A facile route for constructing a graphene-chitosan-ZrO2 composite for direct electron transfer and glucose sensing
Chang-Jun
Cai
a,
Mao-Wen
Xu
b,
Shu-Juan
Bao
*a,
Chao
Lei
a and
Dian-Zeng
Jia
a
aKey Laboratory of Material and Technology for Clean Energy, Ministry of Education; Key Laboratory of Advanced Functional Materials, Xinjiang Autonomous Region; Institute of Applied Chemistry, Xinjiang University, Urumqi, Xinjiang 830046, P. R. China. E-mail: shjbao@xju.edu.cn; Fax: +86 991-8588883; Tel: +86 991-8581183
bTexas Materials Institute, University of Texas at Austin, Austin, TX 78712, USA. E-mail: maowenxu@mail.utexas.edu
First published on 10th July 2012
Abstract
Integrating graphene-based composites with enzymes provides a potent strategy to enhance biosensor performance due to their unique physicochemical properties. Herein we report on the utilization of graphene-chitosan-ZrO2 (ER-GO/CS/ZrO2) composite as an immobilization matrix for glucose oxidase (GOD). In comparison with electrodes modified with the component materials individually, the ER-GO/CS/ZrO2 modified electrode exhibited excellent electron transfer properties for GOD with a rate constant of 3.12 s−1. The obtained glucose biosensor displayed satisfactory performance over an acceptable linear range from 0.2 mM to 1.6 mM with a detection limit of 45.6 μM at −0.4 V and a sensitivity of 7.6 μA mM−1 cm−2. The integration of graphene, chitosan and mesoporous materials can not only be used for immobilizing GOD, but can also be extended to other enzymes and bioactive molecules, thus providing a promising platform for the development of biosensors.
1. Introduction
With both theoretical significance and practical applications, extensive research efforts have striven to achieve direct electron transfer (DET) between redox proteins and electrode surfaces.1–5 Understanding the direct electron transfer process and reactions between redox proteins and the electrode surface, not only helps to elucidate the intrinsic thermodynamic and kinetic properties of proteins, but also serves as a foundation and route for fabricating new electrochemical biosensors and bioreactors.6,7 Unfortunately, owing to the deeply buried redox-active center of the proteins and the ease with which bioactivity is lost upon interaction with electrode surfaces, DET for most redox proteins on electrode surfaces is still a great challenge.8 Therefore, appropriate promoters have been employed to facilitate the electron transfer process and retain the bioactivity of immobilized redox proteins. Up to now, many materials, including nanoparticles,9,10 ionic liquid,11,12 graphene,13–15 conductive polymers,16,17 biopolymers,18–20 and montmorillonite21 have been studied to immobilize enzymes and promote electron transfer of redox enzymes on the surface of electrodes.In the last few decades, due to their high specific surface area, large pore volume, narrow pore size distribution, tunable pore size and good stability, mesoporous materials have become a research highlight in constructing electrochemical biosensors.22–25 In addition, the porous framework structure, size selective ability, and modest conductivity make them more suitable as enzyme immobilization hosts and “electronic wires”9 to enhance the electron transfer between the redox center in proteins and electrode surfaces. Previous research demonstrated that encapsulation or entrapment of enzymes in mesoporous materials can improve the performance of biocatalytic processes, such as high bioactivity and long-term stability of the biocatalysts, as well as low mass-transfer resistance.22 Among the multifarious mesoporous materials for immobilization of proteins, mesoporous ZrO2 has become an ideal candidate26,27 for high biomolecule activity, chemical inertness, lack of toxicity and affinity for groups containing oxygen. These properties render it a potential material for the construction of enzyme-based electrochemical biosensors.
When it comes to the realm of electrochemical biosensors, graphene, one of the most promising candidates in material research, has exhibited several remarkable characteristics including high surface area-to-volume ratio, excellent electrical conductivity and fast electron mobility at room temperature and good biocompatibility.14 The high surface area-to-volume ratio is helpful in increasing the surface loading of the target enzyme molecules on the surface, the excellent conductivity and small band gap are favorable for conducting electrons from the biomolecules.28,29 Several biosensors30–35 based on graphene have achieved direct electron transfer of GOD, maintained the bioactivity of the enzyme and showed excellent electrocatalysis towards glucose. However, the unique properties of graphene are only associated with individual sheets,36 which put forwards the importance of surmounting its inherent disadvantages, such as hydrophobicity and tendency to aggregate in aqueous solution. This challenge has been well addressed through covalent and noncovalent functionalization methods, such as attachment of other molecules or polymers onto the graphene surface, particularly, using biopolymers.37
Chitosan is a natural biopolymer product derived from chitin via deacetylation with alkali. It shows excellent film forming and adhesion ability, together with nontoxicity and good biocompatibility. Chitosan has been used in combination with nanoparticles as a stabilizing agent to immobilize biomolecules in the last several years. Recently, it has been reported that CS could form a stable nanocomposite with graphene oxide (GO) through electrostatic attraction and hydrogen bonding.38 The plentiful amino groups and hydroxyl groups of CS can provide a hydrophilic environment compatible with the biomolecules, more significantly, it offers a reasonable route to improve both the hydrophilicity and biocompatibility of graphene via covalent grafting strategies.20 Furthermore, the existence of CS on graphene makes it exhibit a positive charge, which is favorable for the further immobilization of negatively charged biomolecules.
A possible route to harness the excellent properties of graphene, chitosan and mesoporous ZrO2 for applications is through incorporating them into composite materials. In this study, we report a facile route to prepare an ER-GO/CS/ZrO2 composite, the fabrication process is shown in Fig. 1. ER-GO was obtained by electrochemical reduction of a homogeneous GO solution on a glassy carbon electrode (GCE, 3 mm in diameter), then ER-GO was grafted with CS by electro-deposition. In the resulting ER-GO/CS/ZrO2 composite, mesoporous ZrO2 acted as the enzyme immobilization host. It was hoped that CS would create a pleasant microenvironment for the GOD and overcome the shortcomings of graphene. ER-GO played a positive role in increasing the surface area and enhancing the electron transfer process to collect current. It has been reported that the integration of carbon-based materials and other nanoparticles liked graphene–CdS, CNT–Pt, graphene–Au can offer synergistic effects in electrocatalytic applications.34,39,40 Similarly, it is fair and reasonable to speculate that the integration of graphene, chitosan and mesoporous ZrO2 can provide an ideal platform for the fabrication of biosensors. Electrochemical measurements revealed that the ER-GO/CS/ZrO2 composite provided a favorable microenvironment for GOD to realize DET. The prepared glucose biosensor exhibited high sensitivity, low detection limit and acceptable stability.
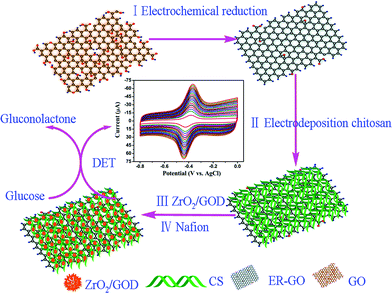 | ||
| Fig. 1 Schematic of the construction of ER-GO/CS/ZrO2 composite modified GCE. | ||
2. Experimental
2.1 Chemicals and materials
Glucose oxidase (Aspergillus Niger) was purchased from Sigma, and stored at −20 °C when not in use. All other chemicals were of analytical grade and used without further purification. All solutions were prepared using deionized Millipore-Q water (18.1 MΩ cm).2.2 Synthesis of mesoporous ZrO2 and graphene oxide
Mesoporous ZrO2 was synthesized using carbon sphere as a template, based on a literature method.41 The starting materials of 0.1 g of colloidal carbon spheres, 100 ml of 0.05 M zirconium chloride solution and 0.02 g of PEG-1000 were sonicated for 10 min to get a stable emulsion. The mixture was then transferred into a flask and stirred during the experiment followed by the dropwise addition of ammonia solution up to pH 7. Subsequently, the obtained colloid solution was sonicated for 30 min, it was found that the carbon spheres coated with Zr(OH)4 subsided on the bottom of the flask. After centrifugation, the carbon spheres coated with Zr(OH)4 were washed 3 times with deionized water and anhydrous ethanol respectively and dried at 60 °C for 5 h. Mesoporous ZrO2 was obtained by calcination of the precursors at 600 °C for 1 h. GO was synthesized from natural graphite by a modified Hummer's method.42–44 In brief, 3 g of graphite powder was put into a 80 °C solution of 12 mL concentrated H2SO4, 2.5 g of K2S2O8 and 2.5 g of P2O5, the mixture was kept at 80 °C for 4.5 h. After cooling to room temperature, the mixture was diluted with 0.5 L of deionized water and left overnight. Then, the mixture was filtered using a 0.2 micron Nylon Millipore filter and dried naturally. The pre-oxidized graphite was re-oxidized according to Hummers' method. Pretreated graphite powder was put into 120 mL of concentrated H2SO4 at 0 °C. After that 15 g KMnO4 was added gradually under stirring and the temperature of the mixture was kept below 20 °C by ice-bath. Successively, the mixture was stirred at 35 °C for 4 h. After 5 days of stirring, 30 mL of 30% H2O2 was injected into the solution to completely react with the excess KMnO4. A bright yellow solution was obtained and washed by 3% H2SO4 and H2O until the pH was 5∼6 and then was freeze dried for further use.2.3 Fabrication of modified electrodes
Prior to use, the glassy carbon electrode was polished to a mirror-like finish with a 100 nm and 30 nm alumina slurry followed by rinsing thoroughly. The electrode was successively sonicated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solution of nitric acid, ethanol solution and deionized water, and dried naturally. ZrO2 and GOD were mixed together rigorously in 0.1 M pH 7.4 PBS solution and storied in a refrigerator for at least 24 h so that the GOD could be sufficiently immobilized, at this pH, GOD (pI = 4.5) bears a net negative charge. ER-GO was obtained by electrochemical reduction of GO. The synthesized GO was exfoliated in electrolytes by ultrasonication for 30 min to form homogeneous GO dispersions with a concentration of 1.0 mg mL−1. The cyclic voltammetric reduction was operated on a CHI 660D electrochemical workstation (CH Instruments, Shanghai) using a three-electrode system: a GCE as the working electrode, platinum wire as the counter electrode, and an SCE as the reference electrode. The scan was performed between −1.5 V and 0.7 V at a rate of 50 mV s−1 for 15 potential cycles.45 Subsequently, the electrode was modified with chitosan by electrodeposition in 0.2% chitosan solution at −2.5 V for a few seconds to form a positively charged surface. After than, 5 μL of ZrO2 and GOD suspension was dropped onto the electrode, thus allowing electrostatic attraction with positively charged chitosan film. Finally, it was coated with a thin film of 0.5% Nafion solution. The resultant electrode was stored at 4 °C when not in use. The same procedure was employed to fabricate other modified electrodes.
:1 solution of nitric acid, ethanol solution and deionized water, and dried naturally. ZrO2 and GOD were mixed together rigorously in 0.1 M pH 7.4 PBS solution and storied in a refrigerator for at least 24 h so that the GOD could be sufficiently immobilized, at this pH, GOD (pI = 4.5) bears a net negative charge. ER-GO was obtained by electrochemical reduction of GO. The synthesized GO was exfoliated in electrolytes by ultrasonication for 30 min to form homogeneous GO dispersions with a concentration of 1.0 mg mL−1. The cyclic voltammetric reduction was operated on a CHI 660D electrochemical workstation (CH Instruments, Shanghai) using a three-electrode system: a GCE as the working electrode, platinum wire as the counter electrode, and an SCE as the reference electrode. The scan was performed between −1.5 V and 0.7 V at a rate of 50 mV s−1 for 15 potential cycles.45 Subsequently, the electrode was modified with chitosan by electrodeposition in 0.2% chitosan solution at −2.5 V for a few seconds to form a positively charged surface. After than, 5 μL of ZrO2 and GOD suspension was dropped onto the electrode, thus allowing electrostatic attraction with positively charged chitosan film. Finally, it was coated with a thin film of 0.5% Nafion solution. The resultant electrode was stored at 4 °C when not in use. The same procedure was employed to fabricate other modified electrodes.
2.4 Material characterizations and electrochemical measurements
The crystal structure of the materials was characterized by powder X-ray diffraction (XRD, Bruker D8 Advance X-ray diffractometer). The morphology and microstructure were examined by field-emission scanning electron microscopy (SEM,JSM-6700F) and transmission electron microscopy (TEM, JEM-2100F). Nitrogen adsorption/desorption was carried out at 77.3 K by means of an ASAP 2050 X tended Pressure Sorption Analyzer. Electrochemical measurements were performed using a CHI 660D electrochemical workstation (CH Instruments Inc.). The measurements were based on a three-electrode system with the as-prepared electrodes as the working electrode, a platinum wire as the auxiliary electrode, and a saturated calomel electrode (SCE) or a Ag/AgCl electrode as the reference electrode. If not specified, all tested solutions were purged with highly purified argon for 30 min before experiments and an argon environment was kept over the solution during electrochemical measurements.3. Results and discussion
3.1 Material characterizations
The synthesized GO, ER-GO, ZrO2 and ERGO-ZrO2 were first characterized with XRD. As displayed in Fig. 2a, the pattern of GO has a typical characteristic peak centered at 2 θ = 10.1°, corresponding to the (001) inter-planar spacing of 8.75 Å. The most intense peak for graphite at 2 θ = 26.4° (corresponding to a d-spacing of 3.35 Å) is absent in the GO sample, confirming the high quality of GO. In the pattern of ER-GO, the reappearance of the (002) diffraction line at 24.4° and disappearance of the diffraction peak at 10.1° give evidence that the GO was reduced to ER-GO and as such restored the ordered crystal structure. Fig. 2b gives the XRD patterns of as-synthesized ZrO2 and ERGO-ZrO2. Through search and contrast via the JADE 5.0 database, it was found that the XRD pattern matched the standard XRD pattern (JCPDS No. 65-1022), the corresponding diffraction is indexed in Fig. 2b, and no characteristic peaks of impurities are observed, suggesting pure ZrO2 was prepared. Notably, no typical diffraction peaks of the ER-GO are observed in the ERGO-ZrO2. The reason can be attributed to the fact that the (011) plane at 24.1°and (110) plane at 24.5°of ZrO2 maybe shielded the main characteristic peak of ER-GO at 24.4°.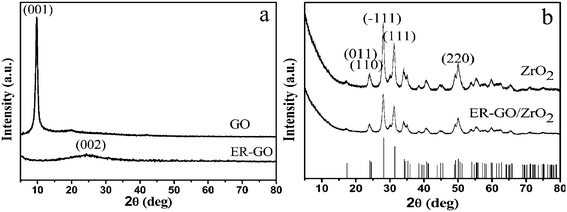 | ||
| Fig. 2 XRD patterns of GO,ER-GO (a) and ZrO2,ER-GO/ZrO2 (b). | ||
Raman spectroscopy is a powerful and widely used technique for characterizing the sp2 and sp3 hybridized carbon atoms contained in graphene to identify disorder and defect structures. As observed in Fig. 3, GO displays two bands at∼1320 and ∼1591 cm−1, corresponding to D and G modes respectively. The reduction of GO to ER-GO leads to an increased in-plane crystalline order of sp2 carbon atoms, and thus increased π-electron conjugation within the sp2 carbon network. As a result, the G band associated with the graphite lattice will shift to a lower value. As seen from Fig. 3, the spectrum of ER-GO shows a red-shift in the G band (∼1581 cm−1) as compared to GO. Also of note, the ID/IG ratio of the ER-GO (1.38) increased compared to that of pristine GO (1.18).These changes indicate that the electrochemical reduction of GO indeed takes place.
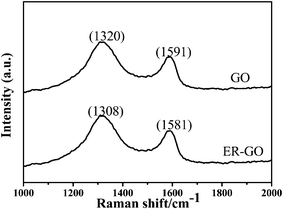 | ||
| Fig. 3 Raman spectra of GO and ER-GO. | ||
Fig. 4a shows a typical TEM image of GO, which clearly indicates its well-packed GO layered structures. The GO appeared transparent and is folded over on one edge, tending to congregate together to form multilayer agglomerates, indicating the high-quality of GO. Fig. 4b is a SEM image of the ER-GO obtained by electrochemical deposited reduction on the GCE surface, which reveals the typical crumpled and wrinkled texture that is associated with the presence of flexible sheets on the rough surface of the film. It has been shown that the edge plane of graphene sheets yield chemical functional groups, such as C–OH and –COOH,46 in the electrochemical reduction process. The functional groups make the graphene sheets more hydrophilic and they are then easier to interact with chitosan and prepare graphene–polymer composites.28 Therefore, it is helpful in increasing the surface loading of the target molecules. From the TEM image of mesoporous ZrO2, it can be seen that mesoporous ZrO2 possesses abundant disordered pores, and the HR-TEM image further reveals that distributed spherical mesopores have a approximately diameter of 10 nm. This indicates that a mesopore structure can be formed using carbon spheres with concentrated diameters as a template.

The nitrogen adsorption–desorption isotherms of ZrO2 (Fig. 5) exhibits large hysteresis loops of a typical type IV curve, implying the mesoporous characteristics with a capillary condensation phenomenon. The BET surface area and pore volume were calculated to be 51.67 m2g−1 and 0.0796 cm3g−1, respectively. The pore size distribution can be calculated from the desorption branch of the isotherms using the BJH method, as shown in Fig. 5, it shows that the ZrO2 has a uniform porous distribution, mainly focused on the range of 5–12 nm, which matches well with the dimensions of GOD (5.2 nm × 6.0 nm × 7.7 nm).47 The BJH pore size distribution of this material determined by N2 adsorption–desorption isotherms is in good agreement with the size observed in the TEM images.
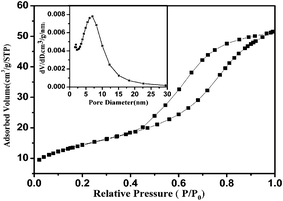 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherms of mesoporous ZrO2. Inset is BJH pore-size distributions of mesoporous ZrO2. | ||
3.2 Electrochemical characteristics and direct electron transfer of the modified electrodes
Cyclic voltammetry was employed to evaluate the performance of the fabricated biosensor during stepwise modification. The direct electrochemistry of GOD immobilized on the modified GCE was investigated in 0.1 M pH 7.4 argon-saturated PBS solution at 200 mV s−1. As shown in Fig. 6a, no redox peak is observed for either the bare electrode or GOD-modified electrode, which indicates they are electro-inactive over this potential window. Slight and poor-defined redox peaks can be seen from the ZrO2/GOD-modified electrode, mainly because ZrO2 is a poor conductor and GOD is deeply seated in a cavity which not easily accessible for conduction of electrons to the electrode surface. Therefore, the ZrO2/GOD-modified electrode only can partially realize the DET between the GOD's electroactive center and the electrode. In contrast, the ER-GO/ZrO2/GOD modified electrode exhibits stable, well-defined and quasi-reversible redox peaks, which are in accordance with FAD to FADH2 conversion, This indicates that the presence of graphene can facilitate the electron transfer between the electroactive center of GOD and the electrode. Most importantly, compared with the ER-GO/ZrO2/GOD modified electrode, the ER-GO/CS/ZrO2/GOD modified electrode displays a much smaller peak-to-peak separation and remarkable increase in peak current which is apparently larger than the sum current of the ER-GO/ZrO2/GOD and ZrO2/GOD modified electrodes. There is also a larger electron transfer rate between the GOD and the electrode, the details of which are listed in Table 1. Based on the excellent electrical conductivity and electron mobility of graphene, the addition of positively charged chitosan can provide a suitable microenvironment for the negatively charged GOD to retain its activity. Finally the combination synergistically enhances the DET process and reactions between the electroactive center of GOD and the electrode surface. The experiment results confirm our assumption of the potential synergistic effect in ER-GO/CS/ZrO2 composites.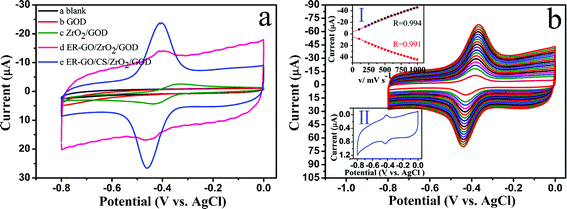 | ||
| Fig. 6 (a) Cyclic voltammograms (CVs) of the different electrodes in 0.1 M pH 7.4 PBS solution at 200 mVs−1. (b) CVs of ER-GO/CS/ZrO2/GOD modified GCE in 0.1 M pH 7.4 PBS solution at different scan rates (from 100 to1000 mV s−1, inside to outside). Inset of (b) is plots (I) of peak currents versus scan rate, and CV(II) of the ER-GO/CS/ZrO2/GOD modified electrode with a peak-to-peak separation of 10 mV in 0.1 M pH 7.4 PBS solution at 10 mVs−1. | ||
| Different modified electrodes | E θ (V) | ΔEp (mV) | I pa/Ipc (μA) | Electron transfer rate constant (s−1) |
|---|---|---|---|---|
| ZrO2/GOD | −0.380 | 106 | −2.390/3.443 | 0.99 |
| ER-GO/ZrO2/GOD | −0.417 | 62 | −4.515/4.157 | 2.30 |
| ER-GO/CS/ZrO2/GOD | −0.437 | 47 | −18.61/21.34 | 3.12 |
To evaluate the reversible electron transfer phenomenon of the ER-GO/CS/ZrO2/GOD modified electrode in more detail, the CVs at different scan rates were recorded in a 0.1 M pH 7.4 PBS solution. As shown in Fig. 6b, as the scan rate increases, the cathodic and anodic peak currents linearly rise simultaneously in the range of 100–1000 mV s−1, indicating a surface controlled electrochemical oxidation/reduction of the FAD/FADH2 involved in the GOD-structure. The linear regression equations are Ipc(μA) = 6.875 + 0.0388v (mV s−1) (n = 18, R = 0.994) and Ipa(μA) = −5.197 − 0.0420v (mV s−1) (n = 18, R = 0.991), respectively. The small ΔEp value stands for the faster direct electron transfer between the redox active site of GOD (the cofactor FAD) without the help of an electron transfer mediator.48 The good linear relationship between the peak currents and the scan rates demonstrates that the electron transfer process for the ER-GO/CS/ZrO2/GOD modified electrode is a surface-confined mechanism, manifesting the characteristics of quasi-reversible surface-controlled electrochemical behaviors.2
It is well known that the DET of GOD is a two-electron coupled with two-proton reaction that undergoes a redox reaction as follows:
| GOD − FAD + 2e + 2H + → GOD − FADH2 | (1) |
Therefore, the pH value of the solution should have an effect on the electrochemical behavior of GOD on the ER-GO/CS/ZrO2/GOD film. As shown in Fig. 7a, a negative shift of both the cathodic and anodic peak potentials occurs when the solution pH value is increasing. The redox potential Eθ changes linearly as a function of solution pH from 5.8 to 8.0 with a slope of −51.4 mV/pH (R = 0.998), which is close to the theoretical value of 59.0 mV/pH. Hence, the direct electrochemical reaction of GOD is a two-proton coupled with two-electron redox reaction process. The pH value also affects the redox peak currents of GOD. The peak current changes as the pH increases until the maximum peak current reaches pH 7.4. With further increases in the pH, the peak current decreases, possibly because of the decrease of proton concentration and bioactivity of the immobilized GOD. Hence, all subsequent studies were carried out in pH 7.4 PBS solutions.
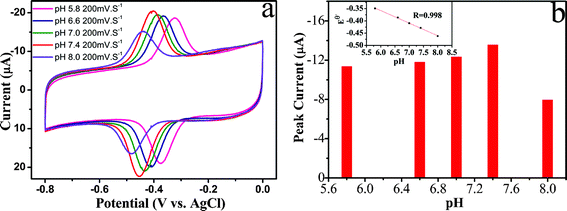 | ||
| Fig. 7 (a) CVs of ER-GO/CS/ZrO2 composite modified GCE in 0.1 M argon-saturated PBS solution with different pH values at 200 mV s−1. (b) Plot of peak current versus pH value. Inset of (b) is the plot of Eθversus pH value. | ||
Fig. 8 displays the CVs of the ER-GO/CS/ZrO2/GOD-modified electrode in 0.1 M pH 7.4 PBS solutions of different atmosphere. A pair of well-resolved redox peaks can be observed in both argon-saturated and air-saturated PBS solutions. In the presence of dissolved oxygen, an increase in reduction current can be observed at the modified electrode, meanwhile, the shape of cyclic voltammograms changed greatly with an increase in the reduction peak current at ER-GO/CS/ZrO2/GOD-modified electrode. The change and difference can be explained as follows:
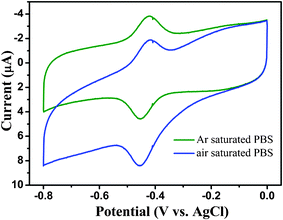 | ||
| Fig. 8 CVs of ER-GO/CS/ZrO2 composite modified GCE in 0.1 M pH 7.4 PBS solution at different atmosphere at 50 mV s−1. | ||
It is well known that in the presence of oxygen the reduced enzyme GOD-FADH2 can be quickly oxidized by oxygen according to the following reaction:
| GOD − FADH2 + O2 → GOD + FAD +H2O2 | (2) |
Obviously, the differences between argon-saturated and air-saturated solutions reveal the electro catalytic reduction of immobilized GOD to dissolved oxygen based on Reaction 1 and 2. Therefore, direct oxidation of GOD (FADH2) at the electrode surface is greatly hindered, leading to the oxidation peak current decreasing.
3.3 Amperometric response of the glucose biosensor
In order to investigate the bioactivity of the adsorbed GOD, the fabricated electrode was employed as a biosensor for electrochemical oxidation of glucose. Fig. 9 shows a typical current–time plot for the ER-GO/CS/ZrO2/GOD modified electrode on successive injections of a glucose solution into argon-saturated and stirred 0.1 M pH 7.4 PBS solutions at −0.4 V. Each successive glucose addition results in a decrease in the steady state current. The peak current of ER-GO/CS/ZrO2/GOD modified electrode increases linearly with increasing glucose concentration over the range 0.2–1.6 mM with a correlation coefficient of 0.997. This demonstrates a sensitivity of 7.6 μA mM−1 cm−2, which is superior to glucose biosensors based on other nanomaterials (Table 2). A low detection limit of 45.6 μM is also readily achieved at a signal-to-noise ratio of 3. The calibration plot in Fig. 9b shows well defined, typical behavior of an enzymatic kinetic reaction. The apparent Michaelis–Menten constant, as an indicator of enzyme–substrate reaction kinetics, can be used to evaluate the biological activity of the immobilized enzyme, which was calculated to be 18.94 mM according to the Lineweaver–Burk equation:Here, Iss is the steady-state current after the addition of substrate, C is the bulk concentration of substrate and Imax is the maximum current measured at the saturated substrate solution. Compared with ER-GO/CS/ZrO2/GOD, ER-GO/ZrO2/GOD exhibits a much higher KM of 28.01 mM (Lineweaver–Burk plot is not shown), which indicates that the presence of CS can provide a favorable microenvironment for GOD to achieve direct electron transfer. The high KM value of ER-GO/CS/ZrO2/GOD may be attributed to the porous structure of ZrO2. The GOD trapped into the pores of the ER-GO/CS/ZrO2 film increases mass transfer resistance, forming a barrier between GOD and glucose reaction. It also can explain the drawback of the glucose biosensor with a relatively long response time. Improvement measures are on the way.
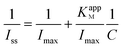
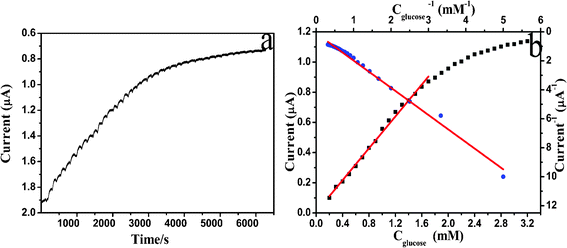 | ||
| Fig. 9 (a) Typical steady state response of the biosensor on successive injection of 0.1 mmol glucose solution into 0.1 M pH 7.4 argon-saturated PBS solution while stirring, at an applied potential of −0.4 V. (b) The calibration curve (current versus glucose concentration) and Lineweaver–Burk plot (current−1versus concentration−1) from the current–time curve. | ||
The stability of a biosensor is also critical. The stability of the glucose biosensor was evaluated by monitoring the CV measurements continuously scanning for 80 cycles. The response dropped by less than 2.5% over this time. The enzyme electrode was stored at 4 °C when not in use. It retained 95% of its initial amperometric response after storage for six weeks, which demonstrates that the sensor has good long-term life time without significant drift.
4. Conclusion
In this paper, a graphene-chitosan-ZrO2 composite provides a suitable microenvironment for the direct electrochemistry of GOD. The immobilized GOD on the modified electrode retains its native structure and electro-catalytic activity. The performance of the resultant electrode is greatly improved compared with electrodes modified by the individual components by virtue of the synergy effect. The integration of graphene, chitosan and mesoporous ZrO2 provides a favorable microenvironment for the enzyme and promotes the DET at the electrode surface. Electrochemical measurements reveal that the prepared glucose biosensor has high sensitivity, low detection limit and good reproducibility. These characteristics suggest that the graphene-chitosan-mesoporous material composite would offer a promising option for the development of various enzyme-based biosensors and the fabrication of stable electrochemical biosensors.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (20963011, 21063014 and 21163021); the Natural Science Foundation of Xinjiang (2011211A001); and the Open Project Program of Xinjiang Laboratory of Advanced Functional Materials (XJDX0902-2010-09).References
- J. Wang, M. Li, Z. Shi, N. Li and Z. Gu, Anal. Chem., 2002, 74, 1993 CrossRef CAS.
- Q. Shen, S. Zhou, X. Zhao, L. P. Jiang, W. Hou and J. J. Zhu, Anal. Methods, 2012, 4, 619 RSC.
- C. Yang, C. Xu and X. Wang, Langmuir, 2012, 28, 4580 CrossRef CAS.
- C. X. Guo, X. T. Zheng, S. R. Ng, Y. Lai, Y. Lei and C. M. Li, Chem. Commun., 2011, 47, 2652 RSC.
- L. Liu, N. Wang and L. Guo, RSC Adv., 2012, 2, 2809 RSC.
- A. Heller, Acc. Chem. Res., 1990, 23, 128 CrossRef CAS.
- C. Léger, S. J. Elliott, K. R. Hoke, L. J. C. Jeuken, A. K. Jones and F. A. Armstrong, Biochemistry, 2003, 42, 8653 CrossRef.
- C. Mu, Q. Zhao, D. Xu, Q. Zhuang and Y. Shao, J. Phys. Chem. B, 2007, 111, 1491 CrossRef CAS.
- X. Luo, A. Morrin, A. J. Killard and M. R. Smyth, Electroanalysis, 2006, 18, 319 CrossRef CAS.
- S. J. Bao, C. M. Li, J. F. Zang, X. Q. Cui, Y. Qiao and J. Guo, Adv. Funct. Mater., 2008, 18, 591 CrossRef CAS.
- Z. Zhu, X. Li, Y. Zeng, W. Sun, W. Zhu and X. Huang, J. Phys. Chem. C, 2011, 115, 12547 CAS.
- X. Lu, J. Hu, X. Yao, Z. Wang and J. Li, Biomacromolecules, 2006, 7, 975 CrossRef CAS.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027 CrossRef CAS.
- Y. Liu, X. Dong and P. Chen, Chem. Soc. Rev., 2012, 41, 2283 RSC.
- Y. Zhang, S. Liu, L. Wang, X. Qin, J. Tian, W. Lu, G. Chang and X. Sun, RSC Adv., 2012, 2, 538 RSC.
- C. X. Guo and C. M. Li, Phys. Chem. Chem. Phys., 2010, 12, 12153 RSC.
- X. M. Feng, R. M. Li, Y. W. Ma, R. F. Chen, N. E. Shi, Q. L. Fan and W. Huang, Adv. Funct. Mater., 2011, 21, 2989 CrossRef CAS.
- X. L. Luo, J. J. Xu, Y. Du and H. Y. Chen, Anal. Biochem., 2004, 334, 284 CrossRef CAS.
- Y. Liu, M. Wang, F. Zhao, Z. Xu and S. Dong, Biosens. Bioelectron., 2005, 21, 984 CrossRef CAS.
- Q. Zeng, J. S. Cheng, X. F. Liu, H. T. Bai and J. H. Jiang, Biosens. Bioelectron., 2011, 26, 3456 CrossRef CAS.
- M. Seleci, D. Ag, E. E. Yalcinkaya, D. O. Demirkol, C. Guler and S. Timur, RSC Adv., 2012, 2, 2112 RSC.
- X. Lu, Y. Xiao, Z. Lei, J. Chen, H. Zhang, Y. Ni and Q. Zhang, J. Mater. Chem., 2009, 19, 4707 RSC.
- M. Zhou, L. Shang, B. Li, L. Huang and S. Dong, Biosens. Bioelectron., 2008, 24, 442 CrossRef CAS.
- G. Zhou, K. K. Fung, L. W. Wong, Y. Chen, R. Renneberg and S. Yang, Talanta, 2011, 84, 659 CrossRef CAS.
- S. J. Bao, C. X. Guo and C. M. Li, RSC Adv., 2012, 2, 1014 RSC.
- B. Liu, Y. Cao, D. Chen, J. Kong and J. Deng, Anal. Chim. Acta, 2003, 478, 59 CrossRef CAS.
- Y. Yang, H. Yang, M. Yang, G. Shen and R. Yu, Anal. Chim. Acta, 2004, 525, 213 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. B. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- X. Kang, J. Wang, H. Wu, I. A. Aksay, J. Liu and Y. Lin, Biosens. Bioelectron., 2009, 25, 901 CrossRef CAS.
- Y. Liu, D. Yu, C. Zeng, Z. Miao and L. Dai, Langmuir, 2010, 26, 6158 CrossRef CAS.
- Y. Chen, Y. Li, D. Sun, D. Tian, J. Zhang and J. J. Zhu, J. Mater. Chem., 2011, 21, 7604 RSC.
- D. A. C. Brownson and C. E. Banks, Analyst, 2011, 136, 2084 RSC.
- J. Y. Sun, K. J. Huang, S. F. Zhao, Y. Fan and Z. W. Wu, Bioelectrochemistry, 2011, 82, 125 CrossRef CAS.
- K. Wang, Q. Liu, Q. M. Guan, J. Wu, H. N. Li and J. J. Yan, Biosens. Bioelectron., 2011, 26, 2252 CrossRef CAS.
- K. Wang, H. N. Li, J. Wu, C. Ju, J. J. Yan, Q. Liu and B. Qiu, Analyst, 2011, 136, 3349 RSC.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101 CrossRef CAS.
- J. D. Qiu, J. Huang and R. P. Liang, Sens. Actuators, B, 2011, 160, 287 CrossRef CAS.
- X. Yang, Y. Tu, L. Li, S. Shang and X. Tao, ACS Appl. Mater. Interfaces, 2010, 2, 1707 CAS.
- Y. Yao, Y. Ding, L. S. Ye and X. H. Xia, Carbon, 2006, 44, 61 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang, J. Luo, C. Li and S. Luan, Electrochim. Acta, 2010, 55, 3055 CrossRef CAS.
- C. Guo, P. Hu, L. Yu and F. Yuan, Mater. Lett., 2009, 63, 1013 CrossRef CAS.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782 CrossRef CAS.
- Y. Xu, H. Bai, G. Lu, C. Li and G. Shi, J. Am. Chem. Soc., 2008, 130, 5856 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- C. Liu, K. Wang, S. Luo, Y. Tang and L. Chen, Small, 2011, 7, 1203 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535 CrossRef CAS.
- S. Wu, H. Ju and Y. Liu, Adv. Funct. Mater., 2007, 17, 585 CrossRef CAS.
- L. Yang, H. Xiong, X. Zhang and S. Wang, Biosens. Bioelectron., 2011, 26, 3801 CrossRef CAS.
- D. Patil, N. Q. Dung, H. Jung, S. Y. Ahn, D. M. Jang and D. Kim, Biosens. Bioelectron., 2012, 31, 176 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |