbons (PAHs). It is found that PAHs which are larger than hexabenzocoronene (C42H18), are not soluble in organic solvents or not sublimable without decomposition because of strong intramolecular π–π interactions.67 One strategy to solubilize the nanographenes is to introduce the long, flexible aliphatic chains to the edge of the planar nanographene.68 However, with the size of graphene increasing, such a strategy makes less difference. Thus, it is seriously needed to develop new methods to solve this problem. Without using aromatic polymers, Sun et al.69 carried out the deposition of large area and high-quality graphene with controllable thickness using different solid carbon sources, such as polymer film (polymethyl methacrylate) molecules, where a metal catalyst substrate and a temperature as low as 800 °C was used. This method stands as a complementary method to CVD growth with the advantage of permitting growth at low temperature and overcoming the limitation in the use of gaseous materials in the CVD method.
Although the bottom-up approach introduces much less defects on the graphene surfaces as compared the top-down approach, the operation is relatively more difficult and costs more. In summary of the two approaches, both of which have their advantages and disadvantages, it is still needed to develop new methods or make some improvements on these known methods for the preparation of high-quantity and high-quality graphene nanomaterials. To clearly understand the synthesis methods for graphene, the mature methods mentioned above are summarized and listed in Table 1, including the used chemicals, and the structures and properties of the resulting graphene nanomaterials.
| Approach | Method | Chemicals used | Structure(s) | Properties or applications |
|---|---|---|---|---|
| Top-down approach | Exfoliation | Graphite, concentrated acid (H2SO4 ,HNO3, H3PO4), KMnO4 | One- or few-layered graphene oxide nanosheets | 1. to be reduced by reductants to form reduced graphene oxide |
| 2. to be blended with other components to synthesize functional composites | ||||
| 3. to remove pollutants from aqueous solutions | ||||
| Unzipping | Carbon nanotubes, concentrated acid (H2SO4, HNO3, H3PO4), KMnO4 | Narrow graphene nanoribbons | 1. to be blended with other components to synthesize functional composites | |
| 2. to be applied as biosensors | ||||
| Bottom-up approach | Chemical vapor deposition | Hydrocarbon gases, metal substrates | Patterned graphene films | 1. to be used as transparent conductive films for device fabrication |
| Epitaxial growth | SiC substrates | Graphene films | 1. to be used in electronic devices such as field effect transistors | |
| Organic synthesis | Large polycyclic aromatic hydrocarbons or solid polymer film and a metal catalyst substrate | Nanographene | 1. to be used as transparent conductive films for device fabrication | |
| 2. to be constructed with other components for application in lithium ion batteries |
3. Application of graphene-based nanomaterials in energy-related areas
Due to the special one-atom thick layer 2D structure and its superb characteristics of large surface area, high electrical conductivity and chemical stability, graphene is emerging as a particular carbon material for application in energy storage device such as supercapacitors, solar cells and lithium ion batteries.3.1. Application as supercapacitor materials
Usually, supercapacitors can be divided into two types, denoted electrochemical double-layer capacitors (EDLCs) and pseudocapacitors due to the different mechanism of charge storage. Different from those conventional high-surface-area materials such as activated carbons and CNTs, the available surface area of graphene nanomaterials as capacitor electrodes is independent of the pore distribution in the solid state.70 Vivekchand et al.71 reported graphene materials prepared from the thermal treatment of graphite oxide and used them as electrodes for EDLCs. At a scan rate of 100 and 1000 mV s−1 with H2SO4 aqueous solution as the electrolyte, the capacitance of graphene was 117 and 100 F g−1, respectively. In Chen's group,72 graphene sheets synthesized by simultaneously exfoliating and reducing graphite oxide under low-temperature plasma also showed high specific capacitance and good electrochemical stability. It is obvious that graphene with less agglomeration, fewer layers, higher effective surface area should be expected to exhibit a better supercapacitor performance.73 Therefore, the chemical modifications of graphene are focused in many research groups.70a,74 According to Zhang et al.,75 the performance of supercapacitors using chemically modified graphene as electrode depends on the flexible graphene sheets, not on the rigid, porous structure of activated carbon to provide its large surface area.75 Du et al.76 synthesized two kinds of functionalized graphene nanosheets through thermal exfoliation of graphite oxide at low temperature in air and at higher temperature in N2 conditions. It was found that the former functionalized graphene showed much higher specific capacitance than the latter. Similar to Du et al., Lv et al.77 reported a high capacitance (up to 264 F g−1) of a functionalized graphene by vacuum-promoted and low-temperature exfoliation of graphite oxide. The high capacitance of this kind of functionalized graphene is due to the oxygen-containing groups on the surface such as hydroxyl, carboxyl and epoxy groups, which can bring in the fast redox processes for the pseudocapacitance. Yang et al.78 reported a multilayered graphene film with highly open pore morphology, which allowed the electrolyte solution to access the individual sheet surfaces using water as an effective ‘spacer’ to prevent the restacking of graphene layers. The as-prepared film gave a specific capacitance as high as 215 F g−1 in aqueous solution. Another porous 3D graphene network was reported by using Ni foam as a sacrificial template in a facile CVD process with ethanol as the carbon source (Fig. 7). The obtained NiO/graphene composites exhibited a high specific capacitance of ∼816 F g−1 at a scan rate of 5 mV s−1, with a stable cycling performance even after 2000 cycles.79 | ||
| Fig. 7 Photographs of (a) Ni foam before and after the growth of graphene, and (b) ∼0.1 g 3D graphene networks obtained in a single CVD process after removal of the Ni foam. SEM images of (c) 3D graphene networks grown on Ni foam after CVD, and (d) 3D graphene networks after removal of Ni foam.79 | ||
One feasible strategy to prevent the aggregation of graphene in the solution and functionalization process is to use other materials such as carbon black,80 CNTs,81 or metal oxide nanoparticles 49,82 to separate 2D graphene nanosheets and further to obtain high surface areas. For instance, Fan et al.83 constructed a novel 3D CNT/graphene sandwich (CGS) and used it as an electrode in supercapacitors, where CNT pillars were grown in the graphene layers. A maximum specific capacitance of 385 F g−1 was obtained with a scan rate of 10 mV s−1 in 6 M KOH solution owing to the comprehensive utilization of pseudocapacitance from a catalyst and the double-layer capacitance from graphene, adding to the rapid transport of the electrolyte ions or electrons throughout the sandwich structure. Wang et al.84 did similar research by using a simple green hydrothermal route to prevent restacking of individual graphene sheets. Guo and Li85 reported a self-assembly approach to synthesize a hierarchical nanostructure comprised of carbon spheres and graphene nanosheets. The carbon spheres act as a nanospacer to separate graphene nanosheets, resulting in a high power density of 15.4 kW kg−1 and long cycle life of the supercapacitor.
The most popular pseudocapacitive materials are transition-metal oxides. Recently, there have been many reports on graphene–metal oxide/hydroxide composites, such as SnO2,49,86 Mn3O4,87 Fe3O4,88 Co3O4,89 Co(OH)2,90 and ZnO.49,82a Chen et al.91 synthesized graphene–Co(OH)2 nanocomposites and found that the specific capacitance of the composites reached a high value of 972.5 F g−1, with a significant improvement relative to the individual components. A spherical α-Ni(OH)2 nanoarchitecture grown on graphene was realized and used as an advanced electrochemical pseudocapacitor material, whose maximum specific capacitance was found to be high up to 1760.72 F g−1 at a scan rate of 5 mV s−1 according to Yang et al.92 Similarly, Zhao et al.44 applied monolayer graphene/NiO nanosheets with two-dimensional structure for supercapacitors. It was found that the NiO nanoparticles (5–7 nm) were uniformly dispersed on the graphene surface, and the two-dimensional structure enhanced the supercapacitive performance, with a high specific capacitance of 525 F g−1 at a current density of 200 mA g−1 (Fig. 8). Other reduced graphene oxide–metal oxide materials have also been applied in supercapacitors such as rGO–Co3O4,93 rGO–ZnO,49 and rGO–SnO2.49,94 In Dai's group, an advanced asymmetrical supercapacitor obtained by coupling Ni(OH)2/graphene and RuO2/graphene hybrids were firstly fabricated, which showed high specific capacitances and high energy and power densities.95 For the first time, Zhao et al.96 synthesized few-layered MnO2 nanosheets using graphene oxide nanosheets as templates. The in situ replacement of carbon atoms on graphene oxide framework by the edge-shared [MnO6] octahedron resulted in this special graphene-based nanomaterial, which showed a notable capacitance of ∼1017 F g−1 at a scan rate of 3 mV s−1, and ∼1183 F g−1 at a current density of 5 A g−1.
 | ||
| Fig. 8 The first three cycles of CV curves of (a) bare NiO and (b) graphene/NiO. (c) Galvanostatic charge/discharge curves of NiO and graphene/NiO at 200 mA g−1. (d) Rate capacitance of NiO and graphene/NiO with increasing current density. (e) Galvanostatic charge/discharge curves of graphene/NiO at 0.2, 2, 4 and 6 A g−1. (f) Cycle life of NiO and graphene/NiO at 200 mA g−1 in 6 M KOH solution.44 | ||
As important synthetic organic capacitor materials, polymers are often complexed with graphene to form graphene/polymer nanocomposites, which have turned out to show excellent performance in supercapacitors.74,97 Graphene oxide and graphene nanosheets were often doped into a polyaniline matrix.7,74c,98 For example, Wang et al.7 prepared a graphene/polyaniline composite paper (GPCP) by in situ anodic electropolymerization of aniline on graphene paper, and its electrochemical capacitance was as high as 233 F g−1. Mini et al.97 have developed high performance supercapacitor electrodes based on graphene/poly(pyrrole). The composites were synthesized by using electrophoretic deposition of graphene and then the poly(pyrrole) layer was electropolymerized upon the graphene. The specific capacitance was found out to be 1510 F g−1, with area capacitance of 151 mF cm−2 and volume capacitance of 151 F cm−3 at 10 mV s−1.97 Another nanocomposite, poly(sodium 4-styrensulfonate) (PSS)-graphene, proved to have very high cycle stability, the specific capacitance (190 F g−1) decreasing by only by 12% after 14860 cycles.99 It is commonly considered that the excellent capacitive performance of these graphene–polymer composites is due to the good EDL capacitance and pseudocapacitance, besides their good electrical conductivity. In Ruoff's group, a high-performance supercapacitor comprising a poly(ionic liquid)-modified reduced graphene oxide (PIL:RG-O) electrode and an ionic liquid (IL) electrolyte showed a stable electrochemical response up to 3.5 V operating voltage, a maximum energy density of 6.5 W h kg−1 and a power density of 2.4 kW kg−1.74a
In summarizing the research on transition-metal oxide/graphene hybrids, the excellent specific capacitance and cycle performance at high charge/discharge current can be attributed to three factors: (i) enhanced electronic conductivity of the composite because of graphene matrix; (ii) uniform dispersion of the transition-metal oxide nanoparticles among the graphene nanosheets ensure good interconnection of transition-metal oxide nanoparticles at the surface and interior of the electrodes; and (iii) graphene can act as a buffer for the volume change between the oxidized form and reduced form in the whole process, which ensures a good cycling performance of the composites. As promising electrode materials for supercapacitors the development of graphene–metal oxide materials is still in its early stage and requires further research.
3.2. Application in lithium ion batteries
In the modern society, rechargeable lithium ion batteries (LIBs) are widely used in portable devices. Thus, a great effort is being made to improve the performance of LIBs, especially their energy density, rate capability and cycling stability, which are largely dependent on the electrode materials since the recharge of LIBs is completed with the lithium ion insertion/extraction process in the electrodes. Because the commonly used graphite has low specific capacity many workers have turned their attention to alternative anode materials. It has been discovered that disordered carbon shows a higher capacitance than the ordered graphitic carbons.100 Thus it is reasonable for randomly organized graphene nanosheets to show excellent lithium storage capacity, due to the high specific surface areas, especially though the facile functionalization of oxygen-containing functional groups in the graphene oxide precursor through the chemical route.101 In order to prevent restacking of graphene nanosheets, CNTs or C60 molecules were introduced as spacers, which led to an increase in capacity of LIBs from 540 mA h g−1, to 730 and 784 mA h g−1, respectively.102Metal oxide nanoparticles are attractive anode materials owing to their high specific capacities.6,103 However, the huge volume variation during the charge/discharge processes causes the pulverization of the electrode, which results in poor reversibility, although by reducing the size of these materials to nanoscale this drawback can be partly improved. Recently, high specific capacity and good cycling performance of graphene–metal oxide hybrids as active materials in LIBs have been reported. The metal oxides mainly include Co3O4,104 NiO,105 Mn3O4,106 CuO,107 TiO2,108 SnO2105,109 and Co(OH)2.110 These nanoparticles not only play an important role as spacers between graphene layers, but also provide as active materials to interact reversibly with lithium ions. Taking SnO2 for example, it has high specific capacity (782 mA h g−1) theoretically as an anode in LIBs. However, its cycling performance is not satisfactory because of pulverization. By incorporating SnO2 with graphene to form a delaminated, 3D, flexible structure, the lithium storage capacity and cycling performance have been improved significantly according to the work from Paek et al.111 In these hybrid materials, graphene nanosheets act as not only lithium storage electrodes, but also electronically conductive channels. At the same time, the volume expansion upon lithium insertion is limited due to the dimensional confinement of SnO2 nanoparticles by the surrounding graphene. Furthermore, the developed pores between graphene and SnO2 act as buffer spaces during charge/discharge processes. However, in fact, this kind of structure is difficult to operate in a controlled fashion. Xu et al.112 synthesized MoO2/graphene oxide composites by a simple solvothermal method. It was found that with the addition of graphene oxide and the increase of graphene oxide content in the precursor solutions, MoO3 rods changed to MoO2 nanorods and further to MoO2 nanoparticles, both of which were uniformly distributed on the graphene surfaces. As shown in Fig. 9, the MoO2/graphene oxide composite with 10 wt% graphene oxide shows a reversible capacity of 720 mA h g−1 with a current density of 100 mA g−1 and 560 mA h g−1 at a high current density of 800 mA g−1 after 30 cycles. A two-step solution-phase reactions to fabricate Mn3O4/graphene hybrids was reported by Dai's group.113 The intimate interaction between the Mn3O4 nanoparticles and graphene gave the hybrid a high specific capacity (∼900 mA h g−1) as an anode material for LIBs. Another kind of nitrogen-doped MnO/graphene nanosheets (N-MnO/GNS) hybrid material was obtained through a hydrothermal method and subsequent annealing by ammonia. The nanostructured hybrid material exhibited a reversible electrochemical lithium storage capacity of 772 mA h g−1 at 100 mA g−1 after 90 cycles and even a high capacity of 202 mA h g−1 at a high current density of 5 A g−1.114 In the work of Peng et al.,115 a facile ultrasonic synthesis of CoO quantum dot/graphene nanosheet composites at room temperature using Co4(CO)12 as cobalt precursor was proposed. The nanosized CoO quantum dots with high dispersity on conductive graphene provide not only large amounts of accessible active sites for lithium-ion insertion but also good conductivity and short diffusion length for lithium ions, resulting in high capacity and rate capability.
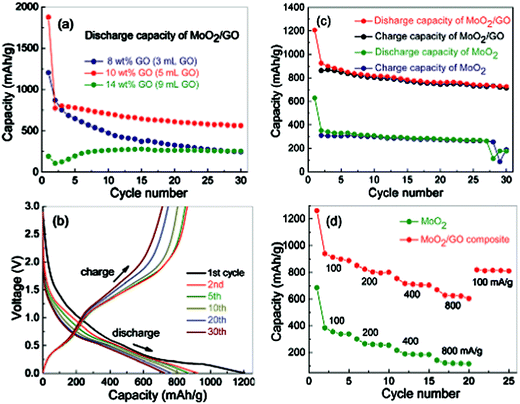 | ||
| Fig. 9 Electrochemical performance of MoO2/GO composites and MoO2. (a) Cycling performance of MoO2/GO composites prepared by adding 3, 5 and 9 mL of GO suspension, respectively, at a current density of 800 mA g−1. (b) Discharge/charge voltage profiles of MoO2/GO composite (with 5 mL of GO suspension) at a current density of 100 mA g−1. (d) Rate performance of MoO2 and MoO2/GO composite (with 5 mL of GO suspension) between 0.005 and 3.00 V with increasing current density from 100 to 800 mA g−1.112 | ||
Interestingly, a hierarchically nanostructured composite of MnO2/conjugated polymer/graphene was fabricated by self-assembly.116 The enhanced performance of the composite can be attributed to: (i) the interlayer spacing of ∼0.72 nm in MnO2 promotes efficient lithium intercalation/deintercalation; (ii) the polymer coating directs the ordered growth of MnO2 and prevents the aggregation, thus large volume expansion can be avoided; and (iii) the highly conductive 3-D graphene material ensures a fast charge/discharge rate. This work provides a universal method to fabricate hierarchically nanostructured composites for energy conversion/storage systems.
Most of the nanomaterials mentioned above are derived from graphene oxide obtained by the chemical top-down approach. There are also some graphene-encapsulated metal or metal oxide nanoparticles synthesized by the in situ, bottom-up method, in which the particle formation and encapsulation process are conducted simultaneously.117 For instance, the carbon/Co3O4 composites were synthesized by solid-state pyrolysis of organic–cobalt precursors.117a The resulting graphene-coated Co3O4 showed a high capacity of 940 mA h g−1 even after 20 cycles.
It is noted that in these LIBs, after a loss in the initial capacity the subsequent electrochemical lithium storage performance is much better, which may be due to the formation of a solid electrolyte interphase (SEI) layer.50,118 Considering the large surface area of graphene-based materials, the effect of the SEI layer may be larger than that of other anode materials.
In brief, graphene-based nanomaterials can largely decrease the volume variation of the active anode materials during the charge/discharge process, thus the high specific capacities of metal oxide nanoparticles (or other active components) can be maintained. As for the graphitic material itself, the high specific surface area compared with graphite and the good electrical conductivity ensure graphene-based materials are good alternative anode materials.
3.3. Application in solar cells
Due to the great potential for transparent and conductive electrodes in solar cells, graphene has been widely studied as different parts of solar cells in terms of their low cost, flexibility and high efficiency. As an ideal 2D material that can be assembled into film electrodes with low roughness and high conductivity, large surface area, thin graphene films are often used as window or/and counter electrodes, electron and hole transport materials, and buffer layers in solar cells.119 Wang et al.120 first reported a 10 nm-thick graphene film with a transparency of more than 70% and a conductivity of 550 S cm−1 by the thermal reduction of a graphene oxide film. This graphene was then used as a window electrode in dye-sensitized solar cells (DSSC), in which the current–voltage characteristics showed a short-circuit photocurrent density (Isc) of 1.01 mA cm−1, with an open-circuit voltage (Voc) of 0.7 V, a calculated fill factor (FF) of 0.36, and an power-conversion efficiency of 0.26%. The excellent performance is considered due to the low transmittance of the electrode and the series resistance of the device. Up to now, many efforts have been made to enhance the transparency and conductivity of such graphene thin films for application as electrodes in solar cells.121 However, these graphene films produced from solution often have many grain boundaries and defects which significantly increases the resistance. One strategy to tackle this problem is to incorporate other conductive filler materials such as silica,122 polymer123 or CNTs124 into the graphene matrix. For example, a nanocomposite of chemical converted graphene and CNTs gave a film resistance of 240 Ω sq−1 at 86% transmittance.124a Su et al.125 used large aromatic molecules such as pyrene-1-sulfonic acid sodium salt (PyS) and the disodium salt of 3,4,9,10-perylene-tetracarboxylic diimide bis-benzenesulfonic acid (PDI) to functionalize graphene, obtaining the enhancement in both the conductivity and electronic structure of graphene. The subsequent thermal reduction of graphene nanosheets led to a great increase of the conductivity and the overall power efficiency of 1.12% of the solar cells.Besides the solution process, a more commonly used route for high-quality graphene is CVD process, especially for their application as transparent conductive electrodes in photovoltaic devices.126 Yao et al.126b synthesized graphene film on Ni film-coated SiO2/Si wafers by the CVD method, and further transferred it to other substrates directly by dry-transfer technology. It was found that the average resistance of the 6–30 nm-thick graphene films varied from 1350 to 210 Ω sq−1 with an optical transparency from 91 to 72% in the visible light wavelength range, which was much lower than that of graphene oxide films processed from solution oxidation. In the report of Yan et al.,127 soluble graphene quantum dots were employed as a sensitizer for dye-sensitized solar cells (DSSC), in which an open-circuit voltage of 0.48 V and a short-circuit current density of 200 μA cm−2 were obtained, with a fill factor of 0.58.
Apart from DSSC, graphene has also been used in organic solar cells.5b,128 Compared with indium tin oxide (ITO), which suffers from rising cost and brittleness, graphene has many advantages in terms of its low cost, transparency, chemical robustness, flexibility and high electrical conductivity. Wang et al.129 reported high-performance organic solar cells by interface engineering of layer-by-layer stacked graphene anodes. The key for the further improvement in PCE is to reduce the sheet resistance of graphene, which can be achieved by increasing the carrier mobility via interface control or by more effective doping.
Differently, another graphene-based film from the nanographene molecules of large polycyclic aromatic hydrocarbons (PAHs) were also employed as a window electrode in organic solar cells.5b The 4 nm-thick films hold a transparency of 90% at a wavelength of 500 nm. Furthermore, the highest external quantum efficiency (EQE) of the solar cell was found to be 43% at a monochromatic light of 520 nm, suggesting that the output voltage of this graphene-based cell is comparable to that of ITO-based solar cells.5b Many applications of graphene-based materials as a transparent conductive electrode in solar cells have been reported in recent years,130 which are not mentioned specifically here. In conclusion, the present research of graphene on solar cells is mainly focused on three aspects: (i) the development of advanced techniques for graphene-based electrodes with high transparency and excellent conductivity; (ii) the synthesis of graphene nanomaterials as efficient acceptors to favor more efficient electron–hole separation; and (iii) the use of graphene-based films as an efficient hole transport layer.
Besides acting as a transparent conductive electrode, graphene has other potential uses for photovoltaic devices. For instance, through incorporation into conjugated polymers, graphene has largely improved the exciton dissociation and charge-transport properties.131 In Chen's group, organic solution-processed graphene material has been applied as an electron acceptor in organic bulk heterojunction (BHJ) photovoltaic devices with P3HT and P3OT as electron donor.131,132 By controlling annealing, the device performance is improved considerably, and under simulated 100 mW cm−2 AM 1.5 G light illumination a best power conversion efficiency of 1.4% was obtained. Solution-processed graphene was further used as a transparent anode to fabricate bulk heterojunction polymer organic photovoltaic devices, which displayed a power-conversion efficiency of 0.13%.133 Wu et al.5a also demonstrated organic light-emitting diodes with these solution-processed graphene thin films as transparent conductive anodes. Guo et al.130c used a simple bottom-up approach to create a novel electron transfer system based on layered graphene/CdS quantum dots, which showed a significantly improved photoresponse and offering a promising path for the development of photovoltaic devices. Organic photovoltaic devices were also fabricated by using graphene-based materials by Yin et al.,121b Park et al.134 and Zhu et al.135
In summary for the published results, graphene-based materials are appropriate for photoconversion due to their strong light absorption, high charge mobility, good stability and good HOMO/LUMO matching.136
4. Application of graphene-based nanomaterials in environmental-related areas
Environmental pollution remediation is another hot issue that needs serious concern in scientific research. Due to the development of economies and the expansion of industrial activities, contamination of heavy metal ions and toxic organic compounds in water has been increased over the past decades. Considering their high specific surface area, which would give the sufficient contact area for pollutants, various studies have been focused on the application of graphene-based nanomaterials for the high-performance removal of different organic and inorganic pollutants from aqueous solutions, such as through adsorption to reduce concentration, decomposition to less toxic molecules, and reduction to low-valent species. In other words, graphene-based materials have been applied as adsorbents, photodegradants and photoreductants in environmental pollution cleanup.4.1. Application as adsorbents for pollutant removal from aqueous solutions
Due to its large surface area, graphene is believed to be a good adsorbent for many pollutants. For example, Li et al.137 obtained monolayer or few-layered graphene nanosheets by ultrasonication and centrifugation of a 1-methyl-2-pyrrolidinone (NMP) suspension, and further removed NMP for the adsorption of fluoride from aqueous solutions, resulting an adsorption capacity of up to 35 mg g−1. To improve the dispersion of graphene in the aqueous solutions, many researches were carried out using the modified graphene as adsorbents for pollutant removal, among which, graphene oxide nanosheets are commonly used as adsorbents for heavy metal ions owing to the abundant functional groups such as hydroxyl, epoxide, carboxyl and carbonyl on the surfaces, which are expected to form strong surface complexes with toxic metal ions. According to the reports from Wang's group, the synthesized few-layered graphene oxide showed a high adsorption ability toward many heavy metal ions such as Pb(II),138 Cd(II),139 Co(II)139 and U(VI).140 In their reports, the maximum adsorption capabilities were 842 mg g−1 for Pb(II), 106.3 mg g−1 for Cd(II), 68.2 mg g−1 for Co(II) and 97.5 mg g−1 for U(VI), which are all much higher than that of other adsorbents. Fig. 10 shows the sorption isotherms of Cd(II) and Co(II) on graphene oxide nanosheets, and from their report, one can see that the sorption of Cd(II) and Co(II) on graphene oxide nanosheets is far from saturation. Deng et al.141 also reported the removal of Pb(II) and Cd(II) ions from wastewater using one-step synthesized ionic-liquid-functionalized graphene nanosheets directly from flake graphite, with adsorption capacities up to 406 mg g−1 for Pb(II) and 73 mg g−1 for Cd(II), respectively. | ||
| Fig. 10 Sorption isotherms of Cd(II) (A) and Co(II) (B) on graphene oxide nanosheets (0.1 g L−1) at different temperatures: pH = 6.0 ± 0.1, I = 0.01 M NaClO4. The solid and dashed lines correspond to Langmuir and Freundlich model simulations.139 | ||
Besides metal ions, there are also many reports on the adsorption of organic pollutants. Reduced graphene oxide was used to remove anionic dyes by Ramesha et al.,142 which turned out to be a good adsorbent because of the high surface area and the lack of negative surface charge. Zhao et al.9 used sulfonated graphene for the removal of naphthalene and 1-naphthol from aqueous solutions, and obtained maximum adsorption capacities of 2.33 mmol g−1 for naphthalene and 2.41 mmol g−1 for 1-naphthol (Fig. 11 and 12), which are much higher than for other reported materials.
 | ||
| Fig. 11 (A) The synthesis processes of sulfonated graphene from graphite. (B) SEM image of sulfonated graphene on Si substrates. (C) AFM image of sulfonated graphene on Si/SiO2 substrates (SiO2ca. 300 nm). The SEM and AFM images show that few-layered sulfonated graphene sheets are formed.9 | ||
 | ||
| Fig. 12 (A) Effect of sulfonated graphene content on the adsorption of naphthalene and 1-naphthol on sulfonated graphene. C(naphthalene/1-naphthol)initial = 0.2 mmol L−1, pH = 7.0 ± 0.1, I = 0.01 M NaClO4, T = 293 K. (B) Adsorption isotherms of naphthalene and 1-naphthol on sulfonated graphene (0.04 g L−1 for naphthalene; 0.08 g L−1 for 1-naphthol); pH = 7.0 ± 0.1, I = 0.01 M NaClO4, T = 293 K, C(naphthalene)initial = 0.10–0.39 mmol L−1, C(1-naphthol)initial = 0.17–0.70 mmol L−1.9 | ||
For the full utilization of graphene building blocks in environment remediation, some functional nanoparticles are combined with graphene in composites to give some additional properties to graphene. Typically for the adsorption of pollutants, magnetic Fe3O4 nanoparticles are incorporated to facilitate separation in the pollutant treatment. For example, Shen et al.143 synthesized graphene oxide–magnetic nanocomposites through a high temperature reaction of ferric triacetylacetonate with graphene oxide in 1-methyl-2-pyrrolidone. Many similar magnetic graphene nanocomposites have been proposed through solvothermal method or in situ chemical coprecipitation.144 Wang et al.144 applied graphene/Fe3O4 composites for the removal of organic dyes from aqueous solutions, with the adsorption capacity for fuchsine as high as 89 mg g−1, and He et al.145 obtained an adsorption capacity of 190 mg g−1 for methylene blue and 140 mg g−1 for neutral red cationic dyes using as-prepared magnetic graphene hybrids. Very similar to He et al., Xie et al.146 reported the adsorption capacities of superparamagnetic graphene oxide–Fe3O4 composites for methylene blue and neutral red were 167.2 and 171.3 mg g−1, respectively. The magnetic graphene hybrids were also used to adsorb As(III) and As(V), resulting in arsenic removal to as low as 1 ppb in aqueous solutions.147 These hybrids also showed an endothermic and spontaneous adsorption process toward Co(II) from aqueous solutions, with the maximum adsorption capacity up to 12.98 mg g−1 at room temperature according Liu et al.148 The growth of noble metal nanoparticles such as Au, Pt and Pd on graphene showed enhanced electronic properties due to the spatial confinement and synergetic electric interactions between the metal and graphene. These kinds of composites are mainly applied as catalysts in organic synthesis and transformation and less so for water purification.149 Only Sreeprasad et al.150 reported the use of graphene-Au composites supported on river sand for the excellent uptake of Hg(II) from aqueous solutions. More research about the application of graphene–metal composites in pollutant removal is expected. Besides the usual composites mentioned above, complex composite components such as CoFe2O4 and ZnFe2O4, also play an important role in the application of nanomaterials in environmental pollution cleanup. For example, magnetic CoFe2O4-functionalized graphene nanosheets were used to adsorb methyl orange from aqueous solution with an adsorption capability of 71 mg g−1.79 In conclusion, the excellent adsorption capabilities of graphene-based nanomaterials toward pollutants are due to: (i) the inherent high specific surface area of graphene; (ii) the decreased restacking and agglomeration of graphene layers; and (iii) the strong interaction between the adsorbates and the surface of the modified or functionalized graphene nanosheets. It should be noted that the interaction mechanism of organic pollutants with graphene-based materials is different to that of inorganic pollutants with graphene-based materials. Thereby, in the application of graphene-based materials in the removal of organic or inorganic pollutants from aqueous solutions, different types of graphene-based materials should be selected, and this will lead to much future research.
4.2. Application as degradants for pollutants in aqueous solutions
Except for the adsorption of the pollutants in water, many toxic compounds can be eliminated by photocatalytic degradation with the aid of photocatalysts such as ZnO, TiO2 and CdS incorporated with graphene, which shows prominent activity in photocatalytic application due to the novel electronic property of graphene and its zero-band gap. Furthermore, the nanoparticles on the graphene surface prevent the aggregation between the graphene layers in some extent, so that it is understandable to expect efficient properties in the pollutant treatment. For instance, Liang et al.45 synthesized graphene/TiO2 nanocrystal hybrids through the growth of TiO2 on graphene oxide nanosheets by hydrolysis and further hydrothermal treatment, and studied their photocatalytic activity on the degradation of rhodamine B. It is demonstrated that the as-prepared graphene/TiO2 nanocrystal hybrids show an impressive three-fold photocatalytic enhancement relative to P25 particles (Fig. 13). TiO2–graphene nanocomposites were also used for the efficient photocatalytic degradation of butane in the gas phase by Štengl et al.48 Zhao et al.10 also synthesized graphene/TiO2 composites and the results of photocatalytic degradation of methylene blue over UV and visible-light spectrum regions indicated that the enhanced photocatalytic activity was due to e− adsorption and transportation in the presence of graphene, as a result of which, graphene/TiO2 was able to absorb a high amount of photo-energy in the visible-light region and drive efficient photochemical degradation reactions. According to Zhang et al.,151 the enhanced degradation performance was originated from three attributes: (i) the increasing adsorption of pollutants on graphene-based nanomaterials; (ii) the extended light absorption range; (iii) facile charge transportation and separation. Li and Cao152 synthesized ZnO/graphene composites via a chemical deposition route and demonstrated their efficient photosensitized electron injection, slow electron recombination and enhanced photocatalytic activity under UV and visible light. Similar research was reported by Xu et al.153 As a well-known II–VI semiconductor, CdS has attracted extensive attention in photocatalytic research because of its suitable band gap (2.4 eV), which corresponds well with the spectrum of sunlight, and extensive attention has been paid to CdS–graphene composites.154 Liu et al.154d utilized as-prepared CdS-reduced graphene oxide composites for photocatalytic reduction of Cr(VI), and they found that: (i) CdS-graphene composites exhibited a better photocatalytic performance than CdS; (ii) the performance was dependent on the proportion of graphene in the composites, with the composites containing 1.5 wt% graphene achieving the highest Cr(VI) removal rate of 92%. The more complex composites, such as CoFe2O4 and ZnFe2O4, have also been studied for the degradation of organic pollutants. According to Wang's research group, the combination of CoFe2O4 or ZnFe2O4 nanoparticles with graphene leads to high activity for organic pollutant degradation.155 They noticed that CoFe2O4 showed an excellent degradation ability toward methylene blue, rhodamine B, methyl orange, active black BL-G and active red RGB without the presence of H2O2 under visible light irradiation.155a For the magnetic ZnFe2O4-graphene nanocomposites, the experimental results showed that these composites can serve as both photoelectrochemical degrader for organic molecules and the generator for hydroxyl radicals via photoelectrochemical decomposition of H2O2 under visible light irradiation with no noticeable change in the structure and composition.155b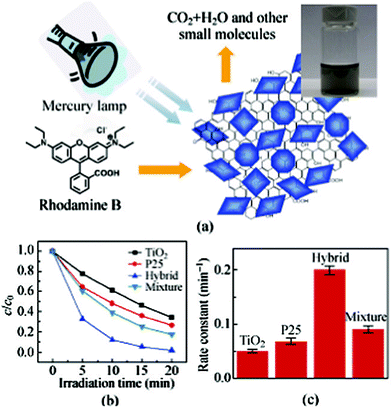 | ||
| Fig. 13 (a) Schematic illustration of the photodegradation of rhodamine B molecules by graphene/TiO2 nanocrystals hybrid under irradiation by a mercury lamp. The inset shows the solution of the graphene/TiO2 nanocrystals hybrid. (b) Photocatalytic degradation of rhodamine B monitored as normalized concentration change vs. irradiation time in the presence of free TiO2, P25, graphene/TiO2 nanocrystals hybrid and a graphene/TiO2 mixture. (c) Average reaction rate constant (min−1) for the photodegradation of rhodamine B with free TiO2, P25, graphene/TiO2 nanocrystals hybrid, and graphene/TiO2 mixture. The error bars are based on measurements on at least four different samples.45 | ||
The photocatalytic degradation of organic pollutants is an environmental friendly method to eliminate organic pollutants from aqueous solutions in environmental pollution cleanup. First, the high surface area assures the high adsorption of organic pollutants on the surface of graphene-based nanomaterials. Furthermore, the high dispersion of metal oxides on graphene nanosheets and the graphene as electron transfer channels make the degradation process occur efficiently on these graphene-based nanomaterials. Therefore, the synthesis of graphene-based composites with high photocatalytic capacity is important for the degradation of organic pollutants.
5. Summary and outlook
As a unique 2D carbon material, graphene has shown a continuously growing research upsurge since its appearance. Very soon, more researches will be focused on the modification of graphene and the incorporation of graphene with other functional nanoparticles and their further applications in multidisciplinary areas. In this critical review, we have covered recent researches on the synthesis of graphene-based materials and their applications in energy-related and environmental pollution remediation areas, both of which are the issues of most concern. Specifically, the utilization of graphene-based materials in supercapacitors, lithium ion batteries, solar cells, adsorption and degradation for the toxic pollutants from large volumes of aqueous solutions are covered. It is concluded that graphene-based materials, synthesized through ‘top-down’ or ‘bottom-up’ methods, show prominent properties in the energy-related and environmental-related areas, compared with other nanomaterials.However, there are still many difficulties waiting for efficient solutions. Firstly, the ‘top-down’ synthesis often results in large amounts of defects on the graphene framework, while the ‘bottom-up’ method is difficult to realize for mass production in practical applications. New approaches for the synthesis of high-quality graphene in large scale at low price are thus required. Secondly, although graphene-based materials are used as active materials for charge collection and transportation in electronic devices, there are still numerous critical problems requiring further study for the improvement in quality and function, which include the morphology of graphene, defects, functional groups, assembly behavior between graphene and other functional particles. Thirdly, for the application in environmental pollutant remediation, although the functionalized graphene-based nanomaterials show prominent ability in the removal of many pollutants from aqueous solutions, the selective adsorption for specific pollutants is rarely reported; and degradation reactions mainly focus on organic dyes, and relatively less on other toxic organic pollutants or high-valent metal ions. Furthermore, the research for the understanding of the relationship between graphene-based nanomaterials and improved performance is still at its early stage, which also hinders the further design for the effective functional graphene-based materials. It is necessary to point out that the synthesis of graphene-based materials is still expensive and in low scale at present, which limits the application of graphene-based nanomaterials in practical environmental pollution cleanup. With the development of technology, no doubt that these problems will be solved and more functional graphene-based materials will find extensive use in the near future.
Acknowledgements
Financial support from 973 projects of MOST (2011CB933700), NSFC (20971126, 21071147, 21071107, 91126020), and open foundation of State Key Lab of Pollution Control and Resource Reuse are acknowledged.References
- S. Park and S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS.
- (a) J. Wu, M. Agrawal, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen and P. Peumans, ACS Nano, 2010, 4, 43–48 CrossRef CAS; (b) X. Wang, L. Zhi, N. Tsao, Ž. Tomović, J. Li and K. Müllen, Angew. Chem., Int. Ed., 2008, 47, 2990–2992 CrossRef CAS.
- Z. Chen, M. Zhou, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2012, 2, 95–102 CrossRef CAS.
- D. W. Wang, F. Li, J. Zhao, W. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745–1752 CrossRef CAS.
- R. Kou, Y. Shao, D. Wang, M. H. Engelhard, J. H. Kwak, J. Wang, V. V. Viswanathan, C. Wang, Y. Lin and Y. Wang, Electrochem. Commun., 2009, 11, 954–957 CrossRef CAS.
- G. Zhao, L. Jiang, Y. He, J. Li, H. Dong, X. Wang and W. Hu, Adv. Mater., 2011, 23, 3959–3963 CrossRef CAS.
- D. Zhao, G. Sheng, C. Chen and X. Wang, Appl. Catal., B, 2012, 111–112, 303–308 CrossRef CAS.
- F. Schedin, A. Geim, S. Morozov, E. Hill, P. Blake, M. Katsnelson and K. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS.
- (a) O. Akhavan and E. Ghaderi, ACS Nano, 2010, 4, 5731–5736 CrossRef CAS; (b) Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS.
- (a) W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339–1339 CrossRef CAS; (b) S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- (a) L. Zhang, J. Liang, Y. Huang, Y. Ma, Y. Wang and Y. Chen, Carbon, 2009, 47, 3365–3368 CrossRef CAS; (b) L. Zhang, X. Li, Y. Huang, Y. Ma, X. Wan and Y. Chen, Carbon, 2010, 48, 2367–2371 CrossRef CAS.
- (a) H. C. Schniepp, J.-L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS; (b) X. Li, H. Wang, J. T. Robinson, H. Sanchez, G. Diankov and H. Dai, J. Am. Chem. Soc., 2009, 131, 15939–15944 CrossRef CAS.
- S. Dubin, S. Gilje, K. Wang, V. C. Tung, K. Cha, A. S. Hall, J. Farrar, R. Varshneya, Y. Yang and R. B. Kaner, ACS Nano, 2010, 4, 3845–3852 CrossRef CAS.
- X.-Y. Peng, X.-X. Liu, D. Diamond and K. T. Lau, Carbon, 2011, 49, 3488–3496 CrossRef CAS.
- L. Xie, L. Jiao and H. Dai, J. Am. Chem. Soc., 2010, 132, 14751–14753 CrossRef CAS.
- P. Kumar, K. S. Subrahmanyam and C. N. R. Rao, ArXIV, 2010, 1009.1028 Search PubMed , preprint arXiv.
- B. Luo, S. Liu and L. Zhi, Small, 2012, 8, 630–646 CrossRef CAS.
- W. Chen, L. Yan and P. R. Bangal, J. Phys. Chem. C, 2010, 114, 19885–19890 CAS.
- I. Kaminska, M. R. Das, Y. Coffinier, J. Niedziolka-Jonsson, J. Sobczak, P. Woisel, J. Lyskawa, M. Opallo, R. Boukherroub and S. Szunerits, ACS Appl. Mater. Interfaces, 2012, 4, 1016–1020 CAS.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2008, 4, 25–29 CrossRef.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370–12372 RSC.
- M. Liang, J. Wang, B. Luo, T. Qiu and L. Zhi, Small, 2012, 8, 1180–1184 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS.
- A. O'Neill, U. Khan, P. N. Nirmalraj, J. Boland and J. N. Coleman, J. Phys. Chem. C, 2011, 115, 5422–5428 CAS.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS.
- J. H. Lee, D. W. Shin, V. G. Makotchenko, A. S. Nazarov, V. E. Fedorov, Y. H. Kim, J.-Y. Choi, J. M. Kim and J.-B. Yoo, Adv. Mater., 2009, 21, 4383–4387 CrossRef CAS.
- (a) V. Abdelsayed, S. Moussa, H. M. Hassan, H. S. Aluri, M. M. Collinson and M. S. El-Shall, J. Phys. Chem. Lett., 2010, 1, 2804–2809 CrossRef CAS; (b) D. A. Sokolov, K. R. Shepperd and T. M. Orlando, J. Phys. Chem. Lett., 2010, 1, 2633–2636 CrossRef CAS.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872–876 CrossRef CAS.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877–880 CrossRef CAS.
- A. G. Cano-Márquez, F. J. Rodíguez-Macías, J. Campos-Delgado, C. G. Espinosa-González, F. Tristán-Loópez, D. Ramírez-González, D. A. Cullen, D. J. Smith, M. Terrones and Y. I. Vega-Cantuú, Nano Lett., 2009, 9, 1527–1533 CrossRef.
- (a) A. L. Elías, A. s. R. Botello-Méndez, D. Meneses-Rodríguez, V. Jehová González, D. Ramírez-González, L. Ci, E. Muñoz-Sandoval, P. M. Ajayan, H. Terrones and M. Terrones, Nano Lett., 2010, 10, 366–372 CrossRef; (b) M. Terrones, Nature, 2009, 458, 845–846 CrossRef CAS.
- A. L. Higginbotham, D. V. Kosynkin, A. Sinitskii, Z. Sun and J. M. Tour, ACS Nano, 2010, 4, 2059–2069 CrossRef CAS.
- (a) L. Jiao, X. Wang, G. Diankov, H. Wang and H. Dai, Nat. Nanotechnol., 2010, 5, 321–325 CrossRef CAS; (b) L. Xie, H. Wang, C. Jin, X. Wang, L. Jiao, K. Suenaga and H. Dai, J. Am. Chem. Soc., 2011, 133, 10394–10397 CrossRef CAS.
- D. B. Shinde, J. Debgupta, A. Kushwaha, M. Aslam and V. K. Pillai, J. Am. Chem. Soc., 2011, 133, 4168–4171 CrossRef CAS.
- L. M. A. Perdigão, S. N. Sabki, J. M. Garfitt, P. Capiod and P. H. Beton, J. Phys. Chem. C, 2011, 115, 7472–7476 Search PubMed.
- (a) Y. Miyamoto, H. Zhang and D. Tománek, Phys. Rev. Lett., 2010, 104, 208302 CrossRef; (b) M. E. Garcia and H. O. Jeschke, Appl. Surf. Sci., 2003, 208–209, 61–70 CrossRef CAS; (c) C. Carey, W. J. Cantwell, G. Dearden, K. R. Edwards, S. P. Edwardson, J. D. Mullett, C. J. Williams and K. G. Watkins, Laser Assisted Net Shape Engineering 5, Proceedings of the LANE, 2007, pp. 673–686 Search PubMed; (d) G. Zhao, D. Shao, C. Chen and X. Wang, Appl. Phys. Lett., 2011, 98, 183114 CrossRef.
- P. Solís-Fernández, J. I. Paredes, S. Villar-Rodil, L. Guardia, M. J. Fernández-Merino, G. Dobrik, L. P. Biró, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2011, 115, 7956–7966 Search PubMed.
- J. M. Englert, C. Dotzer, G. Yang, M. Schmid, C. Papp, J. M. Gottfried, H.-P. Steinrück, E. Spiecker, F. Hauke and A. Hirsch, Nat. Chem., 2011, 3, 279–286 CrossRef CAS.
- A. Chidembo, S. H. Aboutalebi, K. Konstantinov, M. Salari, B. Winton, S. A. Yamini, I. P. Nevirkovets and H. K. Liu, Energy Environ. Sci., 2012, 5, 5236–5240 CAS.
- B. Zhao, J. Song, P. Liu, W. Xu, T. Fang, Z. Jiao, H. Zhang and Y. Jiang, J. Mater. Chem., 2011, 21, 18792–18798 RSC.
- Y. Liang, H. Wang, H. Sanchez Casalongue, Z. Chen and H. Dai, Nano Res., 2010, 3, 701–705 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS.
- (a) G. Zhou, D.-W. Wang, F. Li, L. Zhang, N. Li, Z.-S. Wu, L. Wen, G. Q. Lu and H.-M. Cheng, Chem. Mater., 2010, 22, 5306–5313 CrossRef CAS; (b) H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CrossRef CAS.
- V. Štengl, D. Popelková and P. Vláčil, J. Phys. Chem. C, 2011, 115, 25209–25218 Search PubMed.
- T. Lu, Y. Zhang, H. Li, L. Pan, Y. Li and Z. Sun, Electrochim. Acta, 2010, 55, 4170–4173 CrossRef CAS.
- Z. S. Wu, W. Ren, L. Wen, L. Gao, J. Zhao, Z. Chen, G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS.
- V. Eswaraiah, K. Balasubramaniam and S. Ramaprabhu, Nanoscale, 2012, 4, 1258–1262 RSC.
- H. Tang, G. J. Ehlert, Y. Lin and H. A. Sodano, Nano Lett., 2012, 12, 84–90 CrossRef CAS.
- J. R. Potts, S. Murali, Y. Zhu, X. Zhao and R. S. Ruoff, Macromolecules, 2011, 44, 6488–6495 CrossRef CAS.
- X. Wang, W. Xing, P. Zhang, L. Song, H. Yang and Y. Hu, Compos. Sci. Technol., 2012, 72, 737–743 CrossRef CAS.
- (a) B. Zhang, W. H. Lee, R. Piner, I. Kholmanov, Y. Wu, H. Li, H. Ji and R. S. Ruoff, ACS Nano, 2012, 6, 2471–2476 CrossRef CAS; (b) X. Li, C. W. Magnuson, A. Venugopal, R. M. Tromp, J. B. Hannon, E. M. Vogel, L. Colombo and R. S. Ruoff, J. Am. Chem. Soc., 2011, 133, 2816–2819 CrossRef CAS; (c) X. Li, C. W. Magnuson, A. Venugopal, J. An, J. W. Suk, B. Han, M. Borysiak, W. Cai, A. Velamakanni, Y. Zhu, L. Fu, E. M. Vogel, E. Voelkl, L. Colombo and R. S. Ruoff, Nano Lett., 2010, 10, 4328–4334 CrossRef CAS.
- (a) D. Nezich, A. Reina and J. Kong, Nanotechnology, 2012, 23, 015701 CrossRef; (b) A. Reina, S. Thiele, X. Jia, S. Bhaviripudi, M. S. Dresselhaus, J. A. Schaefer and J. Kong, Nano Res., 2010, 2, 509–516 CrossRef; (c) X. Li, W. Cai, L. Colombo and R. S. Ruoff, Nano Lett., 2009, 9, 4268–4272 CrossRef CAS.
- M. Xu, D. Fujita, K. Sagisaka, E. Watanabe and N. Hanagata, ACS Nano, 2011, 5, 1522–1528 CrossRef CAS.
- J. K. Wassei, M. Mecklenburg, J. A. Torres, J. D. Fowler, B. C. Regan, R. B. Kaner and B. H. Weiller, Small, 2012, 8, 1415–1422 CrossRef CAS.
- S. Bhaviripudi, X. Jia, M. S. Dresselhaus and J. Kong, Nano Lett., 2010, 10, 4128–4133 CrossRef CAS.
- S. Bae, H. Kim, Y. Lee, X. Xu, J. S. Park, Y. Zheng, J. Balakrishnan, T. Lei, H. R. Kim, Y. I. Song, Y.J. Kim, K.S. Kim, B. Özyilmaz, J.H. Ahn, B.H. Hong and S. lijima, Nat. Nanotechnol., 2010, 5, 574–578 CrossRef CAS.
- A. Reina, X. Jia, J. Ho, D. Nezich, H. Son, V. Bulovic, M. S. Dresselhaus and J. Kong, Nano Lett., 2009, 9, 3087–3087 CrossRef CAS.
- J. W. Suk, A. Kitt, C. W. Magnuson, Y. Hao, S. Ahmed, J. An, A. K. Swan, B. B. Goldberg and R. S. Ruoff, ACS Nano, 2011, 5, 6916–6924 CrossRef CAS.
- (a) W. A. De Heer, C. Berger, X. Wu, P. N. First, E. H. Conrad, X. Li, T. Li, M. Sprinkle, J. Hass and M. L. Sadowski, Solid State Commun., 2007, 143, 92–100 CrossRef CAS; (b) K. V. Emtsev, A. Bostwick, K. Horn, J. Jobst, G. L. Kellogg, L. Ley, J. L. McChesney, T. Ohta, S. A. Reshanov and J. Röhrl, Nat. Mater., 2009, 8, 203–207 CrossRef CAS; (c) Y. Hu, M. Ruan, Z. Guo, R. Dong, J. Palmer, J. Hankinson, C. Berger and W. A. de Heer, ArXIV, 2012, 1202.5975 Search PubMed, preprint arXiv; (d) H. Huang, W. Chen, S. Chen and A. T. S. Wee, ACS Nano, 2008, 2, 2513–2518 CrossRef CAS.
- (a) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS; (b) L. Zhi and K. Mullen, J. Mater. Chem., 2008, 18, 1472–1484 RSC.
- (a) J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Mullen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS; (b) M. Treier, C. A. Pignedoli, T. Laino, R. Rieger, K. Müllen, D. Passerone and R. Fasel, Nat. Chem., 2010, 3, 61–67 CrossRef.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS.
- A. Sinitskii, A. A. Fursina, D. V. Kosynkin, A. L. Higginbotham, D. Natelson and J. M. Tour, Appl. Phys. Lett., 2009, 95, 253108–253108 CrossRef.
- (a) J. Wu, Ž. Tomović, V. Enkelmann and K. Müllen, J. Org. Chem., 2004, 69, 5179–5186 CrossRef CAS; (b) H. N. Tsao, H. J. Räder, W. Pisula, A. Rouhanipour and K. Müllen, Phys. Status Solidi A, 2008, 205, 421–429 CrossRef CAS.
- Z. Sun, Z. Yan, J. Yao, E. Beitler, Y. Zhu and J. M. Tour, Nature, 2010, 468, 549–552 CrossRef CAS.
- (a) M. F. El-Kady, V. Strong, S. Dubin and R. B. Kaner, Science, 2012, 335, 1326–1330 CrossRef CAS; (b) M. Kaempgen, C. K. Chan, J. Ma, Y. Cui and G. Gruner, Nano Lett., 2009, 9, 1872–1876 CrossRef CAS; (c) L. Diederich, E. Barborini, P. Piseri, A. Podesta, P. Milani, A. Schneuwly and R. Gallay, Appl. Phys. Lett., 1999, 75, 2662–2664 CrossRef CAS; (d) C. M. Yang, Y. J. Kim, M. Endo, H. Kanoh, M. Yudasaka, S. Iijima and K. Kaneko, J. Am. Chem. Soc., 2007, 129, 20–21 CrossRef CAS.
- S. R. C. Vivekchand, C. Rout, K. S. Subrahmanyam, A. Govindaraj and C. N. R. Rao, J. Chem. Sci., 2008, 120, 9–13 CrossRef CAS.
- Q. Zhou, Z. Zhao, Y. Chen, H. Hu and J. Qiu, J. Mater. Chem., 2012, 22, 6061–6066 RSC.
- D.-W. Wang, F. Li, Z.-S. Wu, W. Ren and H.-M. Cheng, Electrochem. Commun., 2009, 11, 1729–1732 CrossRef CAS.
- (a) T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2011, 5, 436–442 CrossRef CAS; (b) S. Bose, N. H. Kim, T. Kuila, K.-t. Lau and J. H. Lee, Nanotechnology, 2011, 22, 369502 CrossRef; (c) Q. Wu, Y. Xu, Z. Yao, A. Liu and G. Shi, ACS Nano, 2010, 4, 1963–1970 CrossRef CAS.
- L. L. Zhang, X. Zhao, M. D. Stoller, Y. Zhu, H. Ji, S. Murali, Y. Wu, S. Perales, B. Clevenger and R. S. Ruoff, Nano Lett., 2012, 12, 1806–1812 CrossRef CAS.
- Q. Du, M. Zheng, L. Zhang, Y. Wang, J. Chen, L. Xue, W. Dai, G. Ji and J. Cao, Electrochim. Acta, 2010, 55, 3897–3903 CrossRef CAS.
- W. Lv, D.-M. Tang, Y.-B. He, C.-H. You, Z.-Q. Shi, X.-C. Chen, C.-M. Chen, P.-X. Hou, C. Liu and Q.-H. Yang, ACS Nano, 2009, 3, 3730–3736 CrossRef CAS.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833–2838 CrossRef CAS.
- N. Li, M. Zheng, X. Chang, G. Ji, H. Lu, L. Xue, L. Pan and J. Cao, J. Solid State Chem., 2011, 184, 953–958 CrossRef CAS.
- J. Yan, T. Wei, B. Shao, F. Ma, Z. Fan, M. Zhang, C. Zheng, Y. Shang, W. Qian and F. Wei, Carbon, 2010, 48, 1731–1737 CrossRef CAS.
- (a) D. Yu and L. Dai, J. Phys. Chem. Lett., 2010, 1, 467–470 CrossRef CAS; (b) J. Y. Huang, L. Zhong, C. M. Wang, J. P. Sullivan, W. Xu, L. Q. Zhang, S. X. Mao, N. S. Hudak, X. H. Liu, A. Subramanian, H. Fan, L. Qi, A. Kushima and J. Li, Science, 2010, 330, 1515–1520 CrossRef CAS.
- (a) Y. Zhang, H. Li, L. Pan, T. Lu and Z. Sun, J. Electroanal. Chem., 2009, 634, 68–71 CrossRef CAS; (b) X. Zhang, Y. Huang, Y. Wang, Y. Ma, Z. Liu and Y. Chen, Carbon, 2009, 47, 334–337 CrossRef CAS.
- Z. Fan, J. Yan, L. Zhi, Q. Zhang, T. Wei, J. Feng, M. Zhang, W. Qian and F. Wei, Adv. Mater., 2010, 22, 3723–3728 CrossRef CAS.
- Y. Wang, Y. Wu, Y. Huang, F. Zhang, X. Yang, Y. Ma and Y. Chen, J. Phys. Chem. C, 2011, 115, 23192–23197 CAS.
- C. X. Guo and C. M. Li, Energy Environ. Sci., 2011, 4, 4504–4507 CAS.
- L. Qiu, X. Yang, X. Gou, W. Yang, Z.-F. Ma, G. G. Wallace and D. Li, Chem.–Eur. J., 2010, 16, 10653–10658 CrossRef CAS.
- B. Wang, J. Park, C. Wang, H. Ahn and G. Wang, Electrochim. Acta, 2010, 55, 6812–6817 CrossRef CAS.
- B. Li, H. Cao, J. Shao, M. Qu and J. H. Warner, J. Mater. Chem., 2011, 21, 5069–5075 RSC.
- J. Yan, T. Wei, W. Qiao, B. Shao, Q. Zhao, L. Zhang and Z. Fan, Electrochim. Acta, 2010, 55, 6973–6978 CrossRef CAS.
- Z.-S. Wu, D.-W. Wang, W. Ren, J. Zhao, G. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS.
- S. Chen, J. Zhu and X. Wang, J. Phys. Chem. C, 2010, 114, 11829–11834 CAS.
- S. Yang, X. Wu, C. Chen, W. Hu and X. K. Wang, Chem. Commun., 2012, 48, 2773–2775 RSC.
- (a) H. W. Wang, Z. A. Hu, Y. Q. Chang, Y. L. Chen, Z. Y. Zhang, Y. Y. Yang and H. Y. Wu, Mater. Chem. Phys., 2011, 130, 672–679 CrossRef CAS; (b) W. Zhou, J. Liu, T. Chen, K. S. Tan, X. Jia, Z. Luo, C. Cong, H. Yang, C. M. Li and T. Yu, Phys. Chem. Chem. Phys., 2011, 13, 14462–14465 RSC.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- H. Wang, Y. Liang, T. Mirfakhrai, Z. Chen, H. S. Casalongue and H. Dai, Nano Res., 2011, 4, 729–736 CrossRef CAS.
- G. Zhao, J. Li, L. Jiang, H. Dong, X. Wang and W. Hu, Chem. Sci., 2012, 3, 433–437 RSC.
- P. A. Mini, A. Balakrishnan, S. V. Nair and K. R. V. Subramanian, Chem. Commun., 2011, 47, 5753–5755 RSC.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004–5006 CrossRef CAS.
- H. K. Jeong, M. Jin, E. J. Ra, K. Y. Sheem, G. H. Han, S. Arepalli and Y. H. Lee, ACS Nano, 2010, 4, 1162–1166 CrossRef CAS.
- K. Sato, M. Noguchi, A. Demachi, N. Oki and M. Endo, Science, 1994, 264, 556–558 CAS.
- (a) S. Wang, P.-J. Chia, L.-L. Chua, L.-H. Zhao, R.-Q. Png, S. Sivaramakrishnan, M. Zhou, R. G. S. Goh, R. H. Friend, A. T. S. Wee and P. K. H. Ho, Adv. Mater., 2008, 20, 3440–3446 CrossRef CAS; (b) T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef; (c) L. R. Radovic and B. Bockrath, J. Am. Chem. Soc., 2005, 127, 5917–5927 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H.-s. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS.
- (a) H. Kim and J. Cho, J. Mater. Chem., 2008, 18, 771–775 RSC; (b) R. Demir-Cakan, Y. S. Hu, M. Antonietti, J. Maier and M. M. Titirici, Chem. Mater., 2008, 20, 1227–1229 CrossRef CAS.
- (a) S. Yang, G. Cui, S. Pang, Q. Cao, U. Kolb, X. Feng, J. Maier and K. Müllen, ChemSusChem, 2010, 3, 236–239 CrossRef CAS; (b) H. Kim, D.-H. Seo, S.-W. Kim, J. Kim and K. Kang, Carbon, 2011, 49, 326–332 CrossRef CAS; (c) S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408–8411 CrossRef CAS.
- D. Wang, R. Kou, D. Choi, Z. Yang, Z. Nie, J. Li, L. V. Saraf, D. Hu, J. Zhang, G. L. Graff, J. Liu, M. A. Pope and I. A. Aksay, ACS Nano, 2010, 4, 1587–1595 CrossRef CAS.
- N. Lavoie, P. R. L. Malenfant, F. M. Courtel, Y. Abu-Lebdeh and I. J. Davidson, J. Power Sources, 2012, 213, 249–254 CrossRef CAS.
- Y. J. Mai, X. L. Wang, J. Y. Xiang, Y. Q. Qiao, D. Zhang, C. D. Gu and J. P. Tu, Electrochim. Acta, 2011, 56, 2306–2311 CrossRef CAS.
- (a) D. Wang, D. Choi, J. Li, Z. Yang, Z. Nie, R. Kou, D. Hu, C. Wang, L. V. Saraf, J. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907–914 CrossRef CAS; (b) S. Yang, X. Feng and K. Müllen, Adv. Mater., 2011, 23, 3575–3579 CrossRef CAS.
- X. Huang, X. Zhou, L. Zhou, K. Qian, Y. Wang, Z. Liu and C. Yu, ChemPhysChem, 2011, 12, 278–281 CrossRef CAS.
- Y.-S. He, D.-W. Bai, X. Yang, J. Chen, X.-Z. Liao and Z.-F. Ma, Electrochem. Commun., 2010, 12, 570–573 CrossRef CAS.
- S.-M. Paek, E. Yoo and I. Honma, Nano Lett., 2009, 9, 72–75 CrossRef CAS.
- Y. Xu, R. Yi, B. Yuan, X. Wu, M. Dunwell, Q. Lin, L. Fei, S. Deng, P. Andersen, D. Wang and H. Luo, J. Phys. Chem. Lett., 2012, 309–314 CrossRef.
- H. Wang, L. F. Cui, Y. Yang, H. Sanchez Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS.
- K. Zhang, P. Han, L. Gu, L. Zhang, Z. Liu, Q. Kong, C. Zhang, S. Dong, Z. Zhang, J. Yao, H. Xu, G. Cui and L. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 658–664 CAS.
- C. Peng, B. Chen, Y. Qin, S. Yang, C. Li, Y. Zuo, S. Liu and J. Yang, ACS Nano, 2012, 6, 1074–1081 CrossRef CAS.
- C. X. Guo, M. Wang, T. Chen, X. W. Lou and C. M. Li, Adv. Energy Mater., 2011, 1, 736–741 CrossRef CAS.
- (a) L. Zhi, Y.-S. Hu, B. E. Hamaoui, X. Wang, I. Lieberwirth, U. Kolb, J. Maier and K. Müllen, Adv. Mater., 2008, 20, 1727–1731 CrossRef CAS; (b) G. Cui, Y.-S. Hu, L. Zhi, D. Wu, I. Lieberwirth, J. Maier and K. Müllen, Small, 2007, 3, 2066–2069 CrossRef CAS; (c) G. Cui, L. Gu, L. Zhi, N. Kaskhedikar, P. A. van Aken, K. Müllen and J. Maier, Adv. Mater., 2008, 20, 3079–3083 CrossRef CAS.
- A. Abouimrane, O. C. Compton, K. Amine and S. B. T. Nguyen, J. Phys. Chem. C, 2010, 114, 12800–12804 CAS.
- C. X. Guo, G. H. Guai and C. M. Li, Adv. Energy Mater., 2011, 1, 448–452 CrossRef CAS.
- X. Wang, L. Zhi and K. Müllen, Nano Lett., 2008, 8, 323–327 CrossRef CAS.
- (a) J. K. Wassei and R. B. Kaner, Mater. Today, 2010, 13, 52–59 CrossRef CAS; (b) Z. Yin, S. Sun, T. Salim, S. Wu, X. Huang, Q. He, Y. M. Lam and H. Zhang, ACS Nano, 2010, 4, 5263–5268 CrossRef CAS.
- S. Watcharotone, D. A. Dikin, S. Stankovich, R. Piner, I. Jung, G. H. B. Dommett, G. Evmenenko, S.E. Wu, S.F. Chen, C.P. Liu, S. T. Nguyen and R. S. Ruoff, Nano Lett., 2007, 7, 1888–1892 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- (a) V. C. Tung, L.-M. Chen, M. J. Allen, J. K. Wassei, K. Nelson, R. B. Kaner and Y. Yang, Nano Lett., 2009, 9, 1949–1955 CrossRef CAS; (b) D. Cai, M. Song and C. Xu, Adv. Mater., 2008, 20, 1706–1709 CrossRef CAS.
- Q. Su, S. Pang, V. Alijani, C. Li, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 3191–3195 CrossRef CAS.
- (a) L. Gomez De Arco, Y. Zhang, C. W. Schlenker, K. Ryu, M. E. Thompson and C. Zhou, ACS Nano, 2010, 4, 2865–2873 CrossRef CAS; (b) J. Yao, X. Shen, B. Wang, H. Liu and G. Wang, Electrochem. Commun., 2009, 11, 1849–1852 CrossRef CAS; (c) Y. Shi, K. K. Kim, A. Reina, M. Hofmann, L.-J. Li and J. Kong, ACS Nano, 2010, 4, 2689–2694 CrossRef CAS.
- X. Yan, X. Cui, B. Li and L.-s. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS.
- H. Park, J. A. Rowehl, K. K. Kim, V. Bulovic and J. Kong, Nanotechnology, 2010, 21, 505204 CrossRef.
- Y. Wang, S. W. Tong, X. F. Xu, B. Özyilmaz and K. P. Loh, Adv. Mater., 2011, 23, 1514–1518 CrossRef CAS.
- (a) H. Chang, G. Wang, A. Yang, X. Tao, X. Liu, Y. Shen and Z. Zheng, Adv. Funct. Mater., 2010, 20, 2893–2902 CrossRef CAS; (b) Q. Liu, Z. Liu, X. Zhang, L. Yang, N. Zhang, G. Pan, S. Yin, Y. Chen and J. Wei, Adv. Funct. Mater., 2009, 19, 894–904 CrossRef CAS; (c) C. X. Guo, H. B. Yang, Z. M. Sheng, Z. S. Lu, Q. L. Song and C. M. Li, Angew. Chem., Int. Ed., 2010, 49, 3014–3017 CrossRef CAS.
- Z. Liu, Q. Liu, Y. Huang, Y. Ma, S. Yin, X. Zhang, W. Sun and Y. Chen, Adv. Mater., 2008, 20, 3924–3930 CrossRef CAS.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS.
- Y. Xu, G. Long, L. Huang, Y. Huang, X. Wan, Y. Ma and Y. Chen, Carbon, 2010, 48, 3308–3311 CrossRef CAS.
- H. Park, P. R. Brown, V. Bulović and J. Kong, Nano Lett., 2012, 12, 133–140 CrossRef CAS.
- H. Zhu, J. Wei, K. Wang and D. Wu, Sol. Energy Mater. Sol. Cells, 2009, 93, 1461–1470 CrossRef CAS.
- X. Wan, G. Long, L. Huang and Y. Chen, Adv. Mater., 2011, 23, 5342–5358 CrossRef CAS.
- Y. Li, P. Zhang, Q. Du, X. Peng, T. Liu, Z. Wang, Y. Xia, W. Zhang, K. Wang, H. Zhu and D. Wu, J. Colloid Interface Sci., 2011, 363, 348–354 CrossRef CAS.
- G. Zhao, X. Ren, X. Gao, X. Tan, J. Li, C. Chen, Y. Huang and X. Wang, Dalton Trans., 2011, 40, 10945–10952 RSC.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS.
- G. Zhao, T. Wen, X. Yang, S. Yang, J. Liao, J. Hu, D. Shao and X. Wang, Dalton Trans., 2012, 41, 6182–6188 RSC.
- X. Deng, L. Lü, H. Li and F. Luo, J. Hazard. Mater., 2010, 183, 923–930 CrossRef CAS.
- G. K. Ramesha, A. Vijaya Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270–277 CrossRef CAS.
- J. Shen, Y. Hu, M. Shi, N. Li, H. Ma and M. Ye, J. Phys. Chem. C, 2010, 114, 1498–1503 CAS.
- C. Wang, C. Feng, Y. Gao, X. Ma, Q. Wu and Z. Wang, Chem. Eng. J., 2011, 173, 92–97 CrossRef CAS.
- F. He, J. Fan, D. Ma, L. Zhang, C. Leung and H. L. Chan, Carbon, 2010, 48, 3139–3144 CrossRef CAS.
- G. Xie, P. Xi, H. Liu, F. Chen, L. Huang, Y. Shi, F. Hou, Z. Zeng, C. Shao and J. Wang, J. Mater. Chem., 2012, 22, 1033–1039 RSC.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS.
- M. Liu, C. Chen, J. Hu, X. L. Wu and X. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CAS.
- (a) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mulhaupt, J. Am. Chem. Soc., 2009, 131, 8262–8270 CrossRef CAS; (b) S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS; (c) H. Zhao, J. Yang, L. Wang, C. Tian, B. Jiang and H. Fu, Chem. Commun., 2011, 47, 2014–2016 RSC.
- T. S. Sreeprasad, S. M. Maliyekkal, K. P. Lisha and T. Pradeep, J. Hazard. Mater., 2011, 186, 921–931 CrossRef CAS.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS.
- B. Li and H. Cao, J. Mater. Chem., 2011, 21, 3346–3349 RSC.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- (a) C. Nethravathi, T. Nisha, N. Ravishankar, C. Shivakumara and M. Rajamathi, Carbon, 2009, 47, 2054–2059 CrossRef CAS; (b) K. Wang, Q. Liu, X.-Y. Wu, Q.-M. Guan and H.-N. Li, Talanta, 2010, 82, 372–376 CrossRef CAS; (c) H. Chang, X. Lv, H. Zhang and J. Li, Electrochem. Commun., 2010, 12, 483–487 CrossRef CAS; (d) X. Liu, L. Pan, T. Lv, G. Zhu, Z. Sun and C. Sun, Chem. Commun., 2011, 47, 11984–11986 RSC.
- (a) Y. Fu, H. Chen, X. Sun and X. Wang, Appl. Catal., B, 2012, 111–112, 280–287 CrossRef CAS; (b) Y. Fu and X. Wang, Ind. Eng. Chem. Res., 2011, 50, 7210–7218 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |