Synthesis of phenylacetonitrile by amination of styrene oxide catalyzed by a bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3
Yuecheng
Zhang
,
Weihua
Xu
and
Jiquan
Zhao
*
School of Chemical Engineering and Technology, Hebei University of Technology, Tianjin 300130, P R China. E-mail: zhaojq@hebut.edu.cn (J. Q. Zhao); Tel: +86-22-60202926; Fax: +86-22-60202926
First published on 13th June 2012
Abstract
A novel method for the synthesis of phenylacetonitrile by amination of styrene oxide catalyzed by a bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3 was established. A yield as high as 87.9% was received when the reaction was conducted on a fixed-bed reactor dosed with 30 ml (21 g) catalyst at 693 K under ammonia at atmospheric pressure by keeping the liquid velocity of styrene epoxide diluted with toluene (styrene oxide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 20:80 (m:m)) at 0.2 ml min−1 and the gas velocity of ammonia at 300 ml min−1. The lifetime of the catalyst was evaluated and the activity of the catalyst could be recovered by continuously blowing air into the fixed-bed at 723 K for 3.5 h online. The catalyst was characterized by XRD, XPS, TEM and IR of adsorbed pyridine. The characterization results indicated that the dehydration reaction in the tandem reaction mainly took place on the Lewis acid sites and revealed that ZnAl2O4 is the active species for the dehydrogenation of imine to nitrile. The doping of chromium on to the γ-Al2O3 supported zinc catalyst Zn29.9/γ-Al2O3 could diminish the size of ZnAl2O4 crystallites, which is in favor of the dehydrogenation reaction. The deactivation of the catalyst is due to the carbonaceous deposits generated from the chemical adsorption of some alkaline substances in the catalytic run.
:toluene = 20:80 (m:m)) at 0.2 ml min−1 and the gas velocity of ammonia at 300 ml min−1. The lifetime of the catalyst was evaluated and the activity of the catalyst could be recovered by continuously blowing air into the fixed-bed at 723 K for 3.5 h online. The catalyst was characterized by XRD, XPS, TEM and IR of adsorbed pyridine. The characterization results indicated that the dehydration reaction in the tandem reaction mainly took place on the Lewis acid sites and revealed that ZnAl2O4 is the active species for the dehydrogenation of imine to nitrile. The doping of chromium on to the γ-Al2O3 supported zinc catalyst Zn29.9/γ-Al2O3 could diminish the size of ZnAl2O4 crystallites, which is in favor of the dehydrogenation reaction. The deactivation of the catalyst is due to the carbonaceous deposits generated from the chemical adsorption of some alkaline substances in the catalytic run.
1 Introduction
Phenylacetonitrile is an important intermediate for a variety of useful compounds, such as phenobarbital, methylphenidate, and phenylacetic acid. Phenylacetic acid is largely used in the production of penicillin G.1 Now, phenylacetonitrile is only produced by the reaction of benzyl chloride with sodium cyanide in industry,2 as it is known that sodium cyanide is hypertoxic, therefore, it is very dangerous both in its production and application. On the other hand, benzyl chloride is prepared industrially by the gas-phase photochemical reaction of toluene with chlorine3 and obtained from Blanc chloromethylation of benzene,4 both of which can contaminate the environment. It is obvious that the traditional method for the production of phenylacetonitrile is not environmentally benign. The environmental pressure provides a strong incentive to search for an environmentally friendly process to prepare phenylacetonitrile. However, no novel methodology for the synthesis of phenylacetonitrile was reported recently, only some related patents5–11 disclosed several decades ago can be retrieved.Recently, we have engaged in the research of the synthesis of acetonitrile from alcohol through catalytic amination. Our research results12,13 and the ones reported by others in the literature14 indicated that the transformation of ethanol to acetonitrile proceeds via the following steps. First, ethanol was transformed to acetaldehyde in the presence of a dehydrogenation catalyst. Once acetaldehyde was generated, it condensed immediately with ammonia to give an imine. Finally, the generated imine underwent dehydrogenation again to afford acetonitrile in the absence of hydrogen.12,14–16 Inspired by the formation mechanism of the acetonitrile described above, we can expect that some nitriles such as phenylacetonitrile can be synthesized from the amination of styrene oxides. Epoxides can undergo ring opening reactions by nucleophilic attack of ammonia to give 2-aminoethanols,17,18 and 2-aminoethanols can undergo dehydration to give enamines, which are tautomerisms of imines in principle. The expected transformation of epoxides to nitriles is shown in Scheme 1. It is obvious that the transformation must be helped by a catalyst. From Scheme 1, it can be concluded that the catalyst should have the following functions: promotion of the epoxide ring opening, the dehydration of 2-aminophenylethanol to enamine, and dehydrogenation of imine to phenylacetonitrile. If the epoxide ring opening, dehydration and dehydrogenation active components are integrated in one catalyst, the transformation of styrene oxide to phenylacetonitrile can be accomplished.
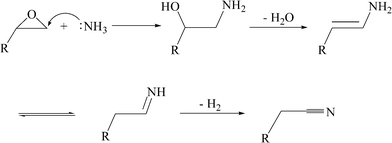 | ||
| Scheme 1 The assumed tandem reaction from epoxides to nitriles. | ||
To achieve the above goal, we have been engaged in the seeking of the catalyst. It has been found that a bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3 can catalyze the amination of styrene oxide to phenylacetonitrile with high activity and selectivity. To our best knowledge, this is the first case where nitriles are synthesized from epoxides by amination up to now. This process of obtaining phenylacetonitrile is environmentally benign compared to the traditional one from the point of view of low toxicity of raw materials. Herein, we report the successful results.
2 Experimental
2.1 Preparation of catalysts
γ-Al2O3 was purchased from Tianjin Research and Design Institute of Chemical Industry, Tianjin, China. The other chemicals were obtained from KRS Chemical Reagent Cooperation, Tianjin, China. All the chemicals were reagent grade and used as received. The monometallic or bimetallic catalysts with γ-Al2O3 as support were prepared by kneading a mixture of γ-Al2O3 with an aqueous solution of the corresponding transition-metal nitrate(s), followed by extruding, drying and calcinating as described in the process to prepare the bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3. For example, the bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3 was prepared as follows: 8.65 g Cr(NO3)3·9H2O and 34.12 g Zn(NO3)2·6H2O were dissolved in 20.0 g H2O, then 16.1 g γ-Al2O3 was added to the solution. The mixture was kneaded for 3 h in a kneader and the resulting kneaded material was then processed in an extruder to obtain extrudates of diameter 2 mm and length 2.5 mm. The extrudates were superficially dried at 383 K for 12 h, followed by calcinating at 823 K for 4 h to give the catalyst. The catalysts with silica and H-ZSM-5 as supports were prepared by respective impregnation of silica and H-ZSM-5 with an appropriate volume of aqueous solution of the corresponding transition-metal nitrate(s), followed by extruding, drying and calcinating. For example, the monometallic catalyst Zn20.1/H-ZSM-5 was prepared as follows: 22.75 g Zn(NO3)2·6H2O was dissolved in 15.0 g H2O, then 20.0 g H-ZSM-5 was added to the solution. The incipient wetness impregnation was maintained at 303 K for 24 h, and then the impregnated H-ZSM-5 was superficially dried at 383 K for 12 h, followed by calcinating at 823 K for 4 h to obtain the catalyst.2.2 Characterization of catalysts
The contents of Zn and Cr were determined by inductively coupled plasma (ICP) spectroscopy. First, accurately weighed samples were respectively dissolved in concentrated HNO3 and HF. Then, the metal contents of the solutions were determined by a T.J.A ICP-9000 (N + M) type ICP-AES instrument. The X-ray diffraction (XRD) patterns of the samples were recorded with a Rigaku D/max 2500 X-ray diffractometer using Cu–Kα radiation (40 kV, 150 mA) in the range 2θ = 10°–90°. X-ray photoelectron spectroscopy (XPS) was performed with a PHI 1600 spectroscope using a Mg–Kα X-ray source for excitation. Transmission electron micrographs (TEM) were obtained on a JEOL 100CX-II instrument equipped with an energy dispersive X-ray (EDX) detector (Oxford Instruments) at an accelerating voltage of 200 kV. The surface area, total pore volume and pore size distribution of the catalysts were measured at 77 K by nitrogen adsorption using a Micromeritics ASAP 2020 Surface Area and Porosity Analyzer.Thermogravimetric-different scanning calorimetry (TG-DSC) measurement was carried out on a Perkin Elmer-7 thermogravimetric analyzer from 313 to 1073 K with the rate of 20 K min−1 under air atmosphere.
The IR spectra of adsorbed pyridine were recorded using a Thermo Nicliet Nicolet Nexus 470 spectrometer equipped with a heatable and evacuatable IR cell with CaF2 windows, connected to a gas dosing–evacuating system. The powdered samples were pressed into self-supporting wafers with a diameter of 20 mm and a weight of 50 mg. Prior to analysis, all samples were pretreated at 673 K for 1 h, under high vacuum conditions (5 × 10−5 Pa), followed by cooling to 473 K. Then, pyridine was adsorbed at this temperature for 15 min. The physisorbed pyridine was removed by evacuating during 1 h at 473 K, under high vacuum conditions (5 × 10−5 Pa). Then the infrared spectra were recorded.
2.3 Catalytic test
Catalytic activity tests were carried out in a continuous fixed-bed reactor. 30.0 ml samples of the catalysts were loaded into the reactor (Id. = 15 mm; length = 1100 mm). The temperature in the catalyst zone was measured using a thermocouple located in the center of the catalyst bed and was regulated by a PID cascade controller. The styrene oxide diluted with toluene was dosed into the reactor at a speed of 0.2 ml min−1 by a syringe pump and the flux of ammonia was 300 ml min−1 (STP). The composition of the reaction mixtures were analyzed by gas chromatography using a 30 m SE-30 capillary column with biphenyl as internal standard compound. Also, the products were identified by gas chromatography-mass spectrometry (HP5971 GC-MS) equipped with a 30 m SE-30 capillary column.3 Results and discussion
3.1 Selection of catalyst
The design of the catalyst is based on the facts that epoxide ring opening and dehydration of alcohols can be conducted by solid acids such as γ-Al2O3, SiO2 and H-ZSM-518–21 and some transition metals such as zinc, nickel etc. can catalyze dehydrogenation of imine to nitriles in the amination of alcohols to nitriles.12,13,16,22,23 (enamine and imine are tautomers). Initially, several metals including zinc, iron, copper, chromium and cobalt were chosen to prepare the catalysts with γ-Al2O3, SiO2, and H-ZSM-5 as the supports, respectively. First, the catalysts with metal content of 20% were prepared and their catalytic performances on the reaction were tested. The results are presented in Table 1. It was found that styrene oxide conversions near 100% were achieved over all the catalysts. However, the selectivity towards phenylacetonitrile changed with different metals. Among all of the supported mono metal oxide catalysts, only supported zinc oxide showed good performance on the reaction. For example, the yield of phenylacetonitrile reached 77.4% with Zn19.8/γ-Al2O3 as catalyst under ammonia at atmospheric pressure at 693 K (Entry 1), whereas, the Cu20.2/γ-Al2O3 only gave 29.4% yield under the same conditions (Entry 6). Besides, it can be concluded that the transition metal is necessary for the transformation of styrene oxide to phenylacetonitrile due to no phenylacetonitrile being detected in the absence of transition metal on the support (Entry 9). On the other hand, γ-Al2O3 exhibited the best performance among all the supports selected (Entries 1,7,8). Therefore, the catalysts with different zinc contents (10%, 20%, 30%, 40%) were prepared with γ-Al2O3 as support and their performances on the reaction were evaluated. It was obtained that the catalyst with zinc content of 30% (Zn29.9/γ-Al2O3) had the highest catalytic activity and selectivity towards phenylacetonitrile. In this case the yield of phenylacetonitrile was 79.3% (Entry 11).| Entry | Catalyst | Yield ab (%) | ||
|---|---|---|---|---|
| PhCH2CN | PhCHCH2 | PhCN | ||
|
a Reaction conditions: catalyst dosage 30 ml (21 g); reaction temperature 693 K; atmosphereic pressure; styrene oxide:toluene = 20:80 (m:m), velocity of liquid mixture 0.2 ml min−1; velocity of ammonia 300 ml min−1.
b PhCH2CN: phenylacetonitrile, PhCHCH2:styrene, PhCN:benzonitrile.
c These catalysts were prepared by impregnation.
|
||||
| 1 | Zn19.8/γ-Al2O3 | 77.4 | 2.6 | 5.6 |
| 2 | Cr20.1/γ-Al2O3 | 53.2 | 2.2 | 2.5 |
| 3 | Co19.9/γ-Al2O3 | 40.8 | 4.7 | 5.8 |
| 4 | Fe20.3/γ-Al2O3 | 35.8 | 9.9 | 8.3 |
| 5 | Ni19.7/γ-Al2O3 | 30.5 | 3.9 | 6.1 |
| 6 | Cu20.2/γ-Al2O3 | 29.4 | 3.4 | 3.8 |
| 7 | Zn20.1/H-ZSM-5c | 65.0 | 4.7 | 10.2 |
| 8 | Zn19.9/SiO2c | 73.7 | 1.3 | 2.0 |
| 9 | Zn0/γ-Al2O3 | — | 19.9 | — |
| 10 | Zn10.2/γ-Al2O3 | 52.8 | 1.2 | 6.0 |
| 11 | Zn29.9/γ-Al2O3 | 79.3 | 1.0 | 4.2 |
| 12 | Zn40.1/γ-Al2O3 | 66.5 | 1.6 | 2.5 |
The performance of Zn29.9/γ-Al2O3 was still not satisfactory. To improve the performance of the catalyst, the Zn29.9/γ-Al2O3 was doped respectively with other metals including Cr, Co, Cu and Ni and the corresponding bimetallic catalysts were obtained. It was found that the catalyst doped with Cr showed good performance compared to the ones doped with other metals (Table 2). Among all the bimetallic catalysts, Zn30.1Cr4.3/γ-Al2O3 exhibited the best performance on the amination of styrene oxide to phenylacetonitrile. The yield of phenylacetonitrile reached 87.9% (Table 2, Entry 1). Also, the catalysis of the catalyst was also related with the content of Cr (Table 2, Entries 1, 6, 7).
| Entry | Catalyst | Yield ab (%) | ||
|---|---|---|---|---|
| PhCH2CN | PhCHCH2 | PhCN | ||
|
a Reaction conditions: catalyst dosage 30 ml (21 g); reaction temperature 693 K; atmosphereic pressure; styrene oxide:toluene = 20:80 (m:m), velocity of liquid mixture 0.2 ml min−1; velocity of ammonia 300 ml min−1.
b PhCH2CN:phenylacetonitrile, PhCHCH2:styrene, PhCN:benzonitrile.
|
||||
| 1 | Zn30.1Cr4.3/γ-Al2O3 | 87.9 | 0.5 | 3.7 |
| 2 | Zn29.8Co4.5/γ-Al2O3 | 84.3 | 1.9 | 3.4 |
| 3 | Zn30.2Cu4.6/γ-Al2O3 | 67.8 | 2.9 | 3.1 |
| 4 | Zn29.9Fe4.4/γ-Al2O3 | 79.1 | 1.2 | 3.3 |
| 5 | Zn30.3Ni4.3/γ-Al2O3 | 60.4 | 1.0 | 3.5 |
| 6 | Zn29.7Cr3.1/γ-Al2O3 | 81.0 | 0.7 | 3.1 |
| 7 | Zn29.5Cr5.8/γ-Al2O3 | 78.6 | 3.4 | 3.7 |
3.2 Performance of catalyst Zn30.1Cr4.3/γ-Al2O3
The influence of reaction temperature on the performance of the catalyst Zn30.1Cr4.3/γ-Al2O3 was investigated under atmospheric pressure by keeping a molar ratio of ammonia to styrene oxide of 20:1 and a contacting time of 4.51 s in the temperature range 633 to 713 K. The results are shown in Fig. 1. It is shown that the conversion of styrene oxide maintained 100% in the experimental temperature range. However, the selectivity towards phenylacetonitrile increased with an increase of temperature from 633 to 693 K, and reached its maximum of 87.9% at 693 K and then decreased with further temperature increases. It was found that low temperature was in favor of the by-product formation but high temperature led to oligomerization of intermediates.
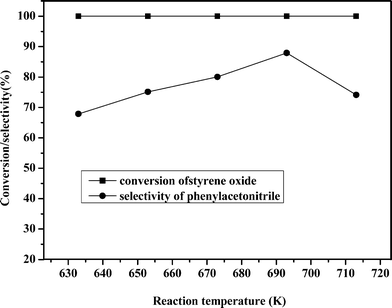 | ||
| Fig. 1 Influence of temperature on the reaction. | ||
The influence of the ammonia:styrene oxide molar ratio on the reaction was also investigated and the results are presented in Fig. 2. It can be seen that the conversion of styrene oxide was always maintained around 100% when the ammonia:styrene oxide molar ratio increased from 8 to 40 under atmospheric pressure, meanwhile, the temperature was kept at 693 K and the residence time was 4.51 s. However, the selectivity of phenylacetonitrile increased first from 70.7% to 87.9% with the ammonia:styrene oxide molar ratio increasing from 8 to 20, but decreased with further increases of the ammonia:styrene oxide molar ratio. The reason may be that appropriate excess ammonia was in favor of the epoxide ring opening and subsequent dehydration and dehydrogenation reaction to phenylacetonitrile. However, when the ammonia excess was too large, for example the ammonia:styrene oxide molar ratio was higher than 20, the ammonia could reduce the acidity leading to low activity on dehydration and harm the dehydrogenation centers of the catalyst retarding the reaction towards phenylacetonitrile.
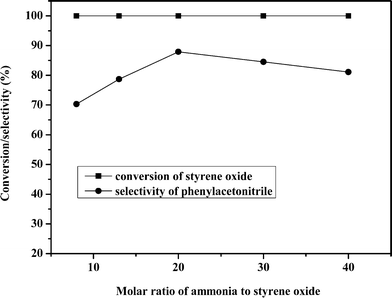 | ||
| Fig. 2 Influence of molar ratio of raw materials. | ||
Residence time is another very important parameter that must be considered. Therefore, the influence of residence time on the catalytic performance of the catalyst was evaluated under atmospheric pressure at 693 K by keeping the ammonia:styrene oxide molar ratio of 20. The results are shown in Fig. 3. It was found that when the residence time was increased from 3.01 s to 8.23 s, the conversion of styrene oxide was kept at 100% but the selectivity towards phenylacetonitrile increased from 71.7% to its maximum of 87.9% with the increase of residence time from 3.01 s to 4.51, and then decreased with the increase of residence time. It is obvious that if the residence time was too short, the intermediates could not transform to phenylacetonitrile thoroughly, however, if the residence time was too long, the generated phenylacetonitrile might be transformed to other substances.
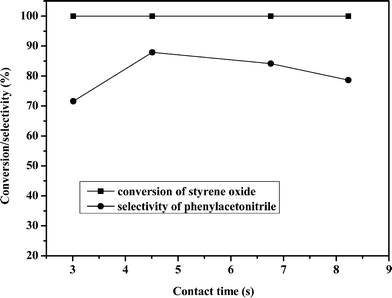 | ||
| Fig. 3 Influence of contact time on the reaction. | ||
The lifetime and regeneration of Zn30.1Cr4.3/γ-Al2O3 on-line were investigated and the results are shown in Fig. 4. It can be seen that the yield of phenylacetonitrile was 87.9% initially and almost maintained within 75 h of the catalyst on stream, then, it decreased gradually and dropped to 71.5% after the catalyst was on stream for 200 h. The catalyst was regenerated by continuously blowing air (STP 250 ml min−1) into the fixed-bed at 723 K for 3.5 h online and the reaction was run once again. The curve representing the changing of the yield of phenylacetonitrile with the running time of the regenerated catalyst almost superposed with that of the fresh one within the first 75 h, which indicated that the catalyst was regenerated online successfully.
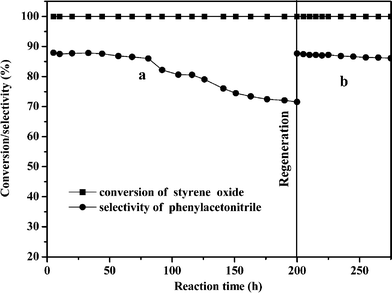 | ||
| Fig. 4 The styrene epoxide conversion and the phenylacetonitrile selectivity as functions of reaction time (a) The reaction process on the fresh Zn30.1Cr4.3/γ-Al2O3 catalyst, (b) the reaction process on the regenerated Zn30.1Cr4.3/γ-Al2O3 catalyst. | ||
The decrease of the selectivity towards phenylacetonitrile with time of the catalyst on stream as shown in Fig. 4 is due to carbonaceous deposit formation inside the pores during the catalytic run. After air regeneration the carbonaceous deposits were removed and active sites were exposed to the reactants, therefore, the selectivity was recovered. Detailed discussion will be given in the characterization section.
The composition of the products was analyzed by GC-MS. Some by-products including styrene, benzonitrile, 3,5-diphenylpyridine (trace) and undetermined compounds were found except for the target product phenylacetonitrile. Due to the absence of hydrogen in the feed, no amine as a major product in the amination of alcohols14 was detected. The possible pathways for the generation of phenylacetonitrile and the by-products are given in Scheme 2 by consulting the literature.12,24–27 In Scheme 2, the intermediates with amino or imine groups could be adsorbed on the Lewis acid sites and transformed subsequently to oligomers as carbonaceous deposits on the surface of the catalyst.
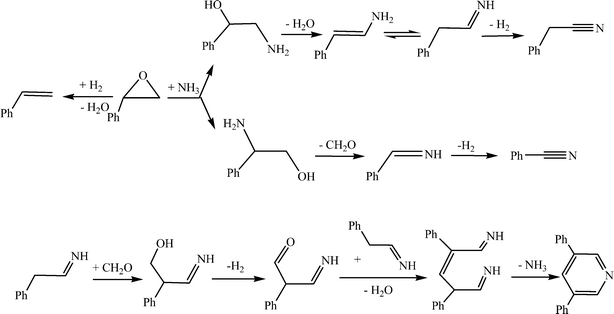 | ||
| Scheme 2 The possible pathways for the generation of phenylacetonitrile and the by-products. | ||
3.3 Characterization of catalysts
The catalysts were characterized by XRD. Fig. 5 shows the diffraction patterns of the samples of γ-Al2O3, Zn19.8/γ-Al2O3, Zn29.9/γ-Al2O3, Zn40.1/γ-Al2O3 and Zn30.1Cr4.3/γ-Al2O3. Except that of γ-Al2O3, all the patterns of the samples have a ZnAl2O4 crystalline phase.28,29 For the samples of the monometallic catalysts, the strength of the peaks representing ZnAl2O4 crystalline phase increased with the increase of the content of Zn of the samples. The sharpness of the XRD peaks indicated the higher crystallinity of ZnAl2O4 in the catalysts. However, the peaks of the ZnAl2O4 crystalline phase in the XRD patterns of bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3 were intensively weakened and broadened, which indicated that the doping of Cr in Zn29.9/γ-Al2O3 sharply decreased the size of ZnAl2O4 crystallites. These results were confirmed by the transmission electron micrographs (TEM) images (Fig. 6). Fig. 6 shows that ZnAl2O4 particle sizes were 2–5 nm and 7–15 nm in the Zn30.1Cr4.3/γ-Al2O3 and Zn29.9/γ-Al2O3 catalysts, respectively. Thus, both the XRD and TEM data are indicative of smaller average ZnAl2O4 particles size in Zn30.1Cr4.3/γ-Al2O3 than in Zn29.9/γ-Al2O3. The results suggest that the doping of Cr has a beneficial effect in suppressing the exaggerated ZnAl2O4 grain growth. A similar phenomenon that the addition of Cr to the supported ZnO decreased the ZnO grain size was observed by others in the literature.30 As reported in the literature,31 the ZnAl2O4 as a heterogeneous catalyst was active in many reactions, such as dehydration, hydrogenation, dehydrogenation, dehydrogenative condensation of normal alcohols, methylation of phenolic compounds and N-alkylation of 2-hydroxypyridine with methanol. It can be concluded that ZnAl2O4 is the active species for the dehydrogenation of imine to nitrile herein, which is the rate-determining step in the transformation of styrene oxide to phenylacetonitrile, and the small ZnAl2O4 particle size is in favor of the dehydrogenation reaction. In the XRD patterns no phase associated with Cr was identified, which indicated that Cr was present either as a highly dispersed Cr phase or as ZnCr2O4 not detectable by XRD.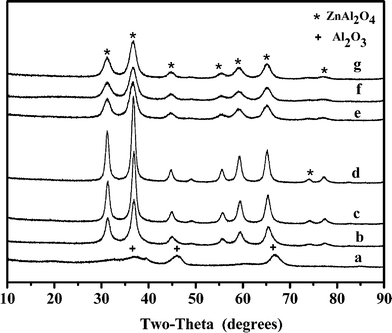 | ||
| Fig. 5 XRD patterns of the catalyst (a) Zn0/γ-Al2O3, (b) Zn19.8/γ-Al2O3, (c) Zn29.9/γ-Al2O3, (d) Zn40.1/γ-Al2O3, (e) Fresh Zn30.1Cr4.3/γ-Al2O3, (f) used Zn30.1Cr4.3/γ-Al2O3, (g) regenerated Zn30.1Cr4.3/γ-Al2O3. | ||
 | ||
| Fig. 6 TEM micrographs of (a) Zn29.9/γ-Al2O3, (b) fresh Zn30.1Cr4.3/γ-Al2O3, (c) used Zn30.1Cr4.3/γ-Al2O3, (d) regenerated Zn30.1Cr4.3/γ-Al2O3, and (e) EDX spectra on points 1–3. | ||
It was observed that the peaks of the ZnAl2O4 crystalline phase in the XRD patterns did not change obviously by comparing the patterns of the sample of Zn30.1Cr4.3/γ-Al2O3 after running on stream for 200 h with those of the fresh one. The patterns of the sample of the regenerated Zn30.1Cr4.3/γ-Al2O3 did not change too, which indicated that the catalyst was stable in the catalytic run and the regeneration process. Therefore, the regenerated catalyst displayed the same catalytic behavior as the fresh one (Fig. 4).
Representative TEM images of Zn29.9/γ-Al2O3, fresh Zn30.1Cr4.3/γ-Al2O3, used Zn30.1Cr4.3/γ-Al2O3 and regenerated Zn30.1Cr4.3/γ-Al2O3 are shown in Fig. 6. The image of Zn29.9/γ-Al2O3 shows ZnAl2O4 particles with an average diameter of about 7–15 nm (Fig. 6a). The ZnAl2O4 particle size displayed in the image of the fresh Zn30.1Cr4.3/γ-Al2O3 is with a diameter of 2–5 nm, much smaller than that of Zn29.9/γ-Al2O3 and well dispersed, which is attributed to the doping of chromium into Zn29.9/γ-Al2O3. One can find that there are some amorphous substances covered on the surface of the catalyst by comparing the image of the used Zn30.1Cr4.3/γ-Al2O3 with that of the fresh one, which makes the ZnAl2O4 particles not clearly displayed. After regeneration as described above, the amorphous substances disappeared and the ZnAl2O4 particles clearly displayed again, which indicates that the substances may be carbonaceous deposits. Meanwhile, the ZnAl2O4 particle size in the image of the regenerated catalyst did not change compared to that of the fresh one, indicating that no agglomeration took place in the catalytic run and the regeneration process. The EDX analysis revealed that the elemental carbon content detected at point 3 is much higher than that at point 2 (Fig. 6e), which indicated that the amorphous substance is carbonaceous deposits generated in the catalytic run. The EDX analysis also showed that the elemental compositions are mainly Al, Zn and O at point 1 and 2 (Fig. 6e), confirming that the particles are ZnAl2O4. It should be mentioned that the peak representing Cu in the Fig. 6e is derived from the copper slides in TEM analysis.
The amount of carbonaceous deposits on the catalyst was determined by TG-DSC and the result is shown in Fig. 7. A total weight loss of 17.8% mainly taking place in the temperature range of 723 to 873 K was found, which indicated that the carbonaceous deposits were removed by combustion. We can conclude that the deactivation of the catalyst is due to the carbonaceous deposits generated from the chemical adsorption of some alkaline substances in the catalytic run, which was also confirmed by IR spectrum of adsorbed pyridine and will be discussed later.
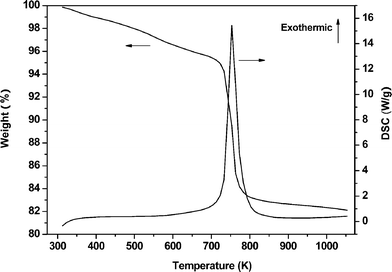 | ||
| Fig. 7 TG-DSC analysis of the used Zn30.1Cr4.3/γ-Al2O3 catalyst. | ||
The surface compositions of the fresh samples of Zn29.9/γ-Al2O3 and Zn30.1Cr4.3/γ-Al2O3 were determined by XPS. The XPS Cr 2p and Zn 2p spectra of the samples of the catalysts are depicted in Fig. 8. The binding energies of the metals are listed in Table 3. The results show that the binding energies of Zn (Zn2p3/2) in Zn29.9/γ-Al2O3 and Zn30.1Cr4.3/γ-Al2O3 are 1021.82 and 1021.99 eV, respectively, indicating that the Zn is present as ZnAl2O4 in the catalysts.32 By comparing the XPS spectra of the fresh, used and regenerated samples of Zn30.1Cr4.3/γ-Al2O3, it was also found that the binding energy of Zn (Zn2p3/2) did not change by much, indicating that the catalyst was stable in the catalytic run as revealed by the XRD analysis. In the case of Zn30.1Cr4.3/γ-Al2O3, the Cr 2p3/2 and Cr 2p1/2 signals showed peaks with a binding energy of 576.96 eV and 586.45 eV corresponding to those of Cr3+, which indicated the presence of Cr as ZnCr2O4.33
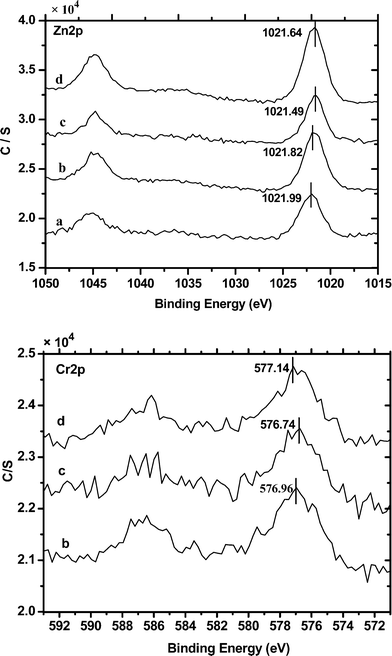 | ||
| Fig. 8 The XPS spectra of Zn 2p and Cr 2p of (a) Zn29.9/γ-Al2O3, (b) fresh Zn30.1Cr4.3/γ-Al2O3, (c) used Zn30.1Cr4.3/γ-Al2O3 and (d) regenerated Zn30.1Cr4.3/γ-Al2O3. | ||
| Catalyst | XPS binding energies (eV) | |||
|---|---|---|---|---|
| Zn 2p3/2 | Zn 2p1/2 | Cr 2p3/2 | Cr 2p1/2 | |
| a Fresh one: Zn30.1Cr4.3/γ-Al2O3 (fresh), used one: Zn30.1Cr4.3/γ-Al2O3 (used), regenerated one: Zn30.1Cr4.3/γ-Al2O3 (regenerated). | ||||
| Zn29.9/γ-Al2O3 | 1021.99 | 1045.70 | — | — |
| Fresh one a | 1021.82 | 1045.20 | 576.96 | 586.45 |
| Used one a | 1021.49 | 1044.74 | 576.74 | 585.74 |
| Regenerated one a | 1021.64 | 1044.89 | 577.14 | 586.14 |
The XPS measurements also showed that the atomic surface ratios Zn:Al of Zn29.9/γ-Al2O3 and Zn30.1Cr4.3/γ-Al2O3 were 0.111 and 0.239, respectively. The results indicated that the doping of Cr increased the amount of active species ZnAl2O4 on the surface of the catalyst, which led to the good performance of Zn30.1Cr4.3/γ-Al2O3 compared to that of Zn29.9/γ-Al2O3. Although ZnCr2O4 was detected in Zn30.1Cr4.3/γ-Al2O3 by XPS, no further information was available either from this experiment or from the literature to support that it is an active species in the dehydration and dehydrogenation steps in the transformation of styrene oxide to phenylacetonitrile.
Table 4 presents the specific surface areas and the range of pore structural parameters of Zn29.9/γ-Al2O3, the fresh, used and regenerated Zn30.1Cr4.3/γ-Al2O3. The fresh Zn30.1Cr4.3/γ-Al2O3 showed a slightly lower surface area compared to Zn29.9/γ-Al2O3 due to the doping of Cr as expected. After the Zn30.1Cr4.3/γ-Al2O3 was run on stream for 200 h, the surface area sharply decreased from 99.88 m2 g−1 to 32.97 m2 g−1 corresponding to the pore volume decrease from 0.270 cm3 g−1 to 0.130 cm3 g−1 indicating the deposition of carbon and other nonvolatile materials on the pores of the catalyst, consistent with the result of TEM. After regeneration, the surface area was increased from 32.97 m2 g−1 to 85.48 m2 g−1 and the pore volume was increased from 0.130 cm3 g−1 to 0.265 cm3 g−1, which indicated that large part of the textural properties was recovered, leading to the recovery of catalytic performance of the catalyst. The used Zn30.1Cr4.3/γ-Al2O3 showed an increase in pore diameter compared to the fresh one being indicative that the pores with small diameters were blocked due to the deposition of carbon and other nonvolatile materials.
| Catalyst | SBETa (m2 g−1) | V b (cm3 g−1) | dpc (nm) |
|---|---|---|---|
| a Calculated from the BET equation. b Calculated from the BJH method desorption. c Average pore diameter = 4 V S−1BET. d Fresh one: Zn30.1Cr4.3/γ-Al2O3 (fresh), used one: Zn30.1Cr4.3/γ-Al2O3 (used), regenerated one: Zn30.1Cr4.3/γ-Al2O3 (regenerated). | |||
| Zn29.9/γ-Al2O3 | 103.09 | 0.347 | 11.05 |
| Fresh one d | 99.88 | 0.270 | 8.87 |
| Used one d | 32.97 | 0.130 | 12.10 |
| Regenerated one d | 85.48 | 0.265 | 9.88 |
Fig. 9 shows the changes of IR spectra of adsorbed pyridine of the samples of Zn29.9/γ-Al2O3 and the fresh, used and regenerated Zn30.1Cr4.3/γ-Al2O3 in the region 1700–1300 cm−1. The peak at 1450 cm−1 is ascribed to pyridine adsorbed on Lewis acid sites, and the peak at 1545 cm−1 is ascribed to pyridine adsorbed on Brønsted acid sites as pyridinium ions.34,35 The spectra show that both Zn29.9/γ-Al2O3 and Zn30.1Cr4.3/γ-Al2O3 have Lewis acidity but very weak Brønsted acidity. It was found that the peak at 1450 cm−1 in the spectrum of the used one almost disappeared and the peak recovered after regeneration by comparing the spectra of the fresh, used and regenerated samples of Zn30.1Cr4.3/γ-Al2O3.
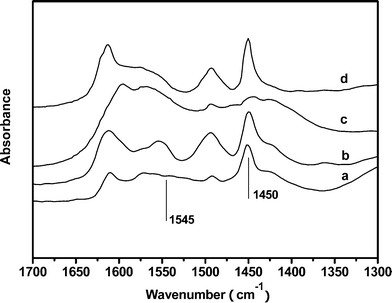 | ||
| Fig. 9 IR spectra of adsorbed pyridine of the samples of the catalysts (a) Zn29.9/γ-Al2O3, (b) fresh Zn30.1Cr4.3/γ-Al2O3, (c) used Zn30.1Cr4.3/γ-Al2O3, (d) regenerated Zn30.1Cr4.3/γ-Al2O3. | ||
The disappearance of the peak at 1450 cm−1 in the spectrum of the used sample indicated that some alkaline substances were absorbed on the Lewis acid sites in the catalytic run, which prevented the dehydration of 2-aminophenylethanol to enamine. Meanwhile, the absorbed alkaline substances as carbonaceous deposits, confirmed by TEM, also masked the active species ZnAl2O4 by blocking the dehydrogenation of imine to phenylacetonitrile, which is the rate determining step of the process, and led to a decrease in the selectivity towards the phenylacetonitrile and deactivation of the catalyst. It is likely that the absorbed alkaline substances were derived from the oligomerization of the intermediates with amino or imine group, which was also found in other amination reactions, such as amination of alcohols to amines.14,36 Meanwhile, it is worthy to mention that water generated from the dehydration of 2-aminophenylethanol was not able to be adsorbed on the catalyst either in chemical or physical form due to its low alkaline and the high reaction temperature. Therefore, the low content of water in the reaction system had little effect on the performance of the catalyst. In the case of ammonia, high content of ammonia in the feed will cause adsorption of it on the Lewis acid sites and led to the deactivation of the catalyst as found in the catalytic test.
The reappearance of the peak at 1450 cm−1 in the spectrum of the regenerated sample indicated that the carbonaceous deposits was removed by air regeneration, therefore, both the Lewis acid sites and the ZnAl2O4 particles were able to be approached by the reaction intermediates again, and the activity of the catalyst resumed.
4 Conclusion
In summary, an environmentally friendly process for the synthesis of phenylacetonitrile by amination of styrene oxide was proposed. A bimetallic catalyst Zn30.1Cr4.3/γ-Al2O3 was prepared and showed good performance in the transformation of styrene oxide to phenylacetonitrile. The characterization results indicated that the dehydration reaction in the tandem reaction mainly took place on the Lewis acid sites and revealed that ZnAl2O4 is the active species for the dehydrogenation of imine to nitrile. The doping of chromium to the γ-Al2O3 supported zinc catalyst Zn29.9/γ-Al2O3 could diminish the size of ZnAl2O4 crystallites, which is in favor of the dehydrogenation reaction. The deactivation of the catalyst is mainly due to the carbonaceous deposits generated from the chemical adsorption of some alkaline substances in the catalytic run. The activation of the catalyst can be resumed by blowing air into the reactor at high temperature.Acknowledgements
We thank the financial support by the National Natural Science Foundation of China (Grant no. 20976034) and the Natural Science Foundation for Young Scientists of Hebei Province, China (Grant no.B2009000009) and the Foundation for Innovative Talents of Hebei Province, China (Grant no. CPRC012).References
- A. A. Brakhage, P. Spröte, Q. Al-Abdallah, A. Gehrke, H. Platter and A. Tüncher, in Molecular biotechnolgy of fungal beta-lactam antibiotics and related peptide synthetases, ed. A. A. Brakhage, Springer Berin, Heidelberg, 2004, vol. 88, pp. 45–80 Search PubMed.
- K. Friedrick and K. Wallensfels, in The chemistry of the cyano group., ed. Z. Rappoport, Wiley-Interscience, New York, 1970 Search PubMed.
- M. Rossberg, W. Lendle, G. Pfleiderer, A. Tögel, E-L. Dreher, E. Langer, H. Rassaerts, P. Kleinschmidt, H. Strack, R. Cook, U. Beck, K-A. Lipper, T. R. Torkelson, E. Löser, K. K. Beutel and T. Mann, in Ullmann's encyclopedia of industrial chemistry, Wiley-VCH, Weinheim, 2006 Search PubMed.
- G. L. Blanc, Bull. Soc. Chim. France, 1923, 33, 313 CAS.
- GB Pat., 645 754, 1947 Search PubMed.
- GB Pat., 1 075 393, 1963 Search PubMed.
- US Pat., 2 553 404, 1950 Search PubMed.
- US Pat., 2606917, 1951 Search PubMed.
- US Pat., 3 002 990, 1961 Search PubMed.
- US Pat., 2 800 496, 1957 Search PubMed.
- US Pat., 2 459 128, 1949 Search PubMed.
- Y. N. Zhang, Y. C. Zhang, C. Feng, C. J. Qiu, Y. L. Wen and J. Q. Zhao, Catal. Commun., 2009, 10, 1454–1458 CrossRef CAS.
- C. Feng, Y. C. Zhang, Y. N. Zhang, Y. L. Wen and J. Q. Zhao, Catal. Lett., 2011, 141, 168–177 CrossRef CAS.
- A. Fischer, T. Mallat and A. Baiker, J. Mol. Catal. A: Chem., 1999, 149, 197–204 CrossRef CAS.
- C. Dume and W. F. Hölderich, Appl. Catal., A, 1999, 183, 167–176 CrossRef CAS.
- R. J. Card and J. L. Schmitt, J. Org. Chem., 1981, 46, 754–757 CrossRef CAS.
- J. March, Advanced organic chemistry. Reactions, mechanisms, and structure,McGraw-Hill Book Company, New York, 2nd edn, 1997, p. 381 Search PubMed.
- L. Saikia, J. K. Satyarthi, D. Srinivas and P. Ratnasamy, J. Catal., 2007, 252, 148–160 CrossRef CAS.
- B. M. Reddy, M. K. Patil, B. T. Reddy and S. E. Park, Catal. Commun., 2008, 9, 950–954 CrossRef CAS.
- M. Perissinotto, M. Lenarda, L. Storaro and R. Ganzerla, J. Mol. Catal. A: Chem., 1997, 121, 103–109 CrossRef CAS.
- R. M. West, D. J. Braden and J. A. Dumesic, J. Catal., 2009, 262, 134–143 CrossRef CAS.
- EP Pat., 0 206 632, 1986 Search PubMed.
- CN Pat., 1 062 303, 1992 Search PubMed.
- A. J. Castro, D. K. Brain, H. D. Fisher and R. K. Fuller, J. Org. Chem., 1954, 19, 1444–1448 CrossRef CAS.
- J. R. Calvin, R. D. Davis and C. H. McAteer, Appl. Catal., A, 2005, 285, 1–23 CrossRef CAS.
- Y. Higashio and T. Shoji, Appl. Catal., A, 2004, 260, 251–259 CrossRef CAS.
- J. Barrault and Y. Pouilloux, Catal. Today, 1997, 37, 137–153 CrossRef CAS.
- X. L. Duan, D. R. Yuan, Z. H. Sun, H. Q. Sun, D. Xu and M. K. Lv, J. Cryst. Growth, 2003, 252, 4–8 CrossRef CAS.
- S. Farhadi and S. Panahandehjoo, Appl. Catal., A, 2010, 382, 293–302 CrossRef CAS.
- H. H. Hng and P. L. Chan, Ceram. Int., 2009, 35, 409–413 CrossRef CAS.
- H. Grabowska, M. Zawadzki and L. Syper, Appl. Catal., A, 2006, 314, 226–232 CrossRef CAS.
- Y. Q. Wu, J. Du, K. L. Choy, L. L. Hench and J. K. Guo, Thin Solid Films, 2005, 472, 150–156 CrossRef CAS.
- F. Simard, U. A. Sedran, J. Sepólveda, N.S. Fígoli and H.I. de Lasa., Appl. Catal., A, 1995, 125, 81–98 CrossRef CAS.
- S. Triwahyono, A. A. Jalil and M. Musthofa, Appl. Catal., A, 2010, 372, 90–93 CrossRef CAS.
- B. Chakraborty and B. Viswanathan, Catal. Today, 1999, 49, 253–263 CrossRef CAS.
- A. Fischer, M. Maciejewski, T. Bürgi, T. Mallat and A. Baiker, J. Catal., 1999, 183, 373–383 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |