DOI:
10.1039/C2RA20383A
(Paper)
RSC Adv., 2012,
2, 6576-6589
Regulatory function of divalent cations in controlling the acidogenic biohydrogen production process
Received
2nd March 2012
, Accepted 5th May 2012
First published on 22nd June 2012
Abstract
The functional role of divalent cations viz., iron (Fe2+), magnesium (Mg2+), nickel (Ni2+), zinc (Zn2+) and manganese (Mn2+) in regulating the biohydrogen (H2) production potential of a biocatalyst was evaluated under varying concentrations. The optimum conditions were evaluated in detail and compared with the principle conditions for enhanced conversion efficiency. Higher concentration of Fe2+ showed an enhanced H2 production efficiency of the biocatalyst (61.94 mmol) due to its role as a component of hydrogenase and ferredoxin. On the contrary, a higher concentration of Mg+2 showed a higher substrate utilization capability (76.46%) of the biocatalyst. The optimum metal concentration showed a higher H2 yield (18.23 mol kg−1 CODR; 3.5 mol mol−1 glucose) over principle operation conditions (14.76 mol kg−1 CODR; 2.84 mol mol−1 glucose). Higher concentrations of acetate (78 ± 6%) as the metabolic intermediate over butyrate (12 ± 2%) supported the observed higher H2 yields. Higher reductive catalytic current over oxidative current along with the enhanced dehydrogenase activity supported the higher H2 production efficiency. Shift in the redox Tafel slope was witnessed near zero in the case of Fe2+ and optimum conditions indicating the simultaneous oxidation and reduction reactions facilitating the availability of higher number of protons through metabolite inter-conversions that can make H2. The presence of divalent cations at an optimum concentration will enhance the H2 production capability of the biocatalyst. However, elevated levels of metal concentration showed a negative influence on the biocatalyst efficiency.
1. Introduction
Fermentative hydrogen (H2) production using wastewater is gaining prominence in the present research scenario of bioenergy generation. H2 production process was exploited in the direction of wastewater nature, operating conditions, influencing factors, biochemical aspects etc.1–9 However, some of the basic understanding such as the biocatalyst behavior and the shift in metabolic pathways with experimental variations were less exploited.10,11 Evaluation of the functional role of trace metals in altering the H2 production efficiency of the biocatalyst and shifting their metabolic activities will play a pivotal role in enhancing the process efficiency.9 Trace metals are required for the activation or function of many enzymes and co-enzymes related to energy metabolism and are also essential for the cell growth of many microorganisms. Several studies have been reported pertaining to the influence of different trace metals on anaerobic fermentation and H2 production processes.12–18 However, the optimum concentrations of various trace metals were inconsistent in different studies because the requirement of metal ions varies depending on other operational conditions. Zhang and Shen13 reported the requirement of decreasing Fe2+ concentration with increasing temperature. Nickel (Ni2+) can enter the cells via the Mg2+–manganese (Mn2+) transport system,19,20 which means that the presence of higher concentrations of any one of the three metal ions (Ni2+/Mg2+/Mn2+) can increase the Ni2+ uptake into the cell. This gives an immense necessity for understanding of the metabolic process of H2 production in correlation with the other operating conditions to enhance the process efficiency.
The detailed evaluation of bioprocesses during H2 production with respect to the biochemical function of the selected divalent metal ions has not yet been fully explored. Each of the trace metals has a specific biochemical function in the cell during the metabolic pathway and the change in its concentration may alter that metabolic function. Ni2+ and Fe2+ are the unique active site components of hydrogenases, which are classified according to the active site metal ion [Fe]-only, [Ni–Fe], and [Ni–Fe–Se] hydrogenases.14,21–23 Mg2+ is also an active site component of several enzymes and acts as a transporter molecule of adenosine triphosphate (ATP) in the cell.24 Mn2+ has a multi-faceted role in biological systems because of its ability to exist in 11 oxidation states (7+ to 3−), but it is generally restricted to Mn2+ complexes.25 Zinc (Zn2+) is involved in a wide variety of cellular processes and maintenance of cellular Zn2+ is essential for the growth and survival which necessitated an improved understanding of acquisition, assimilation and metabolism of Zn2+. In this context, the present study evaluated the biochemical function of divalent cations, viz., iron (Fe2+), magnesium (Mg2+), nickel (Ni2+), zinc (Zn2+) and manganese (Mn2+) in regulating the H2 production process with respect to the bioprocess parameters. Interpretation of datasets pertaining to the bioprocess parameters was done with the enzyme activities and bio-electrochemical profiles to understand the metabolic shifts during operation under varying metal concentrations.
2. Experimental methodology
2.1 Selection and enrichment of the biocatalyst
Anaerobic mixed consortia acquired from an existing full scale treatment plant was used as the biocatalyst in this experiment. The parent culture (sludge) was enriched in designed synthetic wastewater (DSW, glucose 3 g l−1, NH4Cl 0.5 g l−1, KH2PO4 0.25 g l−1, K2HPO4 0.25 g l−1, MgCl2 0.3 g l−1, CoCl2 25 mg l−1, ZnCl2 11.5 mg l−1, CuCl2 10.5 mg l−1, CaCl2 5 mg l−1, MnCl2 15 mg l−1; NiCl2 16 mg l−1; FeCl2 25 mg l−1) for 5 days to activate the consortia. Later, the culture was centrifuged (at 1000 rpm for 5 min) about three times to remove the particulate material (the supernatant was collected) present in the sludge. This was followed by washing with deionized water about three times (8000 rpm for 5 min) to separate the microorganisms (the pellet was collected) from other particulate materials and to avoid the interference of the trace metals present within the sludge. The isolated culture was enriched for about 48 h in DSW and was used for further experiments.
2.2 Experimental design
DSW was used as a substrate in all the experiments at a constant organic loading rate (OLR) of 2.27 kg COD m3 day−1 [pH, 7.2; volatile fatty acids (VFA), 520 mg l−1; chemical oxygen demand (COD), 4800 mg l−1; total dissolved salts (TDS), 140 mg l−1]. Experiments were carried out in three phases, where the initial phase was designed based on varying concentrations (C1–C6) of selected metal ions to identify the optimum concentration of each metal ion (Table 1). The concentrations were selected based on their original concentration present in the DSW, which was used in our previous experiments [Fe2+, 25 mg l−1; Mg+2, 300 mg l−1; Ni+2, 16 mg l−1; Zn2+, 11.5 mg l−1; Mn2+, 15 mg l−1].11 The selected trace metal concentrations were increased by 2 to 5 times of the original concentration keeping all other concentrations constant compared with the original (principle feed) and zero (control) concentrations (Table 1). In the second phase, detailed investigations were carried out at the optimum metal concentrations with respect to biochemical and electrochemical analysis along with the bioprocess parameters. In the third stage, all the metal ions were added at optimum concentrations to the principle feed (optimized conditions) and the performance of the biocatalyst was evaluated in comparison with the operation using principle feed. All the experiments were carried out in batch mode in 250 ml anaerobic flasks, with a working volume of 180 ml filled with 10 ml of enriched mixed culture and 170 ml of DSW at designated trace metal concentrations. The resulting mixture in each flask was adjusted to pH 6 using concentrated orthophosphoric acid (88%) or 1 N NaOH. After adjusting the pH, the flasks were kept in a shaking incubator at 34 ± 1 °C and 100 rpm to provide efficient contact between the substrate and the inoculum. All the flasks were purged with oxygen free nitrogen gas for 60 s to maintain strict anaerobic microenvironment and were then sealed with a rubber septum to prevent gaseous exchange. All the experiments were carried out in triplicate and the average value was considered for the discussion.
Table 1 Design and consolidated results of the experimental variations carried out during the initial phase
| Trace metal |
Concentration (mg l−1) |
CHP (mmol) |
H2 yield (mol kg−1 CODR) |
ξ
COD (%) |
SDR (kg CODR m3 day−1) |
|
Iron
|
0 (C1) |
2.78 ± 0.1 |
0.94 ± 0.05 |
61.46 ± 2.1 |
1.39 ± 0.08 |
| 25 (C2) |
50.64 ± 2.1 |
14.89 ± 0.3 |
70.83 ± 2.3 |
1.61 ± 0.07 |
| 50 (C3) |
54.99 ± 2.3 |
15.76 ± 0.32 |
72.71 ± 2.1 |
1.65 ± 0.06 |
| 75 (C4) |
60.78 ± 3.4 |
17.12 ± 0.28 |
73.96 ± 2.4 |
1.68 ± 0.07 |
| 100 (C5) |
68.28 ± 3.6 |
19.29 ± 0.34 |
73.75 ± 2.2 |
1.67 ± 0.08 |
| 150 (C6) |
18.89 ± 1.8 |
6.74 ± 0.18 |
58.33 ± 2.1 |
1.32 ± 0.06 |
| 200 (C7) |
8.46 ± 0.8 |
3.60 ± 0.12 |
48.96 ± 2.3 |
1.11 ± 0.06 |
|
Nickel
|
0 (C1) |
0.92 ± 0.05 |
0.27 ± 0.03 |
70.83 ± 2.3 |
1.69 ± 0.08 |
| 8 (C2) |
21.83 ± 1.3 |
6.34 ± 0.36 |
71.67 ± 2.4 |
1.63 ± 0.08 |
| 16 (C3) |
50.64 ± 2.8 |
14.89 ± 0.54 |
70.83 ± 2.5 |
1.61 ± 0.09 |
| 32 (C4) |
25.78 ± 1.4 |
7.78 ± 0.42 |
69.58 ± 2.4 |
1.58 ± 0.07 |
| 48 (C5) |
19.76 ± 1.1 |
6.61 ± 0.38 |
62.29 ± 2.3 |
1.41 ± 0.06 |
| 64 (C6) |
0.00 |
0.00 |
58.33 ± 2.1 |
1.32 ± 0.05 |
|
Magnesium
|
0 (C1) |
0.86 ± 0.08 |
0.26 ± 0.04 |
67.50 ± 2.1 |
1.53 ± 0.06 |
| 300 (C2) |
50.64 ± 3.4 |
14.89 ± 0.42 |
70.83 ± 2.3 |
1.61 ± 0.08 |
| 600 (C3) |
60.26 ± 3.6 |
16.42 ± 0.44 |
76.46 ± 2.2 |
1.74 ± 0.09 |
| 1200 (C4) |
20.12 ± 2.2 |
7.13 ± 0.32 |
58.75 ± 2.1 |
1.33 ± 0.07 |
| 1500 (C5) |
12.46 ± 1.1 |
4.76 ± 0.21 |
54.58 ± 2.2 |
1.24 ± 0.06 |
| 2000 (C6) |
3.11 ± 0.2 |
1.36 ± 0.05 |
48.75 ± 2.3 |
1.11 ± 0.06 |
|
Manganese
|
0 (C1) |
1.85 ± 0.09 |
0.58 ± 0.04 |
65.83 ± 2.2 |
1.49 ± 0.05 |
| 7.5 (C2) |
26.79 ± 1.7 |
8.19 ± 0.3 |
68.13 ± 2.1 |
1.55 ± 0.07 |
| 15 (C3) |
50.64 ± 3.8 |
14.89 ± 0.42 |
70.83 ± 2.3 |
1.61 ± 0.08 |
| 30 (C4) |
0.00 |
0.00 |
67.50 ± 2.4 |
1.53 ± 0.06 |
| 45 (C5) |
0.00 |
0.00 |
61.25 ± 2.2 |
1.39 ± 0.05 |
| 60 (C6) |
0.00 |
0.00 |
53.13 ± 1.8 |
1.21 ± 0.05 |
|
Zinc
|
0 (C1) |
0.92 ±.08 |
0.27 ± 0.024 |
70.83 ± 2.2 |
1.61 ± 0.08 |
| 6 (C2) |
47.99 ± 3.1 |
14.16 ± 0.36 |
70.63 ± 2.1 |
1.60 ± 0.09 |
| 12 (C3) |
50.64 ± 3.2 |
14.89 ± 0.28 |
70.83 ± 2.3 |
1.61 ± 0.09 |
| 24 (C4) |
47.46 ± 2.8 |
14.00 ± 0.31 |
70.63 ± 2.4 |
1.60 ± 0.08 |
| 36 (C5) |
43.07 ± 2.1 |
12.74 ± 0.26 |
70.42 ± 2.6 |
1.59 ± 0.08 |
| 48 (C6) |
5.31 ± 1.1 |
1.56 ± 0.08 |
70.83 ± 2.4 |
1.61 ± 0.07 |
2.3 Analysis
H2 gas generated during the experiment was estimated at regular time intervals using a microprocessor based pre-calibrated electrochemical H2 sensor (ATMI GmBH Inc., Germany). Samples from all the flasks were collected every 6 h and analyzed for pH, chemical oxygen demand (COD), volatile fatty acids (VFA) as per the procedures outlined in standard methods.26 Substrate (COD) removal efficiency (ξCOD) was calculated using eqn (1), where, CSO represents the initial COD concentration (mg l−1) in the feed and CS denotes COD concentration (mg l−1) at a defined time.| | | ξCOD = [(CSO − CS)/CSO] × 100 | (1) |
Substrate degradation rate (SDR-kg COD m3 day−1) was calculated to study the pattern of COD removal with respect to time and volume according to eqn (2), where, FR represents feed rate (m3 day−1) and Rv denotes reactor volume (m3).
| | | SDR = {[(Cso − Cs) × FR]/Rv} | (2) |
Bacterial cell growth was monitored in regular time intervals at 660 nm using UV-Vis spectrophotometer.
2.3.1 Qualitative analysis of metabolic intermediates.
Selected samples collected at regular time intervals were centrifuged to remove the particulate material and qualitative estimation of soluble metabolites was carried out by high performance liquid chromatography (HPLC; Shimadzu LC10AD) operated under optimized conditions [UV-VIS detector; C18 column reverse phase column 250 × 4.6 mm and 5 μ particle size; flow rate 0.6 ml h−1; wave length 210 nm; mobile phase 40% of acetonitrile in 1 mN H2SO4 (pH 2.5 to 3.0); sample injection 20 μl].
2.3.2 Phosphatase.
Phosphatase (PT) enzyme activity of mixed culture was estimated by a method based on the reaction of disodium phenyl phospate.27 10 ml anodic mixed culture was added with 2 ml of methyl benzene, 10 ml disodium phenyl phospate, 10 ml citrate buffer (pH 7) in a sequence in 100 ml volumetric flask. After shaking at 200 rpm for 20 mins, the flask was kept for incubation at 37 °C for 24 h and made up to 100 ml with distilled water. The mixture was then filtered with compact filter papers. The filterate was then transfered to 100 ml volumetric flask and 5 ml borate sodium hydroxide buffer (pH 9.6), 19 ml distilled water, 1 ml of 0.2% Gibbs reagent were added slowly, and the solution in the flask was made up to 100 ml with distilled water (37 °C). The mixture was gently stirred and incubated at room temperature for 20 min to develop color and the absorbance was measured at 578 nm.
2.3.3 Dehydrogenase activity.
Dehydrogenase (DH) enzyme activity of mixed culture was determined by a method based on reduction of 2,3,5-triphenyltetrazolium chloride (TTC).11,28 5 ml TTC (5 g l−1) and 2 ml of glucose solution (0.1 mol l−1) were added to 5 ml of culture and the resulting solution was stirred continuously for 20 min at 200 rpm prior to incubation at 37 °C for 12 h. After incubation, 1 ml of concentrated sulfuric acid was added to this reaction mixture to stop the deoxidization and 5 ml of methylbenzene was added to extract the triphenyl formazan (TF) formed in the reaction mixture and the sample was agitated at 200 rpm for 30 min. After keeping flasks aside for 3 min, the reaction mixture was then centrifuged at 4000 rpm for 5 min. The absorbance of the supernatant was measured at 492 nm in a UV-Vis spectrometer.
2.3.4 Bio-electrochemical behavior.
The bio-electrochemical behavior of the mixed consortia was evaluated using cyclic voltammetry (CV) with an electrochemical cell having a platinum and graphite rod as the working and counter electrodes respectively against Ag/AgCl(S) reference electrode. Voltammograms were recorded by a potentiostat–glavanostat system (Autolab, PGSTAT12, Ecochemie) by applying a potential ramp at a scan rate of 30 mV s−1 over a range of +0.5 to −0.5 V to the working electrode. All the electrochemical assays were performed in whole cell form using the substrate itself as the electrolyte. Redox Tafel slopes and polarization resistance was calculated from the voltammetric profiles through Tafel analysis using GPES 4.0 software of the potentiostat–glavanostat system.
3. Results and discussion
3.1 Optimization of trace metal concentrations
Initially each metal ion was studied at different concentrations to identify the optimum concentration for enhanced H2 production and substrate degradation (Fig. 1; Table 1).
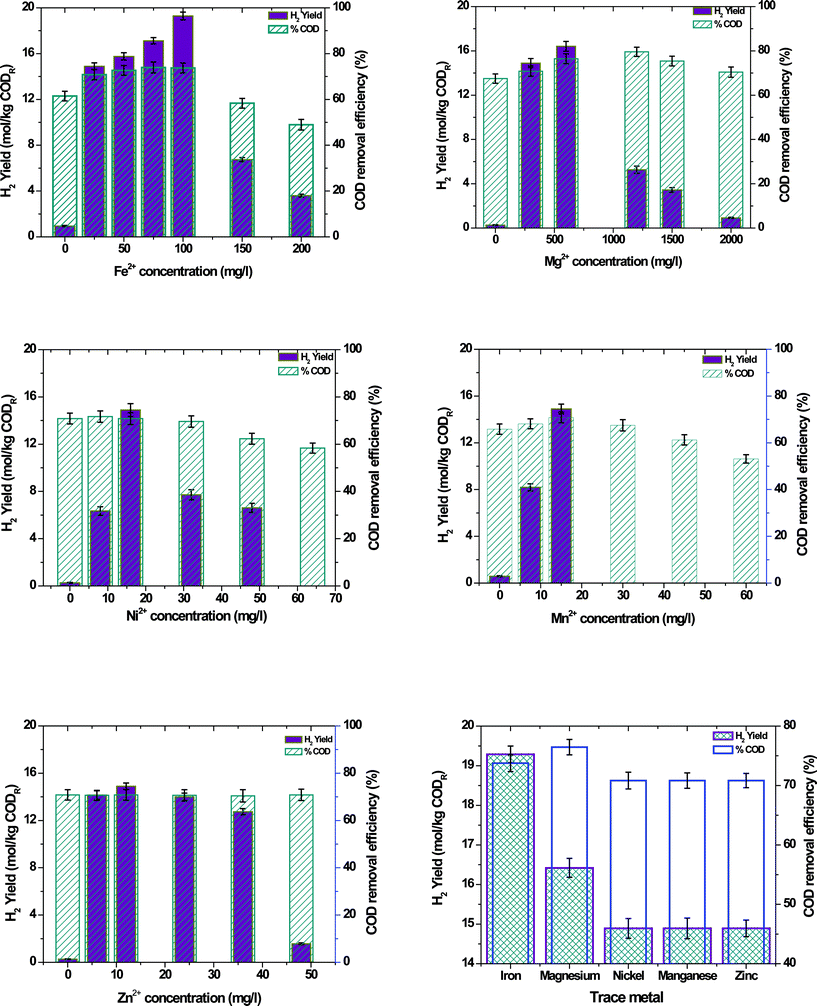 |
| | Fig. 1 H2 yield and substrate removal efficiency with the function of selected divalent trace metals under varying concentrations. | |
3.1.1 Iron (Fe2+).
An increase in Fe2+ concentration to 100 mg l−1 showed an increment in H2 production [C1, 2.78 mmol; C2, 50.64 mmol; C3, 54.98 mmol; C4, 60.77 mmol; C5, 68.28 mmol] indicating its positive role in the metabolic activities. However, a drop in H2 production was observed after 100 mg l−1 [C6, 18.88 mmol; C7, 8.46 mmol], which might be due to the toxic loads of Fe2+ on the biocatalyst. Coagulation of the reactor contents was also observed beyond 100 mg l−1 which might be the reason for the drop in H2 production. Substrate degradation was also increased with increase in Fe2+ concentration up to 75 mg l−1 [C1, 61.46%; C2, 70.83%; C3, 72.71%; C4, 73.96%] and decreased thereafter [C5, 73.75%; C6, 58.33%; C7, 48.96%] indicating its positive influence on substrate conversion also. Overall, the process yield was also increased with an increase in Fe2+ concentration up to 100 mg l−1 and dropped thereafter. Operation at 25 mg l−1 [C2, 14.89 mol kg−1 CODR] showed about 93% increment in H2 yield compared to the operation without Fe2+ [C1, 0.943 mol kg−1 CODR], which indicated the significance of Fe2+ in the H2 production process. Further increments in Fe+2 concentration depicted a 22% increment in H2 yield [C5, 19.29 mol kg−1 CODR] at 100 mg l−1 and dropped thereafter.
3.1.2 Magnesium (Mg2+).
An increment in H2 production was observed with an increase in Mg2+ concentration up to 600 mg l−1 [C1, 0.859 mmol; C2, 50.64 mmol] and dropped thereafter [C3, 60.26 mmol; C4, 20.12 mmol; C5, 12.46 mmol; C6, 3.11 mmol]. Substrate degradation also followed a similar trend for H2 production [C1, 67.50%; C2, 70.83%; C3, 76.46%; C4, 58.75%; C5, 54.58%; C6, 48.75%]. About 98% increment in the H2 yield was obtained with the addition of Mg2+ [C2, 14.89 mol kg−1 CODR] into the reactor when compared to the operation without Mg2+ [C1, 0.265 mol kg−1 CODR], indicating the significant role of Mg2+ in the substrate conversion. Further addition of Mg2+ depicted about 9% increment in the H2 yield [C3, 16.42 mol kg−1 CODR] and dropped thereafter.
3.1.3 Nickel (Ni2+).
H2 production was increased with an increase in Ni2+ concentration up to 16 mg l−1 and dropped thereafter [C1, 0.92 mmol; C2, 21.82 mmol; C3, 50.64 mmol; C4, 25.78 mmol; C5, 19.76 mmol; C6, 0.00 mmol]. On the other hand, negligible influence on the substrate degradation was observed at lower Ni2+ concentrations [C1, 70.83%; C2, 71.67%; C3, 70.83%; C4, 69.58%], while at higher concentrations, a decrement in the substrate utilization was observed [C5, 62.29%; C6, 58.33%]. The cessation of H2 production and low substrate degradation observed at C6 concentration indicated the toxic influence of Ni2+ on the biocatalyst metabolic activities. Overall, the process yield increased by about 98% with the addition of Ni2+ at 16 mg l−1 concentration [14.89 mol kg−1 CODR], compared to the operation without Ni2+ [0.271 mol kg−1 CODR], and further increments showed a negative impact on the process.
3.1.4 Manganese (Mn2+).
Addition of Mn2+ depicted an increment in H2 production up to 15 mg l−1 [C1, 1.85 mmol; C2, 26.79 mmol; C3, 50.64 mmol] concentration and suddenly dropped to zero thereafter. Substrate degradation also showed an increment up to 15 mg l−1 [C1, 65.83%; C2, 68.12%; C3, 70.83%] and decreased thereafter [C4, 67.50%; C5, 61.25%; C6, 53.12%]. H2 yield was increased by about 96% with 15 mg l−1 of Mn2+ concentration [14.89 mol kg−1 CODR] compared with the operation without Mn2+ [0.59 mol kg−1 CODR].
3.1.5 Zinc (Zn2+).
In spite of toxic effects, an increase in Zn2+ concentration depicted a positive role in H2 production compared to the operation without Zn2+. However, negligible variation was observed between 6–36 mg l−1 [C1, 0.92 mmol; C2, 47.99 mmol; C3, 50.64 mmol; C4, 47.46 mmol; C5, 43.06 mmol] and dropped at 48 mg l−1 [C6, 5.31 mmol]. The substrate degradation also didn't show variation with varying Zn2+ concentration [70.75 ± 0.1%]. The H2 yield showed about 98% increment at 12 mg l−1 Zn2+ concentration compared to the operation without Zn2+ [C3, 14.89 mol kg−1 CODR; C1, 0.27 mol kg−1 CODR].
3.2 Comparative evaluation at best metal concentrations
Fe2+ and Mg2+ showed higher performance at 100 mg l−1 and 600 mg l−1 concentrations compared to the principle feed concentrations (Fe2+, 25 mg l−1; Mg2+, 300 mg l−1), while Ni2+ (16 mg l−1), Mn2+ (15 mg l−1) and Zn2+ (12 mg l−1) showed higher performance at concentrations similar to their concentrations in principle feed. Henceforth, detailed investigations were carried out for optimum Fe2+ and Mg2+ concentrations in comparison with the principle feed in the second phase of study. Optimum concentrations of all the selected divalent metal ions was added into the feed (optimum conditions) and evaluated in detail in the third phase. The results obtained in these two stages were comparatively evaluated and depicted here.
3.2.1 Biocatalyst growth.
Significant variation in the biocatalyst growth was observed with the function of operation time and the experimental condition studied (Fig. 2). Maximum cell growth was observed at 24th h in all the cases increasing from a lower value at 0th h. Principle Fe2+ and Mg2+ operations showed a rapid increment in cell growth till 6th h, while optimum condition showed a rapid growth till 12th h. All the cases showed a gradual increment till 18th h followed by a rapid shoot up in cell growth at 24th h. Rapid drop in cell growth was observed after 24th h till 42nd h followed by a gradual drop till 48th h. Higher cell growth was observed in Mg2+ flask (0.536 at 660 nm) followed by optimum (0.494 at 660 nm), Fe2+ (0.436 at 660 nm) and base-line (principle) flasks (0.365 at 660 nm).
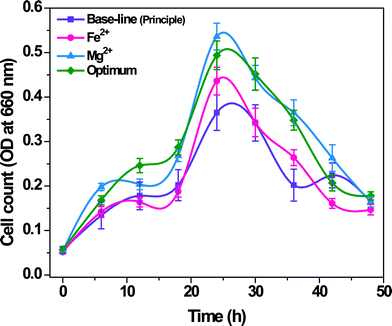 |
| | Fig. 2 Growth curve of the biocatalyst during operation with Fe2+, Mg2+ and optimum conditions in comparison with the Base-line (principle) condition. | |
3.2.2 Hydrogen production.
H2 production efficiency of the biocatalyst depicted variation with the function of operating conditions in terms of production rate (HPR) and cumulative H2 production (CHP) (Fig. 3). H2 production was extended up to the 34th h in the case of Fe2+ and principle feed operations, while it was extended up to the 38th h in the case of Mg2+ operation, which might be due to the higher substrate conversion efficiency. H2 production was started from the 6th h in the case of Fe2+ and Mg2+ operations [Fe2+, 0.38 mmol h−1; Mg2+, 1.16 mmol h−1], while principle feed operation depicted start up from the 8th h [1.62 mmol h−1]. A rapid increment in HPR was observed with Mg2+, while it was gradual with Fe2+ until they reached a maximum HPR at the 12th h [Fe2+, 3.81 mmol h−1; Mg2+, 4.04 mmol h−1]. Principle feed operation also showed a pattern almost similar to the Fe2+ flask and also depicted maximum HPR at the 12th h [3.82 mmol h−1]. However, Mg2+ depicted higher HPR at the 12th h followed by Fe2+ and principle operations. After reaching the maximum HPR, principle operation showed a rapid drop until the 18th h and then showed a gradual decrement thereafter prior to reaching the baseline at the 36th h, while Mg2+ depicted gradual drop in HPR until the 38th h and reached the base line. On the contrary, the Fe2+ flask showed a relatively lower drop until the 28th h and then showed a rapid drop until the 34th h prior to reaching the base line. HPR in the case of optimized conditions was more or less similar to the Fe2+ flask until the 30th h depicting higher HPR at the 12th h [3.82 mmol h−1] but thereafter showed a higher value than the Fe2+ flask resulting in enhanced H2 production. Cell growth observed in the case of the Mg2+ flask was higher suggesting that the reducing equivalents generated during substrate metabolism were mostly utilized for cell growth but not towards H2 production. On the contrary, Fe2+ and optimum flasks showed lower cell growth supporting the observed higher H2 production in these flasks. CHP increased gradually until the 34th h and stabilized thereafter in the case of principle [50.19 mmol] and Fe2+ [61.94 mmol] operations, while it was extended until the 38th h in the case of Mg2+ operation [58.03 mmol] and the 36th h in the case of optimized conditions [64.18 mmol]. The higher CHP observed in the case of optimized conditions might be due to the presence of all the trace metals at required concentrations.
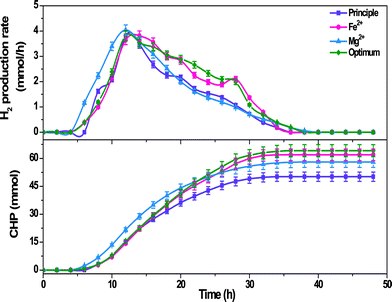 |
| | Fig. 3 H2 production rate and CHP during operation with Fe2+, Mg2+ and optimum conditions in comparison with the principle condition. | |
3.2.3 Substrate degradation.
Glucose was used as the sole carbon source in the experiment and its removal was analyzed at regular time intervals in terms of both carbohydrates and COD removal efficiencies (Fig. 4a&b). On the contrary to H2 production, higher substrate degradation was observed in the Mg2+ flasks (ξCOD, 76.46%; ξCarbohydrate, 79.74%), which might be due to the functional role of Mg2+ in activating the substrate molecules to enter into the metabolic pathway. The higher cell growth observed in this case also supports the same. Principle operation showed higher carbohydrate conversion (ξCarbohydrate, 77.91%) after Mg2+ due to the presence of optimum concentrations of all the metal ions except Fe2+ and Mg2+, playing their respective roles in metabolic activities. However, the COD removal was lower (ξCOD, 70.83%) than Mg2+ and Fe2+ operations. This might be due to the initial rapid conversion of glucose to VFA resulting in a pH drop, which might have lead to a lagging of biocatalytic metabolic activities. Lower substrate degradation was observed with Fe2+ operation (ξCOD, 73.75%; ξCarbohydrate, 74.11%) in spite of higher H2 production which might be due to the higher activity of hydrogenases that maintained the metabolic pathway towards H2 production where the proton reduction was continuous but the carbon removal was less. Lower cell growth observed in this case compared to the Mg2+ flask also supports the same. In the case of optimized conditions, substrate degradation was almost similar with Fe2+ operation (ξCOD, 73.33%), while the carbohydrate degradation was higher over all the other conditions (ξCarbohydrate, 79.92%). This might be due to the presence of Mg2+ at optimum concentrations, which lead to higher glucose removal and an optimum Fe2+ concentration that facilitated controlled conversion to the end product. Substrate degradation rate (SDR) also showed similar trend like removal efficiency (Fig. 4a). A higher SDR was observed in the Mg2+ flask (1.73 kg CODR m3 day−1) and principle operation showed the least SDR (1.61 kg CODR m3 day−1). Fe2+ operation and optimum conditions depicted almost similar SDR (1.669 ± 0.005 kg CODR m3 day−1), which is higher than principle and lower than Mg2+ operations. H2 yield with the function of COD and glucose removal was also calculated which showed a similar pattern to CHP (Fig. 4c; Table 2). Optimized conditions showed a higher H2 yield (18.23 mol kg−1 CODR; 3.5 mol mol−1 glucose) followed by Fe2+ (17.49 mol kg−1 CODR; 3.36 mol mol−1 glucose), Mg2+ (15.81 mol kg−1 CODR; 3.04 mol mol−1 glucose) and principle conditions (14.76 mol kg−1 CODR; 2.84 mol mol−1 glucose). The values obtained were higher and near the theoretical yield which might be due to the inoculation of selective consortia and avoidance of other interferences normally present in sludge.
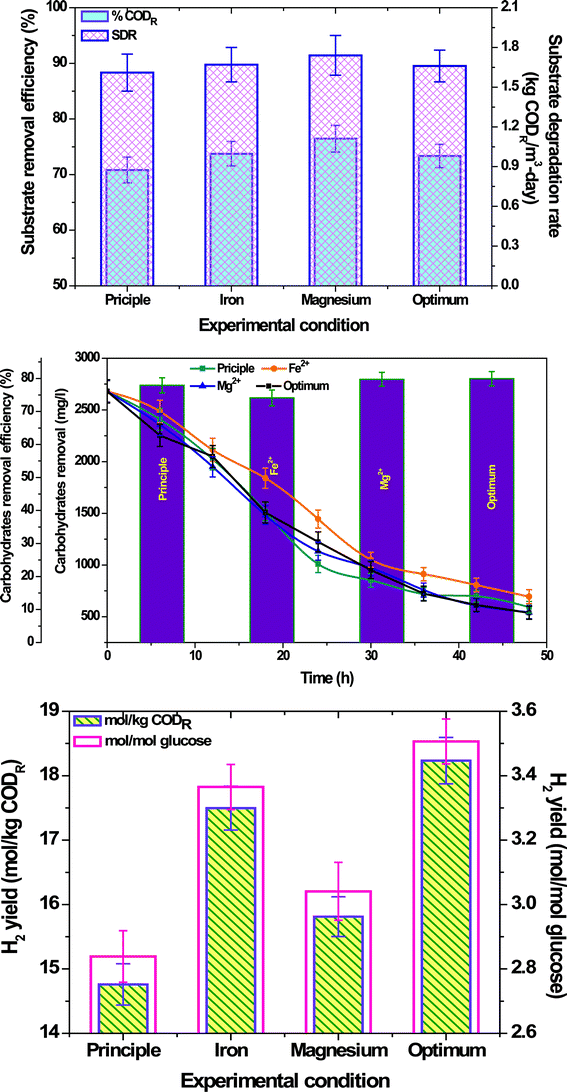 |
| | Fig. 4 (a) Substrate degradation efficiency and substrate degradation rate; (b) Carbohydrates removal efficiency; (c) H2 yield with substrate and glucose, during operation with Fe2+, Mg2+ and optimum conditions in comparison with the principle condition. | |
Table 2 Consolidated table of comparative data pertaining to the optimized conditions
|
|
H2 yield (mol kg−1 CODR) |
ξ
COD (%) |
SDR (kg CODR m3 day−1) |
Oxidative Tafel slope (V dec−1) |
Reductive Tafel slope (V dec−1) |
Polarization resistance (kΩ) |
|
Principle
|
14.76 |
70.83 |
1.61 |
0.041 |
0.933 |
46.320 |
|
Magnesium
|
15.81 |
76.45 |
1.73 |
0.047 |
1.513 |
54.280 |
|
Iron
|
17.49 |
73.75 |
1.67 |
0.33 |
0.451 |
349.20 |
|
Optimum
|
18.23 |
73.33 |
1.66 |
0.373 |
0.503 |
262.30 |
3.2.4 Redox conditions and acid intermediates.
Evaluation of system redox states with the function of experimental conditions and operation time (change in pH) gives a clear understanding of the metabolic shifts because it is one of the governing factors of metabolic pathways of the biocatalyst. Changes in external pH can bring alterations in several physiological parameters, including internal pH, concentration of ions, membrane potential29,30 and proton shuttling11 depending upon the organism. Along with the change in system redox state, quantitative and qualitative analysis of acid intermediates (VFA) at regular intervals is also important to understand the shifts in metabolic activities of the biocatalyst. VFA and pH are considered to be markers for the acidogenic fermentation.10,31 Except principle operation, all the other three experimental conditions showed similar patterns of redox change until the 24th h and then varied (Fig. 5). Principle operation showed a rapid drop in pH compared to the other experimental conditions reaching a lower redox value at the 18th h (4.2) which continued at almost similar values until the 30th h (4.3 ± 0.1) and rapidly increased to higher values at the 36th h (5.4) followed by a gradual increment thereafter. This shows a typical anaerobic fermentation process, where decrement in pH will be noticed initially due to the generation of acid metabolites followed by increment in pH with concomitant VFA consumption towards methanogenesis. The observed VFA profile strongly supports the same. Almost similar concentrations of acetate (42.6 ± 1.8%) and butyrate (45.2 ± 1.2%) were observed during initial phase of operation followed by the generation of propionate (32.6 ± 0.8%) in the later phase. Consumption of acetate (25.4 ± 1%) and butyrate (24 ± 1.3%) was associated with the propionate production after 24th h indicating the typical anaerobic metabolism towards methanogenesis. The drop in system pH was continued till the end of experiment in the case of Mg2+ reaching a lower redox value at 48th h (3.7) which might be due to the higher substrate activation leading to the entrance of more number of glucose molecules into degradation pathway with the generation of acid intermediates. Almost stable and higher VFA observed till 42nd h (850 ± 100 mg l−1) strongly supports the same. Mg2+ condition also showed similar concentrations of acetate (46.2 ± 0.8%) and butyrate (47.6 ± 1.1%) but they remained unchanged even with increasing time indicating the process towards acidogenesis but not towards methanogenesis. On the contrary, Fe2+ condition showed a rapid increment in pH after 24th h (4.4) and continued at stable values until the end of the experiment (5.3 ± 0.1). The VFA profile correlated with the pH profile, where the VFA was lower when compared to principle operation but almost similar for an extended period of time (1258 ± 120 mg l−1) indicating the higher conversion efficiency of the biocatalyst. The optimized system operation showed almost an inverted parabolic curve of redox state indicating the features of anaerobic fermentation similar to principle operation. However, the VFA concentration was initially low (12th h, 640 mg l−1) but increased with time to a maximum value at 24th h (1690 mg l−1) followed by a gradual drop till the end of the experiment (48th h, 1058 mg l−1). In spite of higher VFA generation, the increment in pH than all other experimental conditions during the later phase of operation indicates the higher and controlled substrate degradation along with their effective conversion to H2. The acetate concentration (78 ± 6%) was higher in both the Fe2+ and optimized conditions compared to butyrate concentration (12 ± 2%) indicating the favorable environment for higher H2 yields. However, optimized condition (79.2 ± 1%) showed comparatively higher acetate compared to Fe2+ conditions (74.4 ± 1%). Higher acetate concentrations suggested the metabolic pathway towards higher H2 production which might be the reason for the observed higher H2 yields.
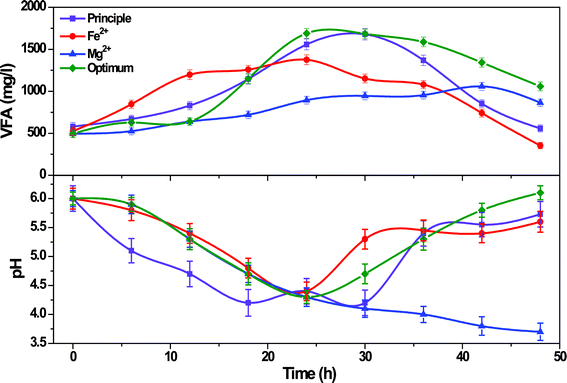 |
| | Fig. 5 (a) Variation in pH and VFA profiles during operation with Fe2+, Mg2+ and optimum conditions in comparison with the principle condition. | |
3.2.5 Enzyme activities.
PT and DH are the important enzymes involved in substrate degradation as well as the inter-conversion of metabolites. PT mainly functions to deactivate the glucose molecule by removing the phosphate (PO43−) group, without which the glucose molecule can not enter metabolic pathways. DH is an intracellular enzyme essential for the transfer of protons (H+) between metabolic intermediates through redox reactions using several mediators (NAD+, FAD+, etc.) which make the H+ available for further reduction to H2. The activity of these two enzymes is inversely proportional to each other during higher H2 production and substrate utilization. PT activity should be low for good substrate utilization and the DH activity should be high for higher H2 production. The activities of PT and DH were assayed at regular time intervals to understand the biochemical functions of system (Fig. 6).
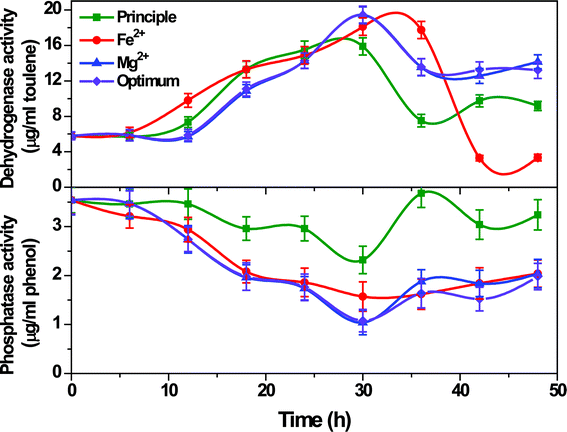 |
| | Fig. 6 Phosphatase and dehydrogenase activities during operation with Fe2+, Mg2+ and optimum conditions in comparison with the principle condition. | |
3.2.6 Bio-electrochemical profiles.
The internal potential of the bacterial cell is maintained by the proton shuttling between the metabolic intermediates, especially under an anaerobic microenvironment.11 The proton shuttling can also be studied from the electron discharge (ED) pattern using cyclic voltammetry (CV). CV permits a direct electrochemical detection of redox signals, and senses the potential difference across the interface and thus helps to elucidate the electrochemical reaction occurring at the electrode surface.10,11,31,32 The potential difference developed between microbial cell and surrounding medium leads to the electron flow towards the working electrode under applied external potential thereby generating oxidation reduction currents in the voltammetric signature.10,11 Oxidation of the substrate in order to obtain energy and synthesis of cellular components from carbon source through energy coupling mechanisms is a general process during bacterial metabolic activities. Protons and electrons, the key precursors for H2 production, are released during the substrate degradation with the involvement of redox reactions catalyzed by the enzymes during anaerobic respiration. Enzyme-catalyzed transfer of electrons from an intracellular electron carrier molecule (redox mediator) to a proton will occur as a part of the metabolic activity during dark fermentation process resulting in H2 production.11 The change in proton and electron transfer mechanisms with the operating conditions can be exploited using cyclic voltammetry. Voltammetric profiles were drawn at regular time intervals during the operation for Fe2+ and Mg2+ flasks along with optimized and principle conditions (Fig. 7).
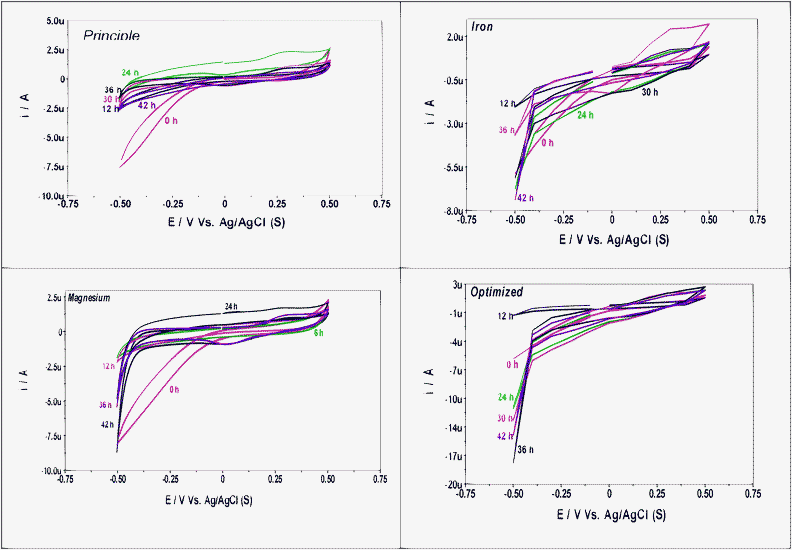 |
| | Fig. 7 Cyclic voltammograms with the function of operation time with Fe2+, Mg2+and optimum conditions in comparison with the principle conditions. | |
Oxidative catalytic currents were lower in all the cases compared to the reductive catalytic currents indicating the prevalence of an anaerobic microenvironment. However, the current varied based on the experimental condition. The oxidative catalytic current in the case of principle and Mg2+ operations showed a similar trend, while Fe2+ and the optimized flask showed a similar pattern. On the contrary, the catalytic reduction currents showed a similar pattern in all the cases until the 12th h and then also except the principle operation, the other three conditions showed similar patterns. Principle and Mg2+ operations showed a marginal drop in oxidative currents from an initial value until the 12th h followed by a rapid increment at the 24th h and then a drop at the 30th h, which continued gradually until the 48th h. In the case of Fe2+ and optimized flasks the oxidative current was increased from initial value at the 6th h followed by a drop and then a marginal increment until the 30th h. A sharp increment in oxidative current was observed in both the cases at the 36th h and again followed by a drop at the 42nd h and continued until the 48th h. The oxidative current in the case of Fe2+ and optimized flasks was almost stable throughout operation except at the 6th and 36th hours where a sharp increment was observed followed by a drop to the previous value. This indicates the similar oxidation phenomenon throughout operation in both the cases leading to the gradual substrate utilization. On the contrary, principle and Mg2+ operations showed typical metabolic patterns of microbes, where at a particular point the oxidation will be more likely to result in higher substrate utilization instead of controlled substrate conversion. However, the oxidative current of Mg2+ was higher when compared with the principle operation indicating the influence of Mg2+ in enhancing the substrate oxidation capability of the biocatalyst.
Reductive catalytic currents showed a clear variation with the function of experimental condition indicating the shift in the metabolic activity of the biocatalyst based on the trace metal composition. Principle operation showed lower reduction currents supporting the lower reduction process that resulted in lower H2 production. Mg2+ and Fe2+ flasks showed almost similar currents throughout operation with marginal variations which was higher than principle operation supporting the observed higher H2 production in both the cases. However, the reduction current in the case of Mg2+ showed a drop from the 36th h almost to the base line, while in the case of Fe2+, the reduction current was higher until the 42nd h followed by a small drop. H2 production observed for extended periods of time in the case of Fe2+ operation strongly supports the same. The optimized operation showed a huge increment in the reduction currents until the 36th h followed by a drop until the 48th h. Higher substrate oxidation along with the increased conversion efficiency of the biocatalyst under optimum trace metal concentration might be the reason for the observed elevated increments in the reduction current.
3.2.7 Bio-electro kinetics.
The bio-electro catalytic ability of the biocatalyst was analyzed through Tafel slope analysis to understand the change in the redox behavior of the biocatalyst with the function of experimental variation. The semi-empirical Tafel equation (eqn (3) and eqn (4)) can be expressed by
Where i represents current (A) and E is applied voltage (V) and βa and βc are the Tafel slopes (αanF/RT or αcnF/RT). Tafel plots provide a visual understanding of the losses present in the system which helps to interpret the biocatalytic activity. The Tafel plot (Fig. 8) helps to derive kinetic parameters viz., oxidative and reductive slopes (βa and βc) and polarization resistance (Rp, Ω).33,34 The Tafel slopes and polarization resistance showed good correlation with the observed H2 production profiles and redox catalytic currents in association with the DH activity of the biocatalyst with the function of trace metal concentration. Significant variation in the redox Tafel slopes were observed with the function of trace metals concentration. In the principle conditions, the oxidative slope was very low (0.041 V dec−1) compared to the reductive slope (0.933 V dec−1) indicating higher oxidation at the working electrode but lower reduction reactions at the counter electrodes. This might be due to the generation of protons and electrons during substrate degradation but their consumption in the metabolic activities instead of reaching the counter electrode. The Mg2+ flask also showed similar redox slope (oxidation, 0.047 V dec−1; reduction, 1.513 V dec−1) trends like the principle conditions, which is also supported by the observed higher substrate degradation. However, the reduction slope was much higher in this case compared to the principle conditions. The shift in the Tafel slope from oxidation to reduction is observed at ∼−0.4 V which is very from to the ideal condition (0 V), indicating the dominance of oxidation over reduction. On the contrary, the iron fed and optimum conditions showed relatively similar oxidation (Fe2+, 0.33 V dec−1c; Optimum, 0.373 V dec−1) and reduction slopes (Fe2+, 0.451 V dec−1; Optimum, 0.503 V dec−1) indicating simultaneous redox behavior of the biocatalyst. The protons and electrons generated during substrate oxidation are consumed during reduction by the hydrogenase. The shift in Tafel slope from oxidation to reduction is also near zero in both the cases indicating simultaneous redox reactions providing more protons. Higher H2 production observed in both these cases supports the same.
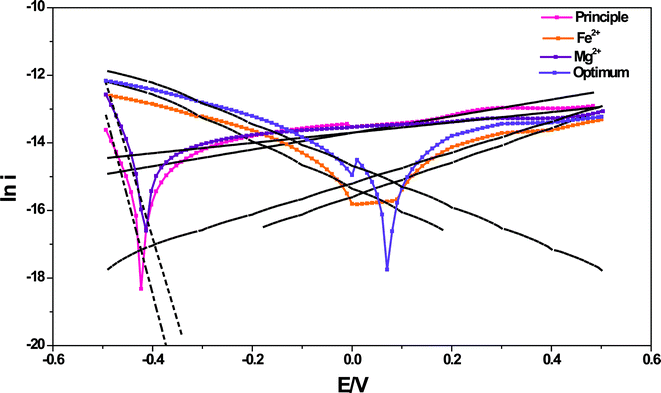 |
| | Fig. 8 Tafel plots depicting shift in the biocatalyst behavior between oxidation and reduction against applied potential with Fe2+, Mg2+ and optimum conditions in comparison with the principle condition. | |
The resistance for the electron transfer from the biocatalyst at the solution–electrode interface can be understood through polarization resistance (Rp), which was calculated using Tafel analysis. Rp depicted significant variation with the function of experimental variation studied. Rp was lower in the case of principle (46.32 kΩ) and Mg2+ conditions (54.28 kΩ), while it was higher in the case of Fe2+ (349.2 kΩ) and optimum conditions (262.3 kΩ). Lower Rp indicates higher electron transfer to the electrode from the biocatalyst which was supported by the observed higher substrate degradation and in turn released more electrons in the case of principle and Mg2+ conditions. A higher function of hydrogenases to convert the released protons and electrons into H2 in the case of Fe2+ and optimum conditions might have resulted in the higher Rp depicting the obstructions for the electron flow to the electrode from the biocatalyst.
3.3 Biochemistry of trace metals in hydrogen metabolism
The functional role of all the selected trace metals in the microbial metabolic activities was depicted in the schematic representation (Fig. 9). Fe2+ is very important for the function of hydrogenase and also as the active site component for the ferridoxin protein, which is a carrier of electrons to the hydrogenase. Fe2+ also acts as mediator for the intra-cellular electron transfer either individually or as a component of prosthetic groups. Higher concentrations of Fe2+ showed enhancement in H2 production efficiency of the biocatalyst due to its significant role as a component of hydrogenase and ferredoxin.23,35–37 However, at elevated levels of Fe2+ concentration, it acts as electron acceptor from the metabolism to defend from its toxic influence. Higher Fe2+ concentration also act as a coagulant which was supported by the observed coagulation of reactor contents at higher concentrations. Higher reduction currents compared to the oxidation currents observed in the case of Fe2+ flask from the voltammetric profiles also supported the same. Negligible variation in oxidation currents throughout the operation indicates the controlled substrate oxidation and higher reduction supports the effective conversion of generated protons and electrons into H2. Increased electron acceptor function of Fe2+ also helped in decreasing the electron loss in the metabolic activities and thus can be utilized for H2 production. Ni2+ is an essential trace element for many microorganisms as an active center of four metallo-enzymes and plays an important role in three major anaerobic processes i.e., uptake and production of H2, methanogenesis and acetogenesis.20–23 Ni2+ is a crucial component of [Ni–Fe] and [Ni–Fe–Se] hydrogenases which are the other classes of hydrogenases. [Ni–Fe] hydrogenases catalyze both the production and utilization of H2 and maintain the proper balance between electron donors or acceptors.21 On the contrary, [Ni–Fe–Se] hydrogenases are meant for the H2 uptake, especially present in the methanogenic archea.14 Optimum Ni2+ concentration (16 mg l−1) showed enhancement in H2 production efficiency of the biocatalyst by activating the [Ni–Fe] hydrogenases, while further increment has shown a drop in H2 production. This might be due to the function of Ni2+ in activating the uptake function of [Ni–Fe] hydrogenases in the H2 producing consortia or activating the [Ni–Fe–Se] hydrogenases in methanogenic consortia at higher concentrations shifting the metabolic pathway distant from the H2 production.
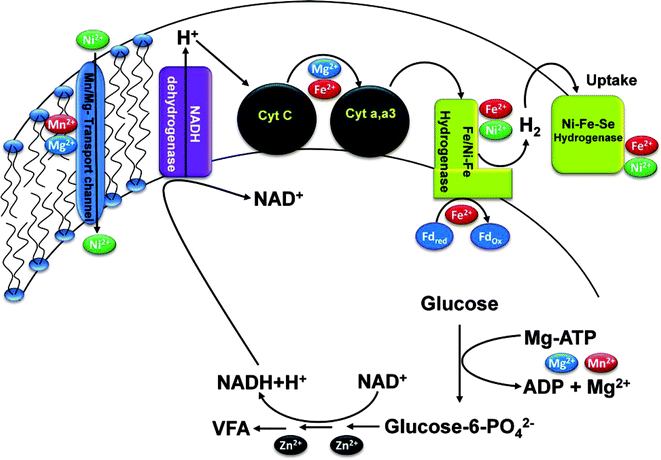 |
| | Fig. 9 Schematic representation of the trace metals functional role in the metabolic activities of the biocatalyst during acidogenic fermentation for H2 production. | |
Mg2+ plays a pivotal role in the metabolic processes for substrate utilization, especially for the activation of substrate molecules without which they cannot enter metabolic pathways. The ATP molecule first attached with the Mg2+ ion (Mg–ATP), which can further react with the glucose, delivering the phosphate group to glucose and forms an active glucose–X–phosphate (X may be 1st or 6th position based on the necessity of the cell metabolism) that can enter the metabolic pathway (Fig. 9). Many electron carriers including cytochromes and protein complexes are containing Mg2+ as central atom.38 Mg2+ ion is also an activator of many enzymes, and component of cell walls and cell membranes. Mg2+ also helps in the transport of different molecules in the cell (ATP transport) and from the medium into the cell (Ni2+ transport).24,38 Higher concentrations of Mg2+ helps in higher substrate degradation that can facilitate higher availability of protons and electrons which can result in higher H2 production. Higher availability of protons also activates the function of hydrogenases which results in higher H2 production. Higher substrate degradation along with higher H2 production observed in the case of Mg2+ strongly supports the same. Higher oxidation currents observed in the case of the Mg2+ flask over all the other flasks witnessed the role of Mg+2 in increasing the substrate utilization capability of the biocatalyst. Almost similar reduction currents with the Fe2+ flask observed in the case of Mg2+ flask throughout the operation is evidence for the higher conversion efficiency of the biocatalyst. This might be due to the activator function of Mg2+ for several electron acceptors in the cell, especially hydrogenases through maintaining the proton gradient. Mn2+ also shows almost similar functions as Mg2+. The ionic radius of Mn2+ is in the middle of Mg2+ and Ca2+ ionic radii and hence it can be possible for an overlapping function of Mn2+ with these metal ions in providing the structural stabilization of several enzymes.25 The redox function of Mn2+ is relevant to the oxygen as the substrate or product in many enzymes, although it is the active site component. Mn2+ also involves in the scavenging of superoxide radicals and hydrogen peroxide as a metal complex or as a component of enzymes such as superoxide dismutase and pseudo catalase to protect the biocatalyst from the reactive oxygen species.39 Endospore formation and regermination in several bacterial species requires Mn+2.15 It also functions as absolute growth factor in some species like Lactobacillus and its relative levels were found to influence the substrate utilization along with the other trace metals.40,41 Higher concentrations of Mn2+ help in the uptake of several trace metals because of its function as a membrane transporter protein. In the present study, the H2 production and substrate degradation were increased until optimum Mn2+ concentration (15 mg l−1) but further increment showed a rapid drop to zero in the H2 production. This might be due to the toxic effects of Mn2+, which can also activate other cell functions due to its multiple roles. The uptake of other metal ions such as Ni2+ might also have increased due to the high Mn2+ concentrations, which results in the shift in the metabolic pathway from the biohydrogen production process. Zn2+ was reported as an important factor that can enhance H2 production using anaerobic microflora, inspite of its toxic effects on biocatalysts at higher concentrations.17 Small amounts of Zn2+ along with the optimum concentrations of other trace metals will enhance the H2 production capability of the biocatalyst. Zn2+ is an essential micronutrient that plays a critical role in various physiological processes as a cofactor in all the six functional classes of enzymes and especially as an important constituent for the maintenance of protein structure.42 Zn2+ is also related with the reactions and transformations of dehydrogenase, dismutase, hydrogenase and methyltransferase based on its concentration.17 H2 production and substrate degradation efficiency did not show significant variation across a wide range of concentrations, which indicates the non-specific role of Zn2+ in the H2 production process. Further increment in Zn2+ concentration showed a rapid drop in H2 production indicating its toxic effect on the metabolic activities of the biocatalyst.
Higher H2 production along with the higher substrate degradation observed in the case of optimized conditions might be due to the presence of all the trace metals at required concentrations. Almost similar oxidative currents observed throughout the operation like the Fe2+ flask and the huge increment in reductive currents over the other flasks supports the controlled and stable substrate degradation releasing protons and electrons accompanied with their effective reduction to H2 by the biocatalyst. In summary, the optimum concentrations of trace metals, especially higher concentrations of Fe2+ and Mg2+ will enhance the electron transfer and H2 production capability of the biocatalyst. They also facilitate proper cell growth and promote different enzyme activities, especially dehydrogenase and hydrogenases that can increase the H2 production.
4. Conclusions
The functional role of divalent metal ions in regulating the H2 production efficiency of a biocatalyst was evaluated in detail using biochemical and electrochemical methods. Fe2+ and Mg2+ showed improved H2 production at higher concentrations of 100 and 600 mg l−1 respectively, while, Ni2+, Mn2+ and Zn2+ showed a positive influence at lower concentrations. On the other hand, Mg2+ showed significant positive influence on substrate degradation compared to other metal ions supporting its role in biocatalytic metabolic activities. The optimized conditions showed higher H2 production and overall process yields compared to the principle operation. Redox catalytic currents observed through cyclic voltammograms showed higher reduction profiles over oxidation in all the cases, however, the optimized conditions showed an elevated increment in reduction current indicating the improved reduction behavior. Higher dehydrogenase activity observed in this case supported the inter-conversion reactions leading to higher proton availability and resulting higher H2 production. Higher VFA maintenance till the end of the experiment also supports the same. Redox Tafel slopes also depicted a shift with the Fe2+ and optimized conditions supporting the simultaneous redox inter conversion reactions providing higher proton concentration in the cell leading to higher H2 production. The presence of divalent trace metals at optimum concentrations will enhance the H2 production capability of the biocatalyst.
Acknowledgements
The authors wish to thank the Director of CSIR-IICT, Hyderabad for his encouragement in carrying out this work. SS wishes to express his gratitude to CSIR for providing a senior research fellowship (SRF). Part of the this research was supported by the National Mission Mode Project on Hydrogen Production through Biological Routes (No. 103/131/2008-NT) and by the Ministry of New and Renewable Energy (MNRE), Government of India.
References
- C. L. Li and H. H. P. Fang, Crit. Rev. Environ. Sci. Technol., 2007, 37, 1–39 CrossRef CAS.
- J. J. Lay, K. S. Fan, J. I. Hwang, J. I. Chang and P. C. Hsu, J. Environ. Eng., 2005, 131, 595–602 CrossRef CAS.
- K. S. Lee, Y. S. Lo, Y. C. Lo, P. J. Lin and J. S. Chang, Enzyme Microb. Technol., 2004, 35, 605–612 CrossRef CAS.
- S. Venkata Mohan, Int. J. Hydrogen Energy, 2009, 34, 7460–7474 CrossRef CAS.
- S. Venkata Mohan, G. Mohanakrishna, S. Veer Raghuvulu and P. N. Sarma, Int. J. Hydrogen Energy, 2007, 32, 3284–3292 CrossRef.
- S. Venkata Mohan, V. Lalit Babu and P. N. Sarma, Bioresour. Technol., 2008, 99, 59–67 CrossRef CAS.
- S. Venkata Mohan, S. Veer Raghuvulu, G. Mohanakrishna, S. Srikanth and P. N. Sarma, Int. J. Hydrogen Energy, 2009, 34, 216–226 CrossRef.
- S. Venkata Mohan, Y. V. Bhaskar and P. N. Sarma, Water Res., 2007, 41, 2652–2664 CrossRef CAS.
- J. Wang and W. Wan, Int. J. Hydrogen Energy, 2009, 34, 799–811 CrossRef CAS.
- S. Srikanth, S. Venkata Mohan, V. Lalit Babu and P. N. Sarma, Int. J. Hydrogen Energy, 2010, 35, 10693–10700 CrossRef CAS.
- S. Venkata Mohan, S. Srikanth, M. Lenin Babu and P. N. Sarma, Bioresour. Technol., 2010, 101, 1826–1833 CrossRef CAS.
- L.-F. Wu, C. Navarro, K. de Pina, M. Quenard and M.-A. Mandrand, Environ Health Perspectives, 1994, 102, 297–300 CAS.
- Y. Zhang and J. Shen, Int. J. Hydrogen Energy, 2006, 31, 441–446 CrossRef CAS.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS.
- N. S. Jakubovics and H. F. Jenkinson, Microbiology, 2001, 147, 1709–1718 CAS.
- C. Y. Lin and C. H. Lay, Int. J. Hydrogen Energy, 2005, 30, 285–292 CrossRef CAS.
- C.-Y. Lin and S.-H. Shei, Int. J. Hydrogen Energy, 2008, 33, 587–593 CrossRef CAS.
- A. Strocchi and M. D. Levitt, J. Clin. Invest., 1992, 89, 1304–1311 CrossRef CAS.
- V. N. Tripathi and S. Srivastava, J. Biosci., 2006, 31, 61–67 CrossRef CAS.
- H. C. Yang, S. L. Daniel, T. D. Hsu and H. L. Drake, Appl Environ Microbiol, 1989, 55, 1078 CAS.
- J. Wang and W. Wan, Bioresour. Technol., 2008, 99, 8864–8868 CrossRef CAS.
- H. J. Yang and J. Q. Shen, Int. J. Hydrogen Energy, 2006, 31, 2137–46 CrossRef CAS.
- D. Karadag and J. A. Puhakka, Int. J. Hydrogen Energy, 2010, 35, 8554–8560 CrossRef CAS.
-
S. Venkata Mohan, G. Mohanakrishna, S. Srikanth, Biohydrogen production from industrial effluents, in Biofuels, Elsevier Science and Technology, ed. A. Pandey, C. Larroche, S. C. Ricke, C.-G. Dussap, and E. Gnansounou, Burlington Academic Press, Burlington, 2011, pp. 499–524 Search PubMed.
- D. G. Kehres and M. E. Maguire, FEMS Microbiol. Rev., 2003, 27, 263–290 CrossRef CAS.
-
APHA, Standard methods for the examination of water and wastewater.American Public Health Association/American water works Association/Water environment federation.Washington DC, USA 1998 Search PubMed.
- M. V. Reddy, S. Srikanth, S. Venkata Mohan and P. N. Sarma, Bioelectrochemistry, 2010, 77, 125–132 CrossRef CAS.
- L. V. Zhenhua, Y. Yanlai and L. V. Zhenmei, Ecotoxicol. Environ. Saf., 2008, 70, 259–265 CrossRef.
- H. R. Kaback, Annu. Rev. Biophys. Biophys. Chem., 1986, 15, 279–319 CrossRef CAS.
- J-H. Shin, J. H. Yoon, S. H. Lee and T. H. Park, Bioresour. Technol., 2010, 101, S53–S58 CrossRef CAS.
- S. Venkata Mohan, V. Lalit Babu, S. Srikanth and P. N. Sarma, Int. J. Hydrogen Energy, 2008, 33, 4533–4546 CrossRef CAS.
-
S. Jayarama Reddy, Studies on electrode processes by cyclic voltammetry, chronoamperometry, chronopotentiometry and controlled potentaial coulometry, Monogram, Sri Venkateswara University, Tirupati 1986 Search PubMed.
- S. V. Raghavulu, P. S. Babu, R. K. Goud, G. V. Subhash, S. Srikanth and S. Venkata Mohan, RSC Adv., 2012, 2, 677–688 RSC.
- S. Venkata Mohan and S. Srikanth, Bioresour. Technol., 2011, 102, 10210–10220 CrossRef CAS.
- J. L. Wang and W. Wan, Int. J. Hydrogen Energy, 2008, 33, 1215–1220 CrossRef CAS.
- Y. J. Lee, T. Miyahara and T. Noike, Bioresour. Technol., 2001, 80, 227–231 CrossRef CAS.
- Y. F. Zhang, G. Z. Liu and J. Q. Shen, Int. J. Hydrogen Energy, 2005, 30, 855–860 CrossRef CAS.
- M. B. C. Moncrief and M. E. Maguire, JBIC, J. Biol. Inorg. Chem., 1999, 4, 523–527 CrossRef CAS.
- M. J. Horsburgh, S. J. Wharton, M. Karavolos and S. J. Foster, Trends Microbiol., 2002, 10, 496–501 CrossRef CAS.
- Y. M. Zhang, T. Y. Wong, L. Y. Chen, C. S. Lin and J. K. Liu, Appl. Environ. Microbiol., 2000, 66, 105–12 CrossRef CAS.
- M. Imbert and R. Blondeau, Curr. Microbiol., 1998, 37, 64–6 CrossRef CAS.
- D. K. Blencowe and A. P. Morby, FEMS Microbiol. Rev., 2003, 27, 291–311 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.