Preparation of diblock copolymer films for the localization of C60 and multiwalled carbon nanotubes on aqueous substrate†
Qifang
Li
*ab,
Jianli
Sun
a,
Jing
Wang
b,
Guang-Xin
Chen
*ab,
Fang
Li
b and
Yajun
Zhang
c
aKey Laboratory of carbon fiber and functional polymers, Ministry of Education, Beijing University of Chemical Technology, Beijing 100029, P. R. China
bCollege of Material Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China
cCollege of Mechanical and Electrical Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: gxchen@mail.buct.edu.cn; Fax: +86 10 64421693; Tel: +86 10 64445680
First published on 25th June 2012
Abstract
Dropping polystyrene-block-poly(ethylene oxide) (PS-b-PEO) solution on a still and clean water surface caused the formation of homogeneous films of diblock copolymer at the organic vapor/water interface. The morphologies of the PS-b-PEO were studied to control and modulate the arrangement of ordered structures by applying different solvents at different annealing times. After 24 h of annealing under toluene vapor, a lamellar morphology was produced, whereas a cylindrical morphology was produced under chloroform vapor. This simple method was used to obtain more than one type of ordered microdomain structure using the same symmetric diblock copolymer. Interestingly, the microdomain size reached 120 nm, much larger than that obtained by bulk or solution self-assembly. The microphase-separated PS-b-PEO copolymer was utilized as a novel template for the selective self-assembly of the polymer-functionalized C60 and the carbon nanotubes (CNTs) by combining aqueous substrate with solvent annealing. The PS-grafted C60 enhanced the PS-b-PEO to form a lamellar microstructure. The PS-grafted C60 accommodated in the PS phase expanded the scale of PS domains. The PEO-covered CNTs were prepared to form a PEO-covered CNT/water solution for application to a complex substrate with diblock copolymer templates. This methodology enables accommodation of a selective assembly of the CNTs onto the PEO phase. This assembly strategy is a versatile approach and may open a new route for the controlled assembly of anisotropic nanostructured materials with desirable patterns on soft substrate.
Introduction
Diblock copolymers are two incompatible homopolymer segments covalently bonded at one end. The incompatibility of these copolymers allows self-assembly into periodic morphologies with nanometer-sized domains ranging from spheres to cylinders to lamellae, depending on the volume fraction of the components.1,2 The nanometer size of the micro-phase separation is tunable between 1–100 nm using traditional top-down microstructure processing methods, such as photoetching. The structure size of the blank region can be formed using emerging bottom-up microstructure processing methods, such as chemical synthesis. Diblock copolymers have attracted considerable attention because of their prospective applications as lithographic masks,3 photonic materials,4 and nano-patterned substrates for microelectronics5 and biomedical devices.6 The polystyrene-block-poly(ethylene oxide) (PS-b-PEO) is of particular interest because of the strong segregation force between the PS and the PEO blocks. Furthermore, PEO is a hydrophilic polymer that has a low tendency for protein adsorption.7Different microstructures of diblock copolymer can be attained using various molecular designs. However, the orientation and the lateral ordering have to be studied to fully realize the potential of diblock copolymers. Several studies have been conducted in combination with external fields, such as mechanical force, electrical field, surface-phase interaction, and temperature gradient, to obtain highly ordered nanometer microdomains.8 Solvent evaporation9 is a simple and effective route for controlling the alignment of microdomain morphology of diblock copolymers. Solvent annealing can markedly enhance the order of copolymer morphologies in very thin films. The solvent can impart mobility to polymeric chains, enabling the orientation and removal of defects.10 The self-assembly of PS-b-PEO films on a water surface has also been studied. The PS-b-PEO polymeric solutions can form homogeneous films when dropped on a water surface. Da Silva et al.11 suggested the existence of PS-b-PEO surface aggregates on a water surface. Devereaux and Baker12 studied the morphology of PS-b-PEO (7% PEO by mass) on a water surface, which showed dots, spaghetti, and continent morphologies.
Diblock copolymers can act as powerful templates to direct the self-assembly of inorganic particles into nanometer-length scale structures.13,14 Significant progress has recently been achieved experimentally and theoretically on the control of spherical nanoparticle location in diblock copolymer templates. Hamdoun et al.15,16 first reported on nanocomposites by dispersing iron oxide nanoparticles into a symmetric polystyrene-poly(butyl methacrylate) diblock copolymer matrix. They positioned the particles in the polystyrene domains by cleverly grafting short chemically identical ligands to the nanoparticles. Lauter-Pasyuk et al.17 concluded that the position was largely controlled by the ratio of the particle diameter to the domain size, with the larger particles favoring the domain centers and the smaller particles favoring the interfaces. This conclusion was supported by the experiments of Bockstaller et al.18 However, subsequent studies by Bockstaller et al.19 and Chiu et al.20 observed counter-examples indicating a more complex condition. Based on systematic experiments, Kramer et al.21 found that the surface affinity of the particles is finely tuned by coating them with either a mixture of ligands or random copolymer ligands. Aside from particle location, experiments were also conducted to investigate other important phenomena,22,23 such as the critical loading beyond which the particles macrophase separate, the effect of particles on the block copolymer period, and particle-induced phase transitions of the block copolymer morphology.
To date, the most studied copolymer-based nanocomposites consist of spherical nanoparticles. Compared with the organized assembly of spherical nanoparticles, the organization of inorganic nanorods (one-dimensional nanomaterials)24,25 is of particular importance because the collective properties of these anisotropic species are strongly affected by rod alignment and internal organization. The self-assembly of hybrid systems consisting of copolymers and nanorods is more complex than those with spherical nanoparticles, caused by their anisotropic shape,26 leading to the formation of liquid crystalline phases. The interplay between the strong and directional van der Waals attraction between the nanorods and the entropy associated with their orientation introduce complexity and challenges to the current research. Investigations on the two-dimensional assembly of nanorods on surfaces concentrated mainly on patterning studies, utilizing diblock copolymer thin films as topographical templates. Russell et al.24 achieved ordered nanorod assembly inside channels of degraded poly(methyl methacrylate) (PMMA) in a film of PS-b-PMMA by controlling the surface chemistry of the nanorods. Char et al.27 showed that surface reconstruction of the film by selective solvents can be used to create the required height contrast to allow for selective patterning of nanoparticles and nanorods. Ploshnik et al.28,29 recently introduced an additional method for obtaining two-dimensional surface patterns of organized nanorods, which is more thermodynamically controlled, utilizing the tendency of nanoparticles to segregate to the film surface while inducing vertical orientation of the diblock copolymer domains. Kenny et al.30 and Mezzenga et al.31 added the carbon nanotubes (CNTs) with high aspect ratio into the block copolymers, and the localization of conducting CNTs inside the domains of a block copolymer matrix was investigated.
In the current study, a simple route was developed using solvent annealing to obtain highly ordered microstructures of PS-b-PEO films on a water surface. The water surface acted as a soft substrate and interacted preferentially with the PEO chain. Annealing in the presence of solvent vapor imparted mobility to the diblock copolymer chains. Consequently, the orientation and ordering of the PS-b-PEO microdomains were better modulated. Under the simultaneous effects of water, solvent vapor, and the two blocks, highly ordered lamellae and cylindrical microphase structures were obtained. The self-assembly of the PS-b-PEO was utilized to organize the fullerene and the CNT on a large scale at the air/water interface. The mutual process of self-assembly, where the block copolymer template affects the organization of the carbon nanomaterials, and the carbon nanomaterials that influence the morphology of the block copolymer and its phase transitions were investigated simultaneously.
Experimental section
Materials
PS-b-PEO with a polydispersity index of 1.03 was purchased from Polymer Source, Inc. The molecular weights of the PS and PEO blocks were 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 and 71000 g mol−1, respectively. The volume fraction of PEO blocks was calculated to be 51.49%, suggesting a symmetrical structure. Polyethylene glycol (Mn = 2000 g mol−1) was purchased from Alfa Aesar, China. C60 (purity > 99.9%) was obtained from the Interuniversity Microelectronics Center, China. Toluene and chloroform, which were bought from Beijing Chemical Reagents Corporation, were used as received. Multiwalled carbon nanotubes (MWNTs) were provided by Aldrich, obtained by chemical vapor deposition and having a purity of >95%. The outer diameter and the length of the MWNTs were about 20 nm and 1–2 μm, respectively.
600 and 71000 g mol−1, respectively. The volume fraction of PEO blocks was calculated to be 51.49%, suggesting a symmetrical structure. Polyethylene glycol (Mn = 2000 g mol−1) was purchased from Alfa Aesar, China. C60 (purity > 99.9%) was obtained from the Interuniversity Microelectronics Center, China. Toluene and chloroform, which were bought from Beijing Chemical Reagents Corporation, were used as received. Multiwalled carbon nanotubes (MWNTs) were provided by Aldrich, obtained by chemical vapor deposition and having a purity of >95%. The outer diameter and the length of the MWNTs were about 20 nm and 1–2 μm, respectively.
Functionalization of C60 with PS-Br
PS-functionalized C60 (C60-PS) was synthesized by atom-transfer radical polymerization in the authors' laboratory.32 The reaction is shown in Scheme S1.† In a typical procedure, a 50 ml round-bottomed flask was charged with C60 (0.0513 g), CuBr (0.0143 g), 2,2-dipyridyl (0.0445 g), and PS-Br (0.420 g), degassed, and refilled with nitrogen thrice. Afterwards, 5 ml 1,2-dichlorobenzene was added using a syringe, and the reaction mixture was degassed thrice using the freeze-pump-thaw cycle. The flask was placed in a thermostated oil bath at 120 °C for 24 h. The reaction mixture was diluted with THF and was centrifuged at 3000 rpm for at least 30 min to separate the unreacted C60. To separate C60-PS from PS, 10 ml benzene was added to dissolve the product, and methanol was slowly added to the solution until the precipitate no longer formed. The precipitate was separated by filtration and dried under vacuum, obtaining C60-PS (brown powder). The product was analyzed by a thermal gravimetric analyzer. Approximately 93.4% of C60-PS was incinerated, indicating an average of three or four PS chains, with a Mn of 2812 g mol−1, grafted to one C60.Functionalization of MWNT with PEO
The PEO-modified MWNT (MWNT-g-PEO) was synthesized using a procedure similar to the previous literature.33 A 250 ml round-bottomed flask was charged with 200 mg acid-treated MWNT and 100 ml SOCl2. The flask was ultrasonicated in a water bath for 30 min. The solution was mixed at 80 °C for 30 h in a water condenser. Afterward, SOCl2 was removed by Heidolph rotary evaporators at 39 °C for 0.5 h. Then, 30 g polyethylene glycol was added to the solid mixture and stirred at 120 °C for 45 h in a water condenser. The resulting reaction medium was dissolved in excess THF and vacuum-filtered three times through a 0.22 μm PTFE membrane and vacuum-dried for 10 h to yield MWNT-g-PEO. The reaction is shown in Scheme S2.†Preparation and annealing of PS-b-PEO films on the surface of water
The solution of PS-b-PEO diblock copolymer in toluene with a concentration of 6.7 or 10 mg ml−1 was prepared. Scheme 1 shows the schematic diagram of the procedure for self-assembly of PS-b-PEO on the solvent vapor/water interface. A clean culture dish (diameter is 75 mm) filled with 60 ml distilled water was placed in a closed container, where an open weighing bottle filled with solvent was also placed. After 1 h, the solvent had evaporated throughout the confined space. Then, 15–20 μl of the polymer solution measured using a micropipettor was dropped on the solvent vapor/water interface. Toluene vaporized immediately upon contact with the solvent/water interface, where the droplet spread. The PS-b-PEO film gradually formed and immersed in the solvent vapor, allowing the movement of the macromolecular chains. Toluene and chloroform were used as annealing vapors, and the annealing time of the film was controlled from 3–24 h. The PS-b-PEO spread over the water surface in the area of the culture dish. The thickness of the PS-b-PEO thin film was calculated to be 34–45 nm (Scheme S3†).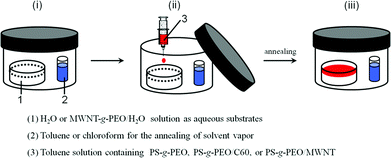 | ||
| Scheme 1 Schematic for the self-assembly of PS-b-PEO on the surface of pure water and water/MWNT-g-PEO solution under solvent vapor annealing: (i) saturated solvent vapor obtained by solvent evaporation, (ii) one drop of solution was placed onto the water surface after 1 h, (iii) PS-b-PEO film formation and solvent annealing. | ||
Preparation and annealing of PS-b-PEO/C60 nanohybrid films on the surface of water
C60 or C60-PS was added into a solution of PS-b-PEO in toluene with a concentration of 10 mg ml−1. A clean culture dish filled with 60 ml distilled water was placed in a closed container, where an open weighing bottle filled with solvent was also placed. After 1 h, a 15 μl PS-b-PEO/C60 solution was dropped on the water surface, and the solvent was allowed to evaporate throughout the confined space. Toluene evaporated immediately upon contact with the solvent/water interface, where the droplet spread. The PS-b-PEO nanohybrid films formed gradually in solvent vapor, allowing the macromolecular chains to move and solvent anneal. Toluene and chloroform were used as annealing vapors. The annealing time was controlled within 2–24 h.Preparation and annealing of PS-b-PEO/MWNTs nanohybrid films on the surface of water/MWNT-g-PEO solution
MWNT-g-PEO (80 mg) was added to 80 ml distilled water and was ultrasonicated for 30 min, obtaining homogeneous aqueous MWNT-g-PEO solution. A clean culture dish filled with 60 ml aqueous MWNT-g-PEO solution was placed in a closed container, where an open weighing bottle filled with solvent was also placed (Scheme 1). After 1 h, 15 μl PS-b-PEO solution was dropped on the surface of the water/MWNT-g-PEO solution, and the solvent was allowed to evaporate throughout the confined space. The PS-b-PEO/MWNTs nanohybrid film formed gradually in the solvent vapor. Toluene was used as solvent vapor. The annealing time was controlled within 3–72 h.Transmission electron microscopy (TEM) imaging
Annealed films were directly collected with carbon-coated copper grids on the water surface and stained with RuO434 at room temperature for 20 min. The morphology of the PS-b-PEO films was characterized using TEM (Hitachi 800) at an accelerating voltage of 200 kV.Results and discussion
Fig. 1 shows the morphologies of the PS-b-PEO films with the water surface as the substrate before and after toluene annealing. The concentration of the polymer solution is 6.7 mg ml−1. The PS-b-PEO films without solvent vapor annealing produces disordered aggregations and difficult to control morphology (Fig. 1a and b). After annealing under toluene vapor for 24 h, the PS-b-PEO films show an ordered lamellar microstructure (Fig. 1c and d). | ||
| Fig. 1 TEM images of PS-b-PEO diblock copolymer films: (a,b) film on water surface without solvent vapor annealing with various magnifications, (c,d) film on water surface under toluene vapor annealing for 24 h with various magnifications. The concentration of the polymer solution is 6.7 mg ml−1. Scale bar = 1700 nm for a, c, scale bar = 400 nm for b, d. | ||
The driving force behind surface aggregation on the water surface is a balance of interaction among air, water, and two polymer blocks.35 The PS domains are preferentially stained dark. When a droplet of polymer is deposited on the water surface without the solvent vapor, the stock solvent vaporizes quickly. As toluene evaporates, a disordered structure results with a higher PS-projected area compared to PEO (Fig. 1a and b) because toluene is selective to PS. While the set-up is under toluene vapor for 24 h, the PS-b-PEO has enough time to self-assemble and reach a lamellar structure. Solvent vapor imparts mobility to the polymeric chains, aiding the dynamic evolution of the chains and improves ordered structures.
Fig. 2 shows the sample morphologies fabricated under toluene vapor for 3, 6, and 24 h at the increased concentration (10 mg ml−1) of the polymer solution. When annealed under toluene vapor for 3 h, the microphase separation is clear and PEO microphase is interconnected to form a network structure (Fig. 2a). After 6 h (Fig. 2b), the network gradually opens up. After 24 h (Fig. 2c), a large portion of the lamellar structure is observed. These results can be attributed to toluene being a selective solvent for PS blocks and the water solubility of PEO. The ordered lamellar structure reflects a state of low entropy. A comparison between Fig. 2c and 1d shows that more uniform structures of long-range lamellae arrays are formed at higher concentrations.
 | ||
| Fig. 2 TEM images of PS-b-PEO diblock copolymer films on water surface under toluene vapor annealing for: (a) 3 h, (b) 6 h, (c) 24 h. The concentration of PS-b-PEO solution is 10 mg ml−1. Scale bar = 400 nm. | ||
Selective toluene and nonselective chloroform solvent vapors were applied to PS-b-PEO. Although toluene is selective to PS, chloroform is a good solvent for both PS and PEO. To observe the dynamic evolution of PS-b-PEO, samples treated with different solvent vapors and annealing times were analyzed for their morphology using TEM. Fig. 3 shows the TEM images of PS-b-PEO under chloroform vapor. After 3 h (Fig. 3a), PS and PEO strips with equivalent microphase sizes are obtained, whereas annealing for 6 h (Fig. 3b) results in a narrow phase of PS strips. After 24 h (Fig. 3c), an ordered cylindrical morphology is produced. Chloroform is a nonselective solvent that gives mobility to both blocks. The PS chains can move in the chloroform vapor, whereas the PEO chains not only extend in the chloroform vapor, but are also driven by the water surface. Compared with toluene vapor, the chloroform system is more suitable for PEO chains than for PS chains.
 | ||
| Fig. 3 TEM images of PS-b-PEO diblock copolymer films on the water surface under chloroform vapor annealing for: a) 3 h, b) 6 h, c) 24 h. The concentration of PS-b-PEO solution is 10 mg ml−1. Scale bar = 400 nm. | ||
Different morphologies are obtained at different annealing times. The morphological ordering is affected by the strong time dependence of film annealing, similar to a study by Kim et al.36 A film of PS-b-PEO was spin-coated under benzene vapor for 48 h. Within 1 h, half of the defects were reduced. The reduction in the number of defects slowed down as time increased. After 48 h, all defects were removed. By applying the proposed method, cylindrical features can be obtained from the symmetric PS-b-PEO. The conditions and methods for self-assembly are important in determining the types of microphase structures, which are analogous to the changes in the block volume fraction of the copolymer.37
The size of the microdomains on the water surface is quite larger than that on the solid substrates. The periodic sizes of PEO and PS are approximately 120 and 80 nm, respectively (Fig. 1d). Spin-coated films on silicon substrates were also prepared from the same PS-b-PEO polymer solution. After annealing under toluene for 24 h, the film was coated onto a TEM grid using 5% hydrofluoric acid solution. The microstructure results in lamellae as shown in Fig. 4. The width of the PS microphase is approximately 45 nm and the distance between PS (PEO microphase) is approximately 40 nm in average. Li and Kaito38 studied bulk PS-b-PEO with the same macromolecular weight and ratio of the two blocks. Under thermal annealing, the periodic sizes of the lamellae structure are 45 and 34 nm for the PEO and PS blocks, respectively. The size of the microphase separation on the water surface is much larger than that in bulk or on the silicon substrate. The soft substrate is presumed to help the macromolecular chains extend and stretch. Thus, a much higher PEO coverage of the PS-b-PEO surfaces can be obtained via this simple method.
 | ||
| Fig. 4 TEM image of PS-b-PEO diblock copolymer films prepared on silica wafer substrate with 24 h toluene vapor annealing and 6.7 mg ml−1 polymer solution. Scale bar = 200 nm. | ||
Combining the water surface with solvent vapor annealing is a simple method to obtain highly ordered structures. The synergistic effect of water and toluene vapor gives equal interaction between PS and PEO blocks. Symmetrical PS-b-PEO films tend to form lamellar morphologies. However, under water and chloroform vapor, the PS block selectively aggregates into cylinder-like structures in thin films on the water surface. The different solubilities between the solvent vapor and the two blocks, as well as water affinity of PEO, play important roles in the resulting ordered structures. This process involves a balance of interaction and repulsion between the solvent and the two blocks. After annealing for a specific time, the film reaches a meso-equilibrium structure. There are several factors related to self-assembly at the solvent vapor/water interface. In the current paper, only the polymer concentration, annealing time, and the type of solvent vapor were investigated. Temperature, film thickness, and interaction parameters between the PS and the water or organic solvent should be considered to precisely control the microstructure. In the present study, the film as a kind of novel soft template was used to direct the self-assembly of C60 (zero dimension) and CNT (one dimension) into nanometer-length scale structures.
The TEM images shown in Fig. 5 exhibit the morphologies of the PS-b-PEO/C60-PS and the PS-b-PEO/C60 nanohybrid films on the water substrate under toluene vapor annealing. The content of the nanofillers is kept at 10 wt%. Comparison between Fig. 5a and 5a* shows that some black domains (aggregations of C60) in Fig. 5a* result from the macrophase separation of C60 from the polymer matrix, indicating the poor interaction between C60 and the two blocks of the PS-b-PEO copolymers. However, when 10 wt% C60-PS is introduced in the PS-b-PEO film (Fig. 5a), there is no aggregation of C60-PS on the surface, indicating that the C60-PS moieties are confined in the PS lamellae. After annealing for 2 h under toluene vapor for the PS-b-PEO/C60-PS nanohybrid film, the nanohybrid film shows a disordered structure. After annealing for 6 h, the irregular lamellar structure of the PS blocks with an approximately 100 nm width in a PEO matrix is formed (Fig. 5b). PEO is hydrophilic compared with the hydrophobic PS. Thus, the PEO blocks swell quickly to occupy the main domains when the film is placed under the organic solvent. Solvent vapor imparts mobility to the polymeric chains, which helps in the dynamic evolution and improvement of the ordered structures. After annealing under toluene vapor for 24 h, the PS-b-PEO/C60-PS nanohybrid films (Fig. 5c) show an ordered lamellar microstructure without aggregation of the C60-PS, attributed to the sufficient assembly time, indicating selective and uniform distribution of the C60-PS moieties within the PS domain during solvent annealing.
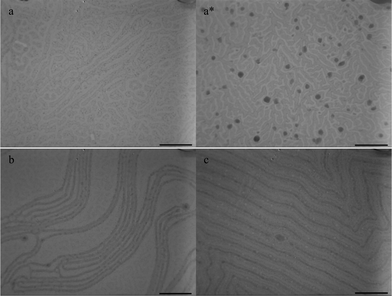 | ||
| Fig. 5 TEM images of PS-b-PEO nanohybrid films on water surface under toluene vapor annealing with 10 wt% C60: (a) PS-b-PEO/C60-PS, 2 h, (a*) PS-b-PEO/C60, 2 h, (b) PS-b-PEO/C60-PS, 6 h, (c) PS-b-PEO/C60-PS, 24 h. Scale bar = 900 nm. | ||
As nanoparticles were added into the PS-b-PEO, more regular microphase structures appeared compared with that in Fig. 2. C60-PS seems beneficial to the formation of the ordered lamellar structure because the size of the PS domain is kept at approximately 100 nm when PS-b-PEO/C60-PS was annealed from 6 h to 24 h under toluene vapor. The addition of C60-PS increases the volume fraction of the PS, causing the PS domains to swell (about 80 nm as shown in Fig. 2d). The transition of the PS-b-PEO from a disordered to an ordered structure seems easier because of the introduction of C60-PS, in which the volume fraction of the PS is increased. The increased volume fraction of the PS can slightly accelerate the formation of a lamellar microstructure. The lamellae emerge when the volume fraction of the PS reaches the limit. Prolonged annealing time enables the PS and the PEO chains to move adequately to form ordered microstructures, such as lamellae.
Fig. 6 shows the TEM images of the microphase-separated PS-b-PEO/C60-PS nanohybrid films on the water surface under solvent annealing under chloroform vapor. Considering the aggregations of the pure C60 in PS-b-PEO (data not shown) during the whole solvent annealing process, only modified C60 (C60-PS) was studied in the current work. As shown in Fig. 6a, after annealing for 2 h under chloroform vapor, the PS-b-PEO/C60-PS nanohybrid film shows a co-continuous microstructure, and the PS block seems to swell quickly and occupy the main domains. After annealing for 6 h (Fig. 6b), the film exhibits a well-organized structure with a high degree of long-range lateral order of lamellar microdomains, and the PS, as well as the PEO with equivalent microphase sizes, are obtained. When the annealing time increases to 24 h (Fig. 6c), the thin film is self-organized into packed PS/C60-PS cylinder-like microdomains embedded in the PEO matrix because of the sufficient time for assembly in the presence of C60-PS.
 | ||
| Fig. 6 TEM images of PS-b-PEO nanohybrid films on water surface under chloroform vapor annealing with 10 wt% C60: (a) PS-b-PEO/C60-PS, 2 h, (b) PS-b-PEO/C60-PS, 6 h, (c) PS-b-PEO/C60-PS, 24 h. Scale bar = 400 nm. | ||
The PS blocks turns cylindrical from the lamellar domains, and the PEO blocks occupies the main domains (Fig. 6c), indicating that chloroform as the annealing solvent can modulate copolymer chains to form the ordered microstructure. Nevertheless, the interaction between the two blocks and water becomes more important because this process results in the self-assembly of amphiphilic diblock copolymers at the air/water interface. Chloroform is nonselective to both PS and PEO chains, whereas water is selective to PEO. When a droplet of polymer solution spreads out to be a nanohybrid thin film on the water surface, the condition is more favorable for PEO chains to form main domains. Interestingly, the addition of C60-PS complicates the phenomenon. When the annealing time is short, such as 2 h, the addition of C60-PS plays a key role, and the microstructure occurs at a higher PS-projected area than at the PEO (Fig. 6a). As the annealing time is increased, such as 6 and 24 h, the selectivity of water to PEO becomes the key factor because the PEO chains have enough time to move and stretch out. This phenomenon is the reason why the diameter of the PS lamellae becomes smaller in favor of the formation of the PS cylinder.
The size of the PS domain becomes larger in the PS-b-PEO/C60-PS nanohybrid film compared with that of the pure PS-b-PEO film (Fig. 3). After adding the C60-PS into the copolymer film, the diameter of the nanoscopic cylindrical PS domains increases from 30 nm (Fig. 3c) to approximately 60 nm (Fig. 6c), and the repeat period of the lattice is kept almost constant (approximately 120 nm). The presence of the solvent mediates the surface energies of the copolymer and imparts a high mobility to the PEO and the PS chains, including PS grafted to C60. The C60 moieties are preferentially confined in the PS lamellae at the effect of the PS chain modification on the surface of C60. When the C60-PS is added into the PS-b-PEO, C60-PS nanoparticles appeared in the PS microdomain to enlarge the size of the PS block. This phenomenon can increase the wt% of the PS and make the PS lamellae swell by reducing the effective diffusion of the PS-b-PEO, making the PS phase larger. Therefore, the transition is proposed to be caused by the increasing wt% of C60-PS, mediating the assembly of the PEO domains by reducing the effective diffusion of PS-b-PEO and induces the swelling of the PS lamellae. George and Cooper-White39 illustrated a simple method to control the domain size of self-assembled block copolymers. They introduced a homopolymer (PS) to the asymmetric (PS-b-PEO) diblock copolymers to self-assemble and found that increasing the PS can reduce the diameter of the PEO domains due to the kinetically constrained phase separation of the system.
CNTs, the pseudo-one-dimensional carbon allotropes with high aspect ratio, high surface area, and excellent material properties, are also studied to gain control over their lateral distribution and positional localization in the PS-b-PEO. The PS- and PEO-modified MWNT have been added into the PS-b-PEO solution to prepare the nanohybrid films. The regular microstructures are not observed in all of the nanohybrid films under solvent annealing for different periods of time. Fig. 7 shows two TEM images representing the annealing time dependence of the structure evolution in the PS-b-PEO/MWNT-g-PEO nanohybrid films. Clearly, the PS-b-PEO film does not exhibit any trace of microphase separation, and the MWNT-g-PEO is uniformly dispersed in the PS-b-PEO film without aggregation after annealing for 2 h under toluene vapor. After 24 h, the PS-b-PEO films annealed under toluene vapor shows a multiple microstructure, and the PS assembles slightly into a lamellar structure. The long MWNT-g-PEO is not confined in the PEO phase, and spans to both phases instead. The confinement of MWNT-g-PEO in the PEO phase is presumed to demand a huge translational entropy penalty, which could not be compensated by the enthalpy gain facilitated by the PEO ligand.
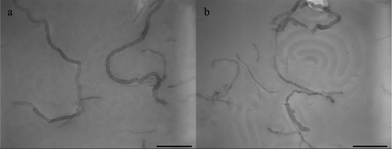 | ||
| Fig. 7 TEM images of PS-b-PEO nanohybrid films on the water surface under toluene vapor annealing with 5 wt% MWNT-g-PEO: (a) PS-b-PEO/MWNT-g-PEO, 2 h, (b) PS-b-PEO/MWNT-g-PEO, 24 h. Scale bar = 400 nm. | ||
Russell et al.24 succeeded in selectively assembling anisotropic PEO-functionalized CdSe nanorods into the defined regions of a patterned surface in the PS-b-PMMA film by allowing the copolymer template to float onto an aqueous solution of the nanorods. In the present study, a modified floating method by dropping the pure PS-b-PEO thin film on the surface of MWNT-g-PEO aqueous solution under solvent vapor is described. The functionalization of the MWNT with the PEO ligands provides the MWNT solubility in water and surface activity with the PEO microphase in the copolymer, enabling the assembly by the flotation method.
The TEM images of microphase-separated PS-b-PEO films annealed under toluene vapor for 24 h on the surface of water/MWNT-g-PEO solution are shown in Fig. 8a and b. The irregular PS lamellae are found in the PS-b-PEO/MWNT-g-PEO nanohybrid films, and the lamellae seem to go along the MWNT-g-PEOs. The benzene ring structures in the PS chains have π–π conjugation with CNTs.40 The π–π conjugation between the PS and the MWNTs induces the MWNTs to penetrate the PS phase. However, the PEO grafted to the MWNTs can guide the MWNTs to remain in the PEO phase of the diblock copolymer. Hence, majority of the MWNT-g-PEOs are found at the boundary between the PS and the PEO phase (Fig. 8b). The annealing time is also increased to study the mutual influence of the MWNT-g-PEOs on the self-assembly of the diblock copolymer. After annealing for 48 h under toluene vapor, the PS microphase lamellae remains irregular and more MWNT-g-PEOs are found to accumulate at the boundary of the two blocks and within the PEO microphase. No orientation of the CNTs in the PEO microphase is observed while a small part of CNTs clung to the PS phase because of the high aspect ratio. Hence, the surface affinity of PEO is required to aid in the segregation of the MWNT-g-PEOs to the water–air interface, where they are absorbed by the PEO part of the soft copolymer templates.
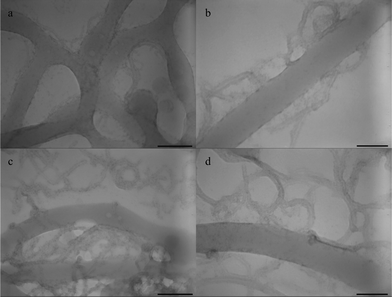 | ||
| Fig. 8 TEM images of PS-b-PEO diblock copolymer films prepared on the surface of water/MWNT-g-PEO solution under toluene vapor annealing for: (a,b) 24 h with various magnifications, (c,d) 48 h with various magnifications. Scale bar = 200 nm for a, c, scale bar = 90 nm for b, d. | ||
Conclusions
An effective and simple method for manipulating and ordering the microphase separation of PS-b-PEO was introduced by combining an aqueous substrate with organic solvent vapor annealing. Using the different solubilities of PS and PEO blocks, highly ordered PS-b-PEO films were obtained under toluene or chloroform vapor on the water surface. After annealing for 24 h, a lamellar microstructure was obtained under toluene vapor, whereas a PS cylinder was obtained under chloroform vapor. The proposed simple preparation generated larger sizes of microdomains. Two kinds of nanohybrid thin films were successfully prepared using the self-assembly of the PS-b-PEO block copolymer on an aqueous substrate combined with solvent vapor annealing, in which the ligand-functionalized C60 and the MWNT were selectively distributed in the block copolymer templates. The polymer ligands covering the carbon nanomaterials were effective in achieving the desired assembly. The C60-PS joined the PS phase, expanding the scale of the PS domains. The lamellar or cylindrical microstructure of the PS-b-PEO/C60-PS nanohybrid film on the water surface was obtained by controlling the annealing solvent and the annealing time. The MWNT-g-PEOs/water solution was prepared as a complex substrate for a film flotation technique with the PS-b-PEO copolymer template. The MWNT-g-PEOs were found to accumulate on the PEO phase of the template.Acknowledgements
The authors gratefully acknowledge financial support of this work from Natural Science Foundation of China (NSFC) (No. 21074009 and 51173009).References
- N. Hadjichristidis, S. Pispas and G. Floudas, Block copolymers: synthetic strategies, physical properties, and applications, John Wiley and Sons, Hoboken, New Jersey, 2003 Search PubMed.
- G. H. Fredrickson and F. S. Bates, Annu. Rev. Mater. Sci., 1996, 26, 501–550 CrossRef CAS.
- M. Park, C. Harrison, P. M. Chaikin, R. A. Register and D. H. Adamson, Science, 1997, 276, 1401–1404 CrossRef CAS.
- Y. Fink, A. M. Urbas, M. G. Bawendi, J. D. Joannopoulos and E. L. Thomas, J. Lightwave Technol., 1999, 17, 1963–1969 CrossRef CAS.
- S. Ouk Kim, H. H. Solak, M. P. Stoykovich, N. J. Ferrier, J. J. de Pablo and P. F. Nealey, Nature, 2003, 424, 411–414 CrossRef.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS.
- P. A. George, B. C. Donose and J. J. Cooper-White, Biomaterials, 2009, 30, 2449–2456 CrossRef CAS.
- I. W. Hamley, Developments in Block Copolymer Science and Technology, Wiley, England, 2004 Search PubMed.
- A. Sidorenko, I. Tokarev, S. Minko and M. Stamm, J. Am. Chem. Soc., 2003, 125, 12211–12216 CrossRef CAS.
- K. Fukunaga, T. Hashimoto, H. Elbs and G. Krausch, Macromolecules, 2002, 35, 4406–4413 CrossRef CAS.
- A. M. Gonçalves da Silva, A. L. Simões Gamboa and J. M. G. Martinho, Langmuir, 1998, 14, 5327–5330 CrossRef.
- C. A. Devereaux and S. M. Baker, Macromolecules, 2002, 35, 1921–1927 CrossRef CAS.
- H. Y. Chen and E. Ruckenstein, Polymer, 2010, 51, 5869–5882 CrossRef CAS.
- H. C. Kim, S. M. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146–177 CrossRef CAS.
- B. Hamdoun, D. Ausserré, V. Cabuil and S. Joly, J. Phys. II, 1996, 6, 503–510 CrossRef.
- B. Hamdoun, D. Ausserré, S. Joly, Y. Gallot, V. Cabuil and C. Clinard, J. Phys. II, 1996, 6, 493–501 CrossRef CAS.
- V. Lauter-Pasyuk, H. J. Lauter, D. Ausserre, Y. Gallot, V. Cabuil, E. I. Kornilov and B. Hamdoun, Phys. B, 1997, 241–243, 1092–1094 CrossRef CAS.
- M. R. Bockstaller, Y. Lapetnikov, S. Margel and E. L. Thomas, J. Am. Chem. Soc., 2003, 125, 5276–5277 CrossRef CAS.
- M. R. Bockstaller and E. L. Thomas, Phys. Rev. Lett., 2004, 93, 166106 CrossRef.
- J. J. Chiu, B. J. Kim, E. J. Kramer and D. J. Pine, J. Am. Chem. Soc., 2005, 127, 5036–5037 CrossRef CAS.
- B. J. Kim, J. J. Chiu, G. R. Yi, D. J. Pine and E. J. Kramer, Adv. Mater., 2005, 17, 2618–2622 CrossRef CAS.
- M. R. Bockstaller, R. A. Mickiewicz and E. L. Thomas, Adv. Mater., 2005, 17, 1331–1349 CrossRef CAS.
- S. W. Sides, B. J. Kim, E. J. Kramer and G. H. Fredrickson, Phys. Rev. Lett., 2006, 96, 250601 CrossRef.
- Q. L. Zhang, S. Gupta, T. Emrick and T. P. Russell, J. Am. Chem. Soc., 2006, 128, 3898–3899 CrossRef CAS.
- R. D. Deshmukh, Y. Liu and R. J. Composto, Nano Lett., 2007, 7, 3662–3668 CrossRef CAS.
- N. R. Jana, Angew. Chem., Int. Ed., 2004, 43, 1536–1540 CrossRef CAS.
- J. G. Son, W. K. Bae, H. M. Kang, P. F. Nealey and K. Char, ACS Nano, 2009, 3, 3927–3934 CrossRef CAS.
- E. Ploshnik, A. Salant, U. Banin and R. Shenhar, Adv. Mater., 2010, 22, 2774–2779 CrossRef CAS.
- E. Ploshnik, A. Salant, U. Banin and R. Shenhar, Phys. Chem. Chem. Phys., 2010, 12, 11885–11893 RSC.
- L. Peponi, A. Tercjak, J. Gutierrez, M. Cardinali, I. Mondragon, L. Valentini and J. M. Kenny, Carbon, 2010, 48, 2590–2595 CrossRef CAS.
- C. X. Li, J. C. Hsu, K. Sugiyama, A. Hirao, W. C. Chen and R. Mezzenga, Macromolecules, 2009, 42, 5793–5801 CrossRef CAS.
- J. Wang, G.-X. Chen, J. Sun and Q. Li, J. Phys. Chem. B, 2011, 115, 2824–2830 CrossRef CAS.
- G.-X. Chen, H. S. Kim, B. H. Park and J. S. Yoon, J. Phys. Chem. B, 2005, 109, 22237–22243 CrossRef CAS.
- Z. Q. Lin, D. H. Kim, X. D. Wu, L. Boosahda, D. Stone, L. LaRose and T. P. Russell, Adv. Mater., 2002, 14, 1373–1376 CrossRef CAS.
- J. K. Cox, K. Yu, B. Constantine, A. Eisenberg and R. B. Lennox, Langmuir, 1999, 15, 7714–7718 CrossRef CAS.
- S. H. Kim, M. J. Misner, T. Xu, M. Kimura and T. P. Russell, Adv. Mater., 2004, 16, 226–231 CrossRef CAS.
- P. M. Simone and T. P. Lodge, ACS Appl. Mater. Interfaces, 2009, 1, 2812–2820 CAS.
- Y. Li and A. Kaito, Eur. Polym. J., 2006, 42, 1986–1993 CrossRef CAS.
- P. A. George and J. J. Cooper-White, Eur. Polym. J., 2009, 45, 1065–1071 CrossRef CAS.
- R. Q. Long and R. T. Yang, J. Am. Chem. Soc., 2001, 123, 2058–2059 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20250f |
| This journal is © The Royal Society of Chemistry 2012 |