Clean synthesis of crystalline p-nitrobenzyl bromide from p-nitrotoluene with zero organic discharge†
Milan
Dinda
a,
Manoj K.
Agrawal
ab,
Mahesh R.
Gandhi
a,
Sumesh C.
Upadhyay
a,
Subbarayappa
Adimurthy
a,
Supratim
Chakraborty
a and
Pushpito K.
Ghosh
*a
aCentral Salt and Marine Chemicals Research Institute, Council of Scientific & Industrial Research, G. B. Marg, Bhavnagar-364 002, India. E-mail: pkghosh@csmcri.org; Fax: +91-278-2567562
bGeneric Division, Lupin Research Park, Survey No. 46/47A, Nande Village, Mulshi Taluka, Pune 411042, Maharashtra, India
First published on 13th June 2012
Abstract
2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 NaBr-NaBrO3-NaCl (obtainable as a low cost and eco-friendly reagent from an alkaline bromine intermediate) has been utilized previously in a number of bromination reactions. One such reaction is the conversion of p-nitrotoluene (PNT) to p-nitrobenzyl bromide (PNBBr) used widely for functional group protection. In the present work, selective cold (0–5 °C) crystallization of PNBBr from the reaction mixture containing ca. 2.5 M PNBBr along with 2.5–5.0 M PNT in ethylene dichloride (EDC) was found to be a winning move which led to gains in two fronts. It allowed the bromination reaction to be carried out cleanly and also enabled direct recycling of the mother liquor in the subsequent batch. Para NO2-Ph-CHBr2 impurity, which built up gradually in the reaction mixture over 8 batches, was converted into PNBBr/PNT through treatment of the mother liquor with NaBH4, thereby helping to recycle the liquor perpetually and eliminate organic waste. EDC was the sole solvent in the entire process and its losses were minimal. The combined yield of isolated and recoverable PNBBr was 98.30% with respect to PNT consumed. The reagent utilization efficiency was 98.26%.
:1:3 NaBr-NaBrO3-NaCl (obtainable as a low cost and eco-friendly reagent from an alkaline bromine intermediate) has been utilized previously in a number of bromination reactions. One such reaction is the conversion of p-nitrotoluene (PNT) to p-nitrobenzyl bromide (PNBBr) used widely for functional group protection. In the present work, selective cold (0–5 °C) crystallization of PNBBr from the reaction mixture containing ca. 2.5 M PNBBr along with 2.5–5.0 M PNT in ethylene dichloride (EDC) was found to be a winning move which led to gains in two fronts. It allowed the bromination reaction to be carried out cleanly and also enabled direct recycling of the mother liquor in the subsequent batch. Para NO2-Ph-CHBr2 impurity, which built up gradually in the reaction mixture over 8 batches, was converted into PNBBr/PNT through treatment of the mother liquor with NaBH4, thereby helping to recycle the liquor perpetually and eliminate organic waste. EDC was the sole solvent in the entire process and its losses were minimal. The combined yield of isolated and recoverable PNBBr was 98.30% with respect to PNT consumed. The reagent utilization efficiency was 98.26%.
1. Introduction
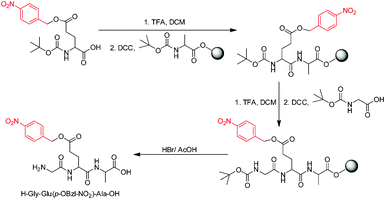
Benzyl esters are of considerable importance as protecting groups in organic synthesis.1 Garbrecht was the first to demonstrate the utility of the p-nitrobenzyl ester in the synthesis of cephalexin through thermal ring expansion of a penicillin sulphoxide under acidic conditions (Scheme 1).2 It was further found by him that this ester “confers on the several intermediate products a strong tendency to crystallize, thus enhancing isolation ease, product purity, and yields.” De-protection was achieved through hydrogenolysis. Baldwin et al. also used the p-nitrobenzyl ester successfully in the synthesis of Penicillin N.3 Being resistant to acidic hydrolysis due to the strong electron withdrawing nature of the nitro group, the p-nitrobenzyl ester was also found to be useful for the protection of side chain –CO2H in solid phase peptide synthesis (Scheme 2).4 The applications of p-nitrobenzyl derivatives (p-nitro benzyl halides, p-nitro benzyl alcohol, p-nitrobenzyl chloroformate) have grown considerably over the years.5 PNBBr is one of the most important of these with 141 global suppliers listed presently.6 Its clean and efficient synthesis is therefore of great importance.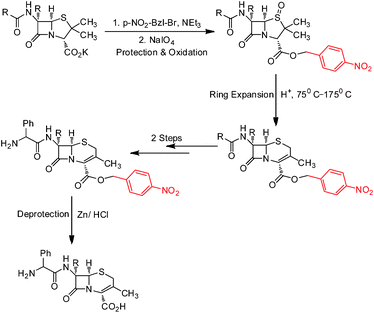 | ||
| Scheme 1 showing the usefulness of the p-nitro benzyl group in the protection–deprotection strategy employed in the synthesis of the Cephalexin intermediate.2 | ||
The synthesis of PNBBr from p-nitrotoluene (PNT) was first reported by Brewster in 71% overall yield and 40% bromine atom efficiency.7 Coleman et al. reported vapour phase bromination of PNT with an isolated yield of 60–70% and 28% bromine atom efficiency.8 Cavill reported the bromination of PNT in the presence of antimony tribromide which gave PNBBr in 76% isolated yield, while the bromine atom efficiency worked out to 38%.9 Whereas the above reactions were conducted with liquid bromine, benzylic bromination with NBS in organic medium,10 aqueous medium,11 supercritical CO2,12 and even under solvent free conditions have been reported.13 However, the emphasis has been mainly on the conversion. In our own hands, a maximum GC-MS yield of 76% (corrected for response factor; Fig. S1, ESI†) was obtained in chlorinated solvent with NBS (Fig. S2, ESI†).
Other reagents include HBr/H2O2,14 NaBrO3/NaHSO3,15 and 2:1 [Br−] : [BrO3−].16 The Br−/BrO3− couple, which generates BrOH upon acidification (eqn (1)), has been of special interest to us as a reagent.
| 2Br− + BrO3− + 3H+ → 3BrOH | (1) |
Whereas ring bromination reactions of amines and phenols with 2:1 [KBr] : [KBrO3]—constituted from the pure salts—were reported by Francis and Hill as early as 1924,17 a crude formulation—2:1:3 NaBr : NaBrO3 : NaCl (designated herein as BR-S16a)—was prepared by us directly in the mixed state from the alkaline intermediate of bromine manufacture by the air-blowing process.18 Consequently, liquid bromine was eliminated from the product cycle in addition to providing a cost-effective reagent.
The benzylic bromination of PNT with BR-S led to the formation of dibromo impurity 1 (pNO2-Ph-CHBr2) besides PNBBr (eqn (2)). The impurity amount was lower at high substrate to reagent ratio.16c However, isolation of the pure product was tedious at these high ratios and there were losses of PNT and PNBBr in the organic waste containing 1, which negated the gains to some extent. We therefore decided to reinvestigate the process to eliminate organic waste and simplify operations.
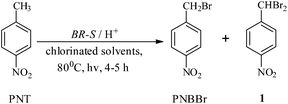 | (2) |
2. Results and discussion
Isolation of a solid product from a reaction mixture is best achieved through crystallization. We recognized that if PNBBr can be made to crystallize selectively from the reaction mixture containing excess PNT–which is required for high selectivity as mentioned above–it would be feasible to recycle the mother liquor containing excess PNT and residual PNBBr. Considering an initial molar ratio of 3:1 of PNT to BR-S to minimize impurity formation, experiments were conducted with 2:1 [PNT] : [PNBBr] to explore the feasibility of selective crystallization of the product through control of the solvent amount and temperature. It was concluded from the study that for 1:4.5 w/v ratio of “reaction mass” (total weight of PNT + PNBBr) to CCl4, and chilling temperature in the range of 0 to 4 °C, selective crystallization is feasible (Table S1, ESI†). Table S2† shows the data generated for the reaction in CCl4 followed by recovery of the product through cold crystallization, filtration and recycling of the mother liquor. After eight such batches, the solvent was removed from the mother liquor followed by recovery of PNT through vacuum distillation. The dark residue remaining comprised 71.1% 1 and 26.9% PNBBr as the main constituents. The isolated yield of PNBBr (98.2% average purity) was found to be 90.2% and the bromine utilization efficiency 88.6%.16d,e
Notwithstanding the encouraging results obtained above, further studies with CCl4 were discontinued on the suggestion of our licensee. With EDC as solvent, selective crystallization was obtained at five-fold higher concentration, i.e., 1.2:1 w/v ratio of “reaction mass” to EDC and chilling temperature of 0–5 °C. These were advantageous as the throughput could be raised and the refrigeration load reduced at the same time.
The reaction was conducted subsequently in EDC and Fig. 1(A) shows the GC-MS of the reaction product obtained with 3.0:1.1 ratio of PNT to BR-S. The reaction was clean and quantitative with respect to the reagent amount taken. Entry 1 in Table 1 provides data on the yield of PNBBr obtained from the reaction mass upon cold crystallization, centrifugation and washing with chilled EDC. Washing was undertaken with a small amount of the chilled solvent (the solvent can be obtained from the reaction mass itself by switching from reflux to distillation mode.) Purified product could be isolated in this manner.
 | ||
| Fig. 1 GC of (A) reaction mixture of Batch 1, Table 1 showing the presence of only PNT (68.53%) and PNBBr (31.47%); (B) composite of the isolated products from Batches 1–8, Table 1 (5.12% PNT and 94.88% PNBBr); (C) mother liquor obtained from Batch 8, Table 1 (composition provided in Table 2); (D) mother liquor of (C) after subjecting to controlled debromination with NaBH4 (composition provided in Table 2). The retention times of PNT, PNBBr and 1 were ca. 10.0, 13.1 and 14.8 min, respectively. | ||
| Batch No. | PNT g/(mmol) | EDC (mL)b | Br/mmol | 98% H2SO4 g/(mmol) | PNBBr g/(mmol)c |
|---|---|---|---|---|---|
| a Batch-wise details are provided as part of the ESI. b 5 mL EDC added along with mother liquor in batch 4 and 8. c crystalline product isolated by centrifugation and washed with small amount of chilled EDC. | |||||
| 1. | 30.0/(219.0) | 30 | 80.3 | 4.0/(41.0) | 11.0/(50.9) |
| 2. | 7.0/(51.1) | Recycled mother liquor | 56.2 | 3.0/28.1 | 13.0/(60.2) |
| 3. | 8.0/(58.4) | Recycled mother liquor | 64.2 | 3.2/(32.2) | 15.0/(69.4) |
| 4. | 10.0/(73.0) | Recycled mother liquor | 80.3 | 4.0/(40.1) | 16.0/(74.1) |
| 5. | 10.0/(73.0) | Recycled mother liquor | 80.3 | 4.0/(40.1) | 16.8/(77.8) |
| 6. | 10.2/(74.5) | Recycled mother liquor | 81.9 | 4.2/(42.8) | 21.0/(97.2) |
| 7. | 13.3/(97.1) | Recycled mother liquor | 106.0 | 5.2/(53.1) | 22.5/(104.2) |
| 8. | 13.7/(100.0) | Recycled mother liquor | 110.0 | 6.0/(61.2) | 11.0/(51.0) |
| Total | 102.2/(746.0) | 40 | 659.2 | 33.6/(338.6) | 126.3/(584.8) Purity: 96.9% (w/w) m.p. 99.2 °C |
The mother liquor together with the washings were recycled in the next batch. Into this mother liquor, a fresh charge of PNT (equivalent to the amount of PNBBr obtained as crystallized product) and BR-S, in the molar ratio of 1:1.1, were added. The process was repeated for eight batches in all and the GC-MS of the composite PNBBr sample prepared from the isolated products obtained in batches 1–8 is shown in Fig. 1(B). PNBBr purity by GC-MS was 94.8% (uncorrected) whereas the purity by weight was 96.9%, corrected for response factor. The melting point was 99.2 °C (lit: 97–100 °C). The purity of the crystalline isolated product can be enhanced further by improving the efficiency of washing with chilled EDC and recycling the washing in the mother liquor given that the only impurity detected was PNT.
The mother liquor from batch 8 was also subjected to GC-MS and there was evidence of 1 in addition to the presence of PNT and PNBBr [Fig. 1(C)]. We ascribe this to the progressive decrease in the overall PNT to BR-S ratio from ca. 3:1 to 2:1 in going from batch 1 to batch 8. To recycle the mother liquor perennially, it was necessary to convert 1 into PNT or preferably PNBBr. Bell & Brown have reported the reduction of benzyl halides to toluene derivatives with NaBH4 in diglyme-water mixture.19 In order to keep operations simple, a similar reaction was attempted with the mother liquor directly, albeit in the presence of a small amount of methanol. Complete conversion of 1 was observed at R.T. with 8 molar equivalents of NaBH4 with respect to the impurity amount present [Fig. 1(D); Table 2]. The overall selectivity towards PNBBr and PNT formation were computed to be 81% and 19%, respectively. Taking into account the PNT and PNBBr amounts in the reusable mother liquor, and also the PNT which would have been recovered by washing the product of Fig. 1(B) more thoroughly with chilled EDC, the combined yield of isolated and recoverable product was found to be 98.30% with respect to PNT consumed (Table S4, ESI†). The less than quantitative yield of PNBBr can be ascribed to handling losses. The “Br” utilization efficiency was also high (98.26%) in view of the clean reaction achieved under the conditions adopted and the selective de-bromination of the small amounts of 1 produced into desired product (Table S4†).
3. Experimental
3.1. Materials and methods
All the reagents were of commercial grade and purified according to established procedure. 1H and 13C NMR spectra of p-nitrobenzyl bromide and 1 were recorded in CDCl3 with TMS as the internal standard using Model Bruker Avance II spectrometer. FT-IR spectrum was recorded in KBr using a Perkin Elmer spectrometer. GC-MS data were acquired on GC-MS QP 2010 Shimadzu instrument. The melting point was recorded using a Mettler Toledo instrument and the data are uncorrected. Reactions were monitored by thin layer chromatography (TLC) on Silica gel 60 F254 (aluminum sheet). For the purpose of assay, 1 was purified by column chromatography using 60–120 mesh silica gel. Centrifugation was performed in KUBOTA 7780 centrifuge with temperature controller at 15000 rpm for 10 min. Unless otherwise stated, all reactions were performed using Tarsons Spinot Digital magnetic stirrer.
3.2. General procedure for preparation of p-nitrobenzyl bromide and recycling of the mother liquor
In a typical reaction (here Batch-1, Table 1), 30.0 g (218.97 mmol) of PNT, 30 mL of EDC, and 10 mL of an aqueous solution of formulated BR-S containing 80.3 mmol available “Br” were taken in a 250 mL RB flask fitted with a reflux condenser. Heat was supplied by immersing the flask in an oil bath (maintained at 80–90 °C) while stirring was undertaken with the help of a magnetic stirrer. The flask was illuminated with a 100 watt tungsten filament light bulb from the outside. To the above reaction flask, 41 mmol of H2SO4 in the form of 1 M solution was added drop wise over a period of 2–2.5 h. After complete addition of acid, the reaction was continued for a further 2 h. The organic layer was separated, cooled to room temperature and kept in the freezer overnight whereupon a crust of white crystalline compound was obtained. After breaking up the crust with a glass rod, the resultant slurry was centrifuged at 4 °C. During the centrifugation process the solution temperature rose up to 10–12 °C. The centrifuged mass was washed with 2–3 mL of chilled EDC. 11 g (50.93 mmol) of PNBBr having GC-MS purity 100% was isolated (Figure S5.1, ESI†).Since 50.93 mmol of PNBBr was obtained in the batch above, an equivalent amount of PNT (51.1 mmol) was added into the mother liquor along with washings obtained above. The “Br” amount taken was 1.1 equivalents with respect to the fresh charge of PNT, i.e., 56.2 mmol of “Br” in the form of BR-S. The sulphuric acid amount added was 28.1 mmol. The rest of the process was the same as above and, once again, after completion of the reaction the organic layer was subjected to cold crystallization to recover PNBBr while recycling the mother liquor along with washings in Batch 3. In this manner, batches 2–8 were carried out.
3.3. General procedure for selective debromination of 1 to PNBBr for perpetual recycling of the mother liquor
After the 8th batch, the mother liquor and washings (ca. 40 mL) containing 40.32%, 30.31% and 29.37% of PNT, PNBBr and 1, respectively, and with total weight of residue amounting to 30 g, were taken in a single neck 250 mL RB flask. Into this solution was added 7.4 g (238.14 mmol) NaBH4, i.e., 8 molar equivalents with respect to the amount of 1 present in the mother liquor (8.811 g; 29.8 mmol). The addition of NaBH4 was made pinch wise along with drop wise addition of 5 mL of MeOH (final MeOH: EDC = 1:8). Stirring at room temperature was continued for 2.5 h. The progress of the reaction was monitored by TLC. After the full conversion of 1 was seen, 10–15 mL of water was added and the EDC layer separated. It was dried over anhydrous Na2SO4, and after keeping aside a small portion for GC-MS analysis, the rest was evaporated under reduced pressure. The total mass weighed 25.56 g having 8.03 g PNT and 17.53 g PNBBr (Table 2, Entry 2; Fig. S7.2, ESI†).
3.4. Preparation of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 [PNT] : [PNBBr] solution to calculate the Response Factor
:1 [PNT] : [PNBBr] solution to calculate the Response Factor
2.408 g (11.15 mmol) of pure PNBBr along with 3.055 g (22.30 mmol) of pure PNT were dissolved in EDC and submitted for GC-MS. The molar percentages were 33.33% and 66.66%, accordingly, whereas the GC-MS area percentages recorded were 29.43% and 70.57% for PNBBr and PNT, respectively (Fig. S1, ESI†). The data were used for the computation of the actual molar proportions of PNT and PNBBr in the reaction mixture, mother liquor and isolated product.
4. Conclusions
We have described in the present study our efforts to evolve a clean protocol for the preparation of crystalline PNBBr from PNT. Our approach laid emphasis both on the reaction and work up. It was shown that selective crystallization of the product was feasible in the presence of the solid substrate under chilled conditions, even with a product to substrate ratio as low as 1:2. As a result, the bromination reaction could be carried out cleanly while still offering the convenience of easy recycling of the excess substrate and unrecovered product. Although the formation of pNO2-Ph-CHBr2 could have been avoided altogether by maintaining a sufficiently high substrate to BR-S ratio throughout, its conversion back into PNBBr/PNT shows that the process is robust to stoichiometric deviations and that organic waste can be eliminated altogether. It is further advantageous that this conversion was effected using the mother liquor directly. “Br” utilization from the purely inorganic reagent was also excellent and the aqueous effluent was nearly free of bromide salts. Another attractive feature was the use of a single solvent for all operations which, additionally, minimized solvent losses. We were successful in extending the methodology to the synthesis of the related o-nitrobenzyl bromide.20 The methodology of cold crystallization and recycling of mother liquor containing excess substrate may also have general applicability. For example, we found the approach useful for selective formation of 5-bromo-2-hydroxy-benzoic acid methyl ester–an intermediate in the synthesis of Labetalol–from 2-hydroxy-benzoic acid methyl ester.16e
Acknowledgements
We are grateful to Mr. Kamal Aggarwal (licensee) for his constructive criticism of our earlier work which motivated us to undertake the present study. We thank the staff of Analytical Division for assistance with analysis and Mr. P. Maiti for design of the graphical abstract. We are also grateful to CSIR India for supporting the study as part of an in-house laboratory project. MD, MKA and SC thank CSIR/UGC for their research fellowships.References
- (a) J. C. Sheehan and D. R. Hoff, J. Am. Chem. Soc., 1957, 79, 237 CrossRef CAS; (b) M. Schelhaas and H. Waldmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2056 CrossRef CAS.
- W. L. Garbrecht, U. S. Patent No. 3,853861, Dec 10, 1974 (to Eli Lily and Company) Search PubMed.
- J. E. Baldwin, S. R. Herchen and P. D. Singh, Biochem. J., 1980, 186, 881 CAS.
- R. L. Prestidge, D. R. K. Harding and W. S. Hancock, J. Org. Chem., 1976, 41, 2579 CrossRef CAS.
- (a) M. J. Bodner, R. M. Phelan and C. A. Townsend, Org. Lett., 2009, 11, 3606 CrossRef CAS; (b) D. Albanese, M. Leone, M. Penso, M. Seminati and M. Zenone, Tetrahedron Lett., 1998, 39, 2405 CrossRef CAS; (c) M. D. Hocker, C. G. Caldwell, R. W. Macsata and M. H. Lytte, Pept. Res., 1995, 8, 310 CAS; (d) P. C. Ray, M. Bandari, M. Qadeeruddin and S. R. Gorantla, US 2009/0062550A1.
- CAS DataBase List; www.chemicalbook.com.
- J. F. Brewster, J. Am. Chem. Soc., 1918, 40, 406 CrossRef CAS.
- G. H. Coleman and G. E. Honeywell, Organic Syntheses, 1943, 2, 443 Search PubMed.
- G. W. K. Cavill, J. Soc. Chem. Ind., 1946, 40, 124 CrossRef.
- (a) K. Ziegler, A. Spath, E. Schooof, W. Schuman and E. Wickelman, Justus Liebigs Ann. Chem., 1942, 551, 80 CrossRef CAS; (b) C. Djerassi, Chem. Rev., 1948, 43, 271 CrossRef CAS; (c) W. Wenner, J. Org. Chem., 1952, 17, 523 CrossRef CAS; (d) W. Offermann and F. Vogtle, Synthesis, 1977, 4, 272 CrossRef; (e) W. Offermann and F. Vogtle, Angew. Chem., Int. Ed. Engl., 1980, 19, 464 CrossRef; (f) M. C. Carreno and J. L. G. Ruano, J. Org. Chem., 1995, 60, 5328 CrossRef CAS; (g) C. H. M. Amijs, G. P. M. van Klink and G. van Koten, Green Chem., 2003, 5, 470 RSC; (h) M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Strusture, 5th ed.; JohnWilley & Sons: New York, 2001, pp 911–914 Search PubMed.
- A. Podgorsek, S. Stavber, M. Zupan and J. Iskra, Tetrahedron Lett., 2006, 47, 1097 CrossRef CAS.
- J. M. Tanko and J. F. Blackert, Science, 1994, 263, 203 CAS.
- (a) M. Zereb, M. Zupan and S. Stavber, Helv. Chim. Acta, 2009, 92, 555 CrossRef ; (long reaction time (16 h), 6% dibromo, no mention of how pure product can be isolated from the mixture of mono bromo, dibromo and succinimide/NBS.); (b) A. N. M. M. Rahman, R. Bishop, R. Tan and N. Shan, Green Chem., 2005, 7, 207 RSC.
- R. Mestres and J. Palenzuela, Green Chem., 2002, 4, 314 RSC.
- D. Kikuchi, S. Sakaguchi and Y. Ishii, J. Org. Chem., 1998, 63, 6023 CrossRef CAS.
- (a) US Patent No: 6,740,253 (CSIR) Search PubMed; (b) S. Adimurthy, G. Ramachandraiah, A. V. Bedekar, S. Ghosh, B. C. Ranu and P. K. Ghosh, Green Chem., 2006, 8, 916 RSC; (c) S. Adimurthy, S. Ghosh, P. U. Patoliya, G. Ramachandraiah, M. Agrawal, M. R. Gandhi, S. C. Upadhyay, P. K. Ghosh and B. C. Ranu, Green Chem., 2008, 10, 232 RSC; (d) PCT Application No. PCT/IB2009/007264 dated 29 Oct, 2009 (CSIR) (claims cleared for National Phase filing); (e) M. K. Agrawal, Ph. D. Thesis, Bhavnagar University (Gujarat, India), 2009 Search PubMed.
- (a) A. W. Francis and A. J. Hill, J. Am. Chem. Soc., 1924, 46, 2498 CrossRef CAS; (b) A. W. Francis, A. J. Hill and J. Johnston, J. Am. Chem. Soc., 1925, 47, 2211 CrossRef CAS.
- A. S. Mehta, Indian J. Chem. Technol., 2010, 17, 204 CAS.
- H. M. Bell and H. C. Brown, J. Am. Chem. Soc., 1966, 88, 1473 CrossRef CAS.
- M. Dinda and P. K. Ghosh, unpublished results.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical Data; NMR, FT-IR and GC-MS data of some selective compounds. See DOI: 10.1039/c2ra20940c |
| This journal is © The Royal Society of Chemistry 2012 |