Synthesis and structural study of an unsymmetrical aliphatic benzamidinato ligand and its use in the formation of selected Cu(I), Mg(II) and Zr(IV) complexes†
Mei
Fan
a,
Qiaokun
Yang
a,
Hongbo
Tong
a,
Shifang
Yuan
a,
Bin
Jia
a,
Donglong
Guo
b,
Meisu
Zhou
*a and
Diansheng
Liu
a
aInstitute of Applied Chemistry, Shanxi University, Taiyuan 030006, People's Republic of China. E-mail: mszhou@sxu.edu.cn; Fax: +86-351-7011688; Tel: +86-351-7010722
bCollege of Life Science, Shanxi University, Taiyuan 030006, People's Republic of China
First published on 17th May 2012
Abstract
The reaction of a Et2O solution of LiN(Cy)SiMe3 (Cy = cyclohexyl) with PhCN in the presence of the Lewis-base donor tetrahydrofuran (THF) or Me2N(CH2)2NMe2 (TMEDA) gave the THF- or TMEDA-solvated unsymmetrical aliphatic lithium benzamidinato compound [{PhC(NCy)(NSiMe3)}Li(THF)]2 (1) or [{PhC(NCy)(NSiMe3)}Li(TMEDA)] (2). Treatment of LiN(Cy)SiMe3 with PhCN in diethyl ether, and further with CuCl, MgBr2(THF)2 or ZrCl4 at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1 and 3:1 allowed the formation of novel metal complexes [{PhC(NCy)(NSiMe3)}Cu]2 (3), [{PhC(NCy)(NSiMe3)}2Mg(OEt2)] (4) and [{PhC(NCy)(NSiMe3)}3ZrCl] (5), respectively. The X-ray structures of complexes 1–4 are presented. Complex 5/MAO system allows the moderate polymerization of ethylene.
:1, 2:1 and 3:1 allowed the formation of novel metal complexes [{PhC(NCy)(NSiMe3)}Cu]2 (3), [{PhC(NCy)(NSiMe3)}2Mg(OEt2)] (4) and [{PhC(NCy)(NSiMe3)}3ZrCl] (5), respectively. The X-ray structures of complexes 1–4 are presented. Complex 5/MAO system allows the moderate polymerization of ethylene.
Introduction
Over the past decades, developing various ancillary ligands as alternatives to cyclopentadienyl ligands has attracted much interest in catalytic processes. Accordingly, the coordination chemistry of the heteroatom-containing non-Cp-spectator ligands, especially the nitrogen-based ligand systems have been recently studied, of which the amidinates and the related guanidinates are largely explored.1–3 The amidinato ligands have been and remain of special interest because of their greater potential in ligand design over Cp ligands by virtue of their different steric and electronic properties due to the variation of the N and C substituents. In addition, they can display a variety of bonding modes to the metals.The majority of previous publications have dealt with C2-symmetric ligands where either N, N′-chelating to a metal occurs or there is bridging between two metal atoms,4 and some of their group 4 complexes are active for the highly stereoregular polymerization of propylene under pressure.5 The C1-symmetric metal amidinates are expected to be catalysts or reagents for regio- or even enantio-selective transformations.4,6 Recently, the synthesis, structures and reactivities of N′-trimethylsilyl 2-pyridylamidinate and fluoro-containing amidinates such as lithium fluoroarylamidinates and lithium N-pentafluorophenyl have been investigated.7–10 Some of them can be used as building blocks in coordination polymers, ladder and cage structures. Very recently, [benzamidinato]3− complexes have been successfully prepared via the reduction of Et2O- or TMEDA-solvated lithium benzamidinates with lithium metal,11 and they showed dynamic behaviour. The aggregation and solution dynamics of some other lithiated amidinates have also been involved.12–14 Unusual bridging μ, η2:η1-benzamidinate ligands were found in bis[bis(1-tert-butyl-3-ethylacetamidinato)magnesium15 and Fe(PhNC(Ph)NPh)4,16 and a μ, η2:η2-benzamidinate ligand was found in K2((Me3Si)NC(Ph)N(PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(tBu)2)2.17
C(tBu)2)2.17
Amongst the many reported amidinato ligands, our group has been interested in developing the coordination chemistry and catalytic behaviour of group 4 transition metal complexes containing unsymmetrical amidinato ligands such as silyl-linked bis(amidinate), silyl-linked amidinate–amidine, mixed silyl-linked bis(amidinate) and cyclopentadienyl.18–21 Some chiral amidinato- and bis(amidinato-) ligands and their metal complexes derived from the well known scaffold (1R, 2R)-1,2-diaminocyclohexane were also documented in our previous publications.22 These ligands were prepared via the nucleophilic reactions of N-centered anions to nitriles free from α-hydrogen, migrations of SiMe3 and isomerization.
Following our results with unsymmetrical aromatic and chiral amidinato ligands, we have decided to extend the ligand system to unsymmetrical aliphatic benzamidinato ligand chemistry and further towards its metal ions. Herein we report the synthesis and structural characterization of the unsymmetrical aliphatic benzamidinato lithium ligand: Lewis-base donor tetrahydrofuran(THF) or Me2N(CH2)2NMe2(TMEDA) solvated compounds [{PhC(NCy)(NSiMe3)}Li(THF)]2 (1) and [{PhC(NCy)(NSiMe3)}Li(TMEDA)] (2). These lithium salts were used as ligand transfer reagents when reacted with metal halides CuCl, MgBr2(THF)2 and ZrCl4 to produce the corresponding complexes [{PhC(NCy)(NSiMe3)}Cu]2 (3), [{PhC(NCy)(NSiMe3)}2Mg(OEt2)] (4) and [{PhC(NCy)(NSiMe3)}3ZrCl] (5), respectively. To the best of our knowledge, this is a rare example of the synthesis and structural characterization of the unsymmetrical aliphatic benzamidinato ancillary ligand system and metal complexes.
Experimental
All manipulations were carried out under an atmosphere of argon using standard Schlenk techniques. The solvent was purchased from commercial sources. Deuterated solvent C6D6, C5D5N, CDCl3 was dried over activated molecular sieves (4 Å) and vacuum transferred before use. Diethyl ether and THF were dried and distilled from sodium/benzophenone and stored over a sodium mirror under argon. Dichloromethane was distilled from activated molecular sieves (4 Å) or CaH2. CuCl, ZrCl4, PhCN, and n-butyllithium in hexane were purchased from Alfa Aesar and used without further purification. Lithiation of cyclohexylamine with n-BuLi in hexane, followed by the reaction of the resulting solution with one equivalent of trimethylsilyl chloride gave the silylated CyNH(SiMe3). Further lithiation of CyNH(SiMe3) with n-BuLi gave the starting material LiN(Cy)SiMe3. Glassware was oven-dried at 150 °C overnight. 1H spectra and 13C spectra were recorded with a Bruker DRX-300 spectrometer. Melting points were measured in sealed capillaries and are uncorrected. Elemental analyses were carried out using a Vario EL-III analyzer (Germany).Synthesis and characterization
Ethylene polymerization
Ethylene polymerization was carried out in a 500 mL autoclave stainless steel reactor equipped with a mechanical stirrer and a temperature controller. Briefly, toluene, the desired amount of cocatalyst, and a toluene solution of the catalytic precursor (the total volume was 100 mL) were added to the reactor in this order under an ethylene atmosphere. When the desired reaction temperature was reached, ethylene at 10 atm pressure was introduced to start the reaction, and the ethylene pressure was maintained by constant feeding of ethylene. After 45 min, the reaction was stopped. The solution was quenched with HCl-acidified ethanol (5%), and the precipitated polyethylene was filtered, washed with ethanol, and dried in vacuum at 60 °C to constant weight.| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Formula | C40H66Li2N4O2Si2·C4H8O | C22H41LiN4Si | C32H50Cu2N4Si2 | C36H60MgN4OSi2 |
| M | 777.13 | 396.62 | 674.02 | 645.37 |
| Crystal system | Orthorhombic | Orthorhombic | Triclinic | Monoclinic |
| Space group | Pna 21 (No.33) | P212121(No.19) | Pī (No.2) | C2/c (No.15) |
| a/Å | 32.031(19) | 10.240(3) | 10.101(12) | 19.456(17) |
| b/Å | 9.999(6) | 12.316(4) | 12.616(15) | 9.444(8) |
| c/Å | 15.155(9) | 20.617(6) | 14.018(17) | 21.882(20) |
| α (°) | 90.00 | 90.00 | 96.910(17) | 90.00 |
| β (°) | 90.00 | 90.00 | 98.657(19) | 93.237(17) |
| γ (°) | 90.00 | 90.00 | 90.190(19) | 90.00 |
| U/Å3 | 4854(5) | 2600.2(14) | 1753(4) | 4015(6) |
| Z | 4 | 4 | 2 | 4 |
| Absorption coeff. (mm−1) | 0.111 | 0.103 | 1.307 | 0.134 |
| Flack parameter | 0.5(3) | 0.2(2) | — | — |
| Unique reflections, Rint | 7313, 0.1433 | 4610, 0.1701 | 6180, 0.0605 | 3562, 0.0762 |
| Reflections with I > 2σ(I) | 3780 | 3344 | 4570 | 2039 |
| Final R indices [I > 2σ(I)] R1, wR2 | 0.0812, 0.1906 | 0.0593, 0.1520 | 0.0394, 0.0867 | 0.0607, 0.1447 |
| R indices (all data) R1, wR2 | 0.1664, 0.2462 | 0.0788, 0.1641 | 0.0616, 0.0977 | 0.1189, 0.1825 |
Results and discussion
Synthesis and characterization
Symmetric amidinato ligands were commonly prepared in situ from an insertion reaction of corresponding carbodiimide into a metal alkyl bond.26–29 Symmetric benzamidinate ligand [PhC(NSiMe3)2]− could be obtained via the reaction of lithium amide LiN(SiMe3)2 with benzonitrile.2,30–32 Unsymmetrical lithium amidinates can be made by first coupling an amine and an isothiocyanate to form a thiourea, followed by sulfur removal with N-bromosuccinimide and treatment of the resulting carbodiimide with an alkyllithium.33 The unsymmetrical aliphatic benzamidinate ligand, [PhC(NCy)(NSiMe3)]Li, was prepared from trimethylsilylcyclohexylamine after lithiation with n-BuLi and the concomitant reaction with benzonitrile. The synthetic routes to complexes 1–5 were illustrated in Scheme 1. This involved the insertion of benzonitrile into the Li–N bond of LiN(Cy)SiMe3 with either a synchronous or subsequent 1,3-SiMe3 N→N′ rearrangement and salt metathesis reactions.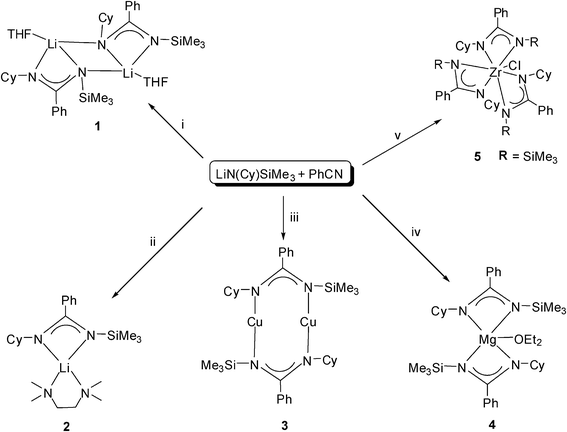 | ||
| Scheme 1 Synthesis of complexes 1–5. Reagents and conditions: (i) Et2O, −78 °C, recrystallization from THF; (ii) Et2O/TMEDA, −78 °C; (iii) CuCl, Et2O, −78 °C; (iv) 1/2 MgBr2(THF)2, Et2O, −78 °C; (v) 1/3 ZrCl4, Et2O, −78 °C, recrystallization from CH2Cl2. | ||
The THF-solvated complex 1 was obtained in 57% yield by treatment of LiN(Cy)SiMe3 with PhCN at the ratio of 1:1 in Et2O and crystallization from THF. TMEDA-solvated complex 2 was obtained also in 57% yield by treatment of LiN(Cy)SiMe3 with PhCN and TMEDA at the ratio of 1:1:1 in Et2O and crystallization from Et2O. Both of them are colorless and highly moisture- and air-sensitive. They quickly changed to a pale-yellow solid when exposed to air. In an inert atmosphere they can be stored without decomposition at room temperature.
Treatment of LiN(Cy)SiMe3 with PhCN at the ratio of 1:1 in diethyl ether, and further with anhydrous CuCl, or MgBr2(THF)2 at ratios of 1:1 or 2:1 allowed the formation of 3 and 4 in yields of 77% and 63%, respectively. Colorless single crystals of 3 and 4 suitable for X-ray analysis were obtained by recrystallization in diethyl ether in situ. Colorless crystalline 3 changes into a white solid in 12 h, and then turns pale-green, dark-green and finally black in 5 d. Crystals of 4 change into white solid in 12 h. Complex 5 was obtained by treatment of LiN(Cy)SiMe3 with PhCN at the ratio of 1:1 and then with one third portion of ZrCl4 in diethyl ether at −78 °C and by recrystallization in dichloromethane. Colorless crystalline 5 was relatively stable in air for several days and finally changed into white solid. The molecular structures of 1–4 are depicted in Fig. 1, 2, 4 and Fig. 5, respectively.
The structure of crystalline lithium benzamidinate 1 is illustrated in Fig. 1 and selected geometric parameters are listed in Table 2. Complex 1 has a ladder-shaped backbone, with a planar Li1N1Li2N4 (mean deviation of 0.0054 Å) rhomboidal core, flanked by the fused planar rings N1C1N2Li2 [mean deviation 0.0905 Å, fold angle to core 53.2°] and Li1N3C17N4 [mean deviation 0.0749 Å, fold angle to core 59.6°]. The angles at the nitrogen atom of the core [69.3(5) and 70.3(4)°] are narrower than the values found for the unsymmetrical lithium guanidinate [(Et2O)LiN(SiMe3)C(NMe2)N(Ph)]2 [73.5(3)°],34 but those at the Li atom [109.6(5) and 110.8(5)°] are wider than in the latter [106.5(3)°]. The N–Li bond lengths [2.118(12) and 2.162(12) Å] of intermetallacyclic LiNCN in 1 are slightly longer than those in [(Et2O)LiN(SiMe3)C(NMe2)N(Ph)]2 [2.057(3) Å]. In complex 1, the amidinato nitrogen atoms act as chelates. It is noteworthy that both the N1 (has a Cy arm) and N4 (possesses a SiMe3 arm) atoms also function as bridges. This novel feature differs from those of [Li{μ:κ2–N(SiMe3)C(Ph)NBut}THF]2,35 [Li{μ:κ2–N(SiMe3)C(C6H4Me–4)NPh}(OEt2)]24 and [(Et2O)LiN(SiMe3)C(NMe2)N(Ph)]234 where the bridging atom is regiospecifically restricted to N(SiNe3) or N(Ph), suggesting that the choice bridging group is governed by steric rather than electronic considerations.
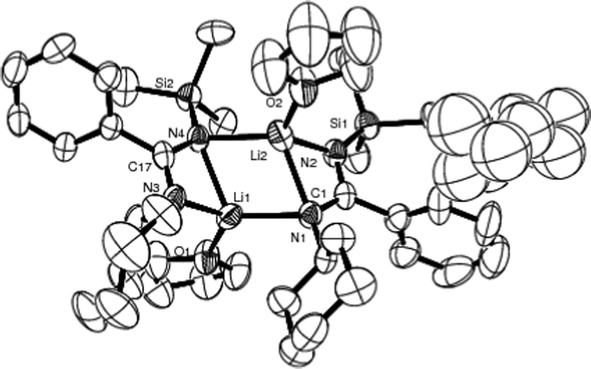 | ||
| Fig. 1 Molecular structure of 1 (50% thermal ellipsoids). | ||
| Li1–Li2 | 2.477(16) | N1–Li1 | 2.118(12) |
| N1–Li2 | 2.184(14) | C1–N1 | 1.306(8) |
| N4–Li1 | 2.194(12) | C1–N2 | 1.349(9) |
| N4–Li2 | 2.162(12) | Li1–O1 | 1.904(12) |
| N1–C1–N2 | 119.7(6) | Li1–N1–Li2 | 70.3(4) |
| N1–Li2–N2 | 66.5(4) | N1–Li1–N4 | 110.8(5) |
| N3–Li1–N4 | 66.2(4) | N1–Li2–N4 | 109.6(5) |
| N3–C17–N4 | 119.5(6) | Li1–N4–Li2 | 69.3(5) |
An X-ray study of crystalline 2 (Fig. 2, Table 3) shows that it has a distorted tetrahedral environment in which the lithium atom is arranged symmetrically between the two benzamidinate and the two TMEDA nitrogen atoms (N1–Li1 = 1.976(5) Å, N2–Li1 = 2.013(5) Å; N4–Li1 = 2.105(6) Å, N5–Li1 = 2.100(6) Å). The dihedral angle between N1Li1N2 and N4Li1N5 is 95.5°. The different C–N distances [N1–C7 = 1.309(3) Å, N2–C7 = 1.343(3) Å] suggest dissimilar electronic delocalized throughout the amidinate moiety. The Li–N(SiMe3) bond length [2.013(5) Å] is slightly longer than that of Li–N(Cy) [1.976(5) Å], suggesting the greater steric nature of the SiMe3 group than the Cy group and the unsymmetrical coordination of the ligand to the lithium atom.
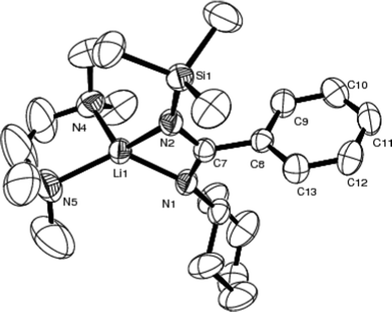 | ||
| Fig. 2 Molecular structure of 2 (50% thermal ellipsoids). | ||
| Si1–N2 | 1.703(2) | N2–Li1 | 2.013(5) |
| N1–C7 | 1.309(3) | N4–Li1 | 2.105(6) |
| N2–C7 | 1.343(3) | N5–Li1 | 2.100(6) |
| N1–Li1 | 1.976(5) | — | — |
| N1–Li1–N2 | 69.50(17) | Si1–N2–Li1 | 144.34(18) |
| N1–C7–N2 | 118.1(2) | Si1–N2–C7 | 131.20(19) |
| C7–N1–Li1 | 86.7(2) | C7–N2–Li1 | 84.30(19) |
The key bond lengths of the NCNLi moiety of 2 and its chiral benzamidinate analogue [N(myrtanyl)C(Ph)N(SiMe3)]Li·TMEDA] I6 and aromatic benzamidinate analogue [LiL(TMEDA)] (L = (2,6–Me2C6H3)NC(Ph)N(SiMe3)) II36 are shown in Fig. 3. The mean Li–N length of 2 [1.994 Å] is comparable to those in the unsymmetrical TMEDA-solvated analogues I [2.022 Å] and II [2.020 Å], and is also comparable to that in the symmetrical benzamidinate analogue Li[PhNC(Ph)NPh](TMEDA) [2.023 Å].14 The Li–N lengths are slightly shorter than those of 2.076(6)–2.188(6) Å in the monomeric PMDIEN-solvated symmetrical benzamidinate complex Li[PhNC(Ph)NPh](PMDIEN),14 but similar to the mean value in Li{(2,6–(p-ButC6H4)2C6H3)C(NPri)2}(TMEDA) [1.997 Å].37
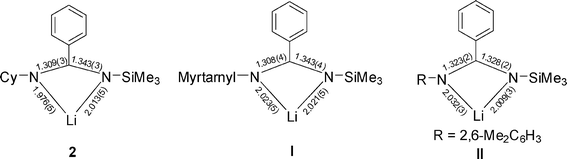 | ||
| Fig. 3 Bond lengths (Å) of NCNLi for 2, I and II. | ||
The X-ray structure of 3 is shown in Fig. 4 and selected bond distances and angles are listed in Table 4. In complex 3, there are two chemically similar independent molecules that lie about the independent inversion centres in the crystal lattice, but only one molecule is shown in Fig. 3 for clarity. Benzamidinate ligands bridge copper atoms in a μ, η1:η1 fashion, which is similar to the structure of [Cu(I) (N, N′-diisopropylacetamidinate)]227 and [CuC(Me)(NsBu)2]2.33 The average distance of Cu–N is 1.902(3) Å, being slightly longer than those of symmetric acetamidinates [Cu(I) (N, N′-diisopropylacetamidinate)]2 [1.869(1) Å]27 and [CuC(Me)(NsBu)2]2 [1.877(2) Å].33 The distance between the two copper atoms is 2.473(2) Å. This is shorter than the “normal” Cu–Cu bond distance of 2.65–2.70 Å, and much shorter than those in nitrogen-based dimeric Cu(I) 1,3,5-triazapentadienyl complexes [Cu{(N(R)C(R′))2N}]2 (R = SiMe3; R′ = dimethylamino or 1-piperidino) [3.636 and 3.536 Å],38 indicating the presence of metal–metal interaction.39–41 The N–Cu–N bonds are essentially linear with 176.41(10)°.
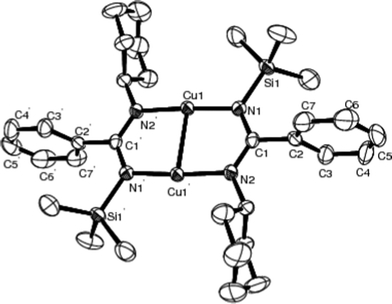 | ||
| Fig. 4 Molecular structure of 3 (50% thermal ellipsoids). Symmetry operations used to generate equivalent atoms: 2 − x, 1 − y, −z. | ||
A perspective view of 4 is presented in Fig. 5, along with selected bond lengths and angles in Table 5. Diethyl ether adduct complex 4 exists as a five-coordinate monomer in which the two amidinato ligands are coordinated in a chelating η2-fashion (Fig. 5) and are arranged around the metal in a butterfly-like fashion. Such coordination affords a much distorted trigonal bipyramidal environment with the metal, the fifth position being occupied by the solvent Et2O. The Mg1 atom and O1 of the diethyl ether ligand lie on a twofold axis. In 4, the pseudo-equatorial sites are taken up by the N2, N2′ (N2 and N2′ bear the Cy substituents) and O atom, and leaving N1 and N1′ (N1 and N1′ bear the SiMe3 substituents) in axial position. When compared to the dimeric amidinate complexes of magnesium15 and some heavy alkaline-earth strontium and barium guanidinates,42 we propose that the monomeric nature of 4 is a result of steric requirements of the substituents of the ligand. This coordination sphere of metal was also found in the five-coordination, C2-symmetric calcium complex [Ca{(Cy)NC{N(SiMe3)2}N(Cy)}2·(Et2O)]43 and the strontium complex [Sr{(iPr)NC{N(SiMe3)2}N(iPr)}2·(Et2O)]42 with bulky guanidinato ligands.
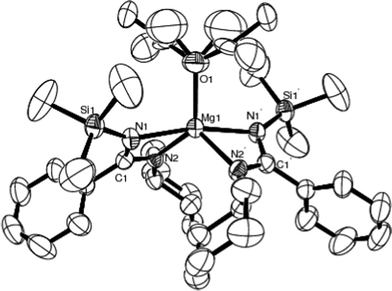 | ||
| Fig. 5 Molecular structure of 4 (50% thermal ellipsoids). Symmetry operations used to generate equivalent atoms: −x, y, 0.5 − z. | ||
| Mg1–N1 | 2.158(3) | Mg1–O1 | 2.078(5) |
| Mg1–N2 | 2.104(3) | N1–Si1 | 1.719(3) |
| C1–N1 | 1.350(5) | C1–N2 | 1.319(4) |
| N1–Mg–N2 | 64.09(12) | Mg1–N2–C1 | 91.1(2) |
| N1–C1–N2 | 115.9(3) | Mg1–N1–Si1 | 141.04(18) |
| Mg1–N1–C1 | 88.0(2) | N1–Mg1–O1 | 96.45(9) |
| N1–Mg–N1′ | 167.10(19) | N2–Mg–N2′ | 114.29(19) |
The bond distances of Mg–N in 4 are in the range of 2.104(3)–2.158(3) Å, shorter than those in the magnesium bis[N,N′-bis(trimethylsilyl)benzamidinate] bis(THF) adduct [PhC(NSiMe3)2]2Mg(THF)2 [2.188(3)–2.208(3) Å],44 but longer than those in the homometallic heteroleptic magnesium amidinate [PhC(NSiMe3)2Mg{μ-NC(CH2SiMe3)Ph}]2 [2.067(1)–2.089(1) Å].45 The bonding distance of Mg1–O1 is 2.078(5) Å. The bond angles of N1–Mg1–N2′, N1–Mg1–N1′ and N2–Mg1–N2′ are 108.37(12)°, 167.10(19)° and 114.29(19)°, respectively. The N–Mg–N angle within the amidinato ligand is 64.09(12)°. For the MgN2C metallacycle, the carbon atom C1 projects 0.0622 Å above the plane defined by Mg1, N1 and N2. The dihedral angle between N1Mg1N2 and N1′Mg1N2′ is 68.0°.
Crystals of 5, obtained from dichloromethane, were of poor quality but the data was adequate to establish its structure of tris(benzamidinato)zirconium chloride, in which the three chelated benzamidinato ligands are disposed around the zirconium atom in a propeller-like fashion. The zirconium atom is the center of a chloride-capped distorted octahedron.
Polymerization experiments
Complex 5 was tested as a procatalyst for ethylene polymerization with methylaluminoxane (MAO) as the activator under varied conditions. The detailed results are summarized in Table 6. At a ratio of Al/Zr 1000, with the temperature from 30 to 70 °C, the activity of 5 increased from no activity to 1.87 × 103 g PE mol−1 h−1. At 50 °C, with an increase of the Al/Zr molar ratio from 1000 to 3000, complex 5 showed relatively high activity (4.13 × 103 g PE mol−1 h−1) at a ratio of Al/Zr 2000. Complex 5 reached its highest activity of 5.44 × 103 g PE mol−1 h−1 with an optimal Al/Zr ratio of 2000 at 70 °C. Overall, complex 5/MAO system displays moderate activity in our case, and the Al/Zr ratio and reaction temperature didn't exert significant influence on the activity.| Entry | T/°C | Al/Zr | Yield (mg) | Activityb |
|---|---|---|---|---|
| a Polymerization conditions: 5 μmol catalyst, Cocat: MAO, 10 atm, 100 ml toluene, 0.75 h. b g mol−1 h−1. | ||||
| 1 | 30 | 1000 | Trace | — |
| 2 | 50 | 1000 | 4.70 | 1.25 × 103 |
| 3 | 70 | 1000 | 7.00 | 1.87 × 103 |
| 4 | 50 | 1500 | 7.40 | 1.97 × 103 |
| 5 | 50 | 2000 | 15.5 | 4.13 × 103 |
| 6 | 60 | 2000 | 16.3 | 4.35 × 103 |
| 7 | 70 | 2000 | 20.4 | 5.44 × 103 |
| 8 | 80 | 2000 | 15.4 | 4.11 × 103 |
| 9 | 50 | 2500 | 14.4 | 3.84 × 103 |
| 10 | 50 | 3000 | 5.70 | 1.52 × 103 |
Compared with the seven-coordination zirconium complexes such as the zirconium benzamidinate complex [N(SiMe3)C(C6H4Me-4)NPh]3ZrCl] (8.6 × 103 g PE mol−1 h−1 bar−1)4 and zirconium guanidinate [PhNC(NMe2)NSiMe3]3ZrCl (1.05 × 104 g PE mol−1 h−1),34 there were only minor differences in activity between complex 5 and its analogues.
Conclusions
We have described here a simple and effective synthesis for a novel unsymmetrical aliphatic benzamidinato ligand system [{PhC(NCy)(NSiMe3)]− and its metal complexes via the addition of lithiated cyclohexylamine to a suitable nitrile and with a concomitant 1,3-SiMe3 N→N′ rearrangement. The coordination about the lithium cation is influenced by the choice of Lewis-base donors. The two ligands in 1 show an interesting feature in that they bind to the metal in different manners. Complex 5 allows the moderate polymerization of ethylene. Effects of the non-donor solvents on the coordination mode of its lithium salts will be published in separate paper. Further work on L2ZrCl2 type complexes that will exhibit high activity for olefin polymerization is underway in our laboratory.Acknowledgements
The authors acknowledge the financial support of the Natural Science Foundation of China (No.20672070, 20772074), Shanxi Scholarship Council of China (No. 201012), and Shanxi International Education Exchange Association.References
- M. P. Coles, Dalton Trans., 2006, 985–1001 RSC.
- F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef.
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and P. G. Merle, Dalton Trans., 2007, 585–594 RSC.
- V. Volkis, M. Shmulinson, C. Averbuj, A. Lisovskii, F. T. Edelmann and M. S. Eisen, Organometallics, 1998, 17, 3155–3157 CrossRef CAS.
- C. Averbuj, E. Tish and M. S. Eisen, J. Am. Chem. Soc., 1998, 120, 8640–8646 CrossRef CAS.
- C. Knapp, E. Lork, P. G. Watson and R. Mews, Inorg. Chem., 2002, 41, 2014–2025 CrossRef CAS.
- S. Aharonovich, M. Botoshanski and M. S. Eisen, Inorg. Chem., 2009, 48, 5269–5278 CrossRef CAS.
- S. Aharonovich, M. Botoshansky, R. M. Waymouth and M. S. Eisen, Inorg. Chem., 2010, 49, 9217–9229 CrossRef CAS.
- S. Aharonovich, M. Botoshanski, Z. Rabinovich, R. M. Waymouth and M. S. Eisen, Inorg. Chem., 2010, 49, 1220–1229 CrossRef CAS.
- M. P. Coles, P. B. Hitchcock, A. V. Khvostov, M. F. Lappert and L. Maron, Dalton Trans., 2011, 40, 3047–3052 RSC.
- M. S. Eisen and M. Kapon, J. Chem. Soc., Dalton Trans., 1994, 3507–3510 RSC.
- A. Lisovskii, M. Botoshansky and M. S. Eisen, J. Chem. Soc., Dalton Trans., 2001, 1692–1698 RSC.
- J. Barker, D. Barr, N. D. R. Barnett, W. Clegg, I. Cragg-Hine, M. G. Davidson, R. P. Davies, S. M. Hodgson, J. A. K. Howard, M. Kilner, C. W. Lehmann, I. Lopez-Solera, R. E. Mulvey, P. R. Raithby and R. Snaith, J. Chem. Soc., Dalton Trans., 1997, 951–955 RSC.
- A. R. Sadique, M. J. Heeg and C. H. Winter, Inorg. Chem., 2001, 40, 6349–6355 CrossRef CAS.
- F. A. Cotton, L. M. Daniels, A. Matonic and C. A. Murillo, Inorg. Chim. Acta, 1997, 256, 277–282 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert and M. Layh, J. Chem. Soc., Dalton Trans., 1998, 3113–3117 RSC.
- S. D. Bai, H. B. Tong, J. P. Guo, M. S. Zhou and D. S. Liu, Polyhedron, 2010, 29, 262–269 CrossRef CAS.
- S. D. Bai, H. B. Tong, J. P. Guo, M. S. Zhou and D. S. Liu, Inorg. Chim. Acta, 2009, 362, 1143–1148 CrossRef CAS.
- H. B. Tong, M. Li, S. D. Bai, S. F. Yuan, J. B. Chao, S. P. Huang and D. S. Liu, Dalton Trans., 2011, 40, 4236–4241 RSC.
- S. D. Bai, F. Guan, M. Hu, S. F. Yuan, J. P. Guo and D. S. Liu, Dalton Trans., 2011, 40, 7686–7688 RSC.
- (a) J. F. Li, S. P. Huang, L. H. Weng and D. S. Liu, J. Organomet. Chem., 2006, 691, 3003–3010 CrossRef CAS; (b) J. F. Li, L. H. Weng, X. H. Wei and D. S. Liu, J. Chem. Soc., Dalton Trans., 2002, 1401–1405 RSC; (c) J. F. Li, S. P. Huang, L. H. Weng and D. S. Liu, Eur. J. Inorg. Chem., 2003, 810–813 CrossRef CAS; (d) J. F. Li, L. H. Weng, S. P. Huang, H. B. Tong and D. S. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, E58, o1078–o1080 CAS; (e) J. F. Li, L. H. Weng, S. P. Huang, H. B. Tong and D. S. Liu, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2002, E58, m510–m512 CAS.
- G. M. Sheldrick, Correction Software, University of Göttinggen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Program for the Solution of Crystal Structures, University of Göttinggen, Germany, 1997 Search PubMed.
- Program for Crystal Structure Refinement, Bruker Analytical X-ray Instruments Inc., Madison, WI, USA, 1998 Search PubMed.
- C. B. Wilder, L. L.Reitfort, K. A. Abboud and L. McEIwee-White, Inorg. Chem., 2006, 45, 263–268 CrossRef CAS.
- B. S. Lim, A. Rahtu, Jin-Seong. Park and R. G. Gordon, Inorg. Chem., 2003, 42, 7951–7958 CrossRef CAS.
- S. R. Foley, Y. Zhou, G. P. A. Yap and D. S. Richeson, Inorg. Chem., 2000, 39, 924–929 CrossRef CAS.
- M. P. Coles, D. C. Swenson, R. F. Jordan and V. G. Jr. Young, Organometallics, 1997, 16, 5183–5194 CrossRef CAS.
- Y. Zhou and D. S. Richeson, Inorg. Chem., 1997, 36, 501–504 CrossRef CAS.
- D. G. Dick, J. J. H. Edema, R. Duchateau and S. Gambarotta, Inorg. Chem., 1993, 32, 1959–1962 CrossRef CAS.
- (a) E. Nelkenbaum, M. Kapon and M. S. Eisen, Organometallics, 2005, 24, 2645–2659 CrossRef CAS; (b) V. Volkis, E. Nelkenbaum, A. Lisovskii, G. Hasson, R. Semiat, M. Kapon, M. Botoshansky, Y. Eishen and M. S. Eisen, J. Am. Chem. Soc., 2003, 125, 2179–2194 CrossRef CAS.
- Z. W. Li, S. T. Barry and R. G. Gordon, Inorg. Chem., 2005, 44, 1728–1735 CrossRef CAS.
- M.-S. Zhou, H.-B. Tong, X.-H. Wei and D.-S. Liu, J. Organomet. Chem., 2007, 692, 5195–5202 CrossRef CAS.
- (a) A. Antolini, P. B. Hitchcock, A. V. Khvostov and M. F. Lappert, Can. J. Chem., 2006, 84, 269–276 CrossRef; (b) P. B. Hitchcock, M. F. Lappert and R. Sablong, Dalton Trans., 2006, 4146–4154 RSC.
- H.-K. Lee, T. S. Lam, C.-K. Lam, H.-W. Li and S.-M. Fung, New J. Chem., 2003, 27, 1310–1318 RSC.
- J. A. R. Schmidt and J. Arnold, J. Chem. Soc., Dalton Trans., 2002, 2890–2899 RSC.
- M.-S. Zhou, P. Li, H.-B. Tong, Y.-P. Song, T. Gong, J.-P. Guo, L.-H. Weng and D.-S. Liu, Inorg. Chem., 2008, 47, 1886–1888 CrossRef CAS.
- S. Franzen, V. M. Miskowski, A. P. Shreve, S. E. Wallace-Williams, W. H. Woodruff, M. R. Ondrias, M. E. Barr, L. Moore and S. G. Boxer, Inorg. Chem., 2001, 40, 6375–6382 CrossRef CAS.
- A. Schaefer and R. Ahlrichs, J. Am. Chem. Soc., 1994, 116, 10686–10692 CrossRef CAS.
- B. Ni and J. R. Kramer, J. Phys. Chem. A, 2003, 107, 8949–8954 CrossRef CAS.
- T. M. Cameron, C.-Y. Xu, A. G. Dipasquale and A. L. Rheingold, Organometallics, 2008, 27, 1596–1604 CrossRef CAS.
- F. Feil and S. Harder, Eur. J. Inorg. Chem., 2005, 4438–4443 CrossRef CAS.
- D. Walther, P. Gebhardt, R. Fischer, U. Kreher and H. Görls, Inorg. Chim. Acta, 1998, 281, 181–189 CrossRef CAS.
- R. Forret, A. R. Kennedy, J. Klett, R. E. Mulvey and S. D. Robertson, Organometallics, 2010, 29, 1436–1442 CrossRef CAS.
Footnote |
| † CCDC reference numbers 865606–865609. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20709e |
| This journal is © The Royal Society of Chemistry 2012 |