Catalytic materials that improve selectivity of biomass conversions
Saikat
Dutta
*
Department of Chemistry, University of Florida, Gainesville, Florida 32611-7200, USA. E-mail: saikatdutta2008@gmail.com; saikatdutta@ufl.edu; Tel: +1-352-213-7546
First published on 26th July 2012
Abstract
Lignocellulosic biomass is composed of cellulosic and hemicellulosic components. Degradation of these components leads to short-chain aqueous soluble glucan with β-1,4-glycosidic linkages and its further degradation produces hexose and pentose sugars. Dehydration of these sugar units result in building blocks (platform chemicals) which upon further chemical transformation produces fine chemicals and liquid fuels. Materials with considerable surface acidity and suitable microstructure play crucial roles as catalysts at different steps of the biomass transformation to chemicals and fuels. This review focuses on explaining the roles of the catalytic materials that are employed for improving selectivity of biomass conversions. The article also advances issues such as the morphology and preparation method of materials as a function of their performance. In attempt to keep the focus on materials aspects, mechanistic aspects of biomass conversion has also been discussed in relevant cases. Apart from conventional acidic materials, applications of recently introduced new materials with functionalized surfaces are highlighted with suitable examples. Factors which govern solid materials to perform as catalysts for biomass conversion have been expanded to inform readers with a possible correlation of the reactivity patterns of the materials for different substrates and their macro- and micro-structures.
 Saikat Dutta | Dr Saikat Dutta obtained his PhD in organometallic chemistry from the Indian Institute of Science, Bengaluru in 2008. After a couple of postdoctoral appointments in Taiwan and in India, he was awarded a Fulbright–Nehru Postdoctoral Fellowship in 2012. Dr Dutta is the co-author of more than 20 research publications in scientific journals. His research experiences include transition-metal organometallics, polymerization catalysis, materials development for photophysical-catalytic applications and biomass conversion. He is currently working as a Fulbright research fellow at the Department of Chemistry, University of Florida, USA. His current research is focused on materials for photochemical conversions and organometallics for environment protection. |
1. Introduction
Easily exploitable fossil fuel reserves will run out eventually. Furthermore, extensive CO2 emissions linked to the use of fossil fuels have been associated with global climate changes, which is another major concern. Thus, a great extent of academic and industrial initiatives in exploring renewable lignocellulosic biomass for liquid fuels and chemicals have been witnessed in the last decade since an increased demand for fuels and chemicals has resulted from the growth of world population and development. A considerable increase in world production of bioethanol from 17 billion liters in 2000 to more than 46 billion liters in 20071 indicates that a shift to a biorenewable based production technology for transportation fuels and chemicals is underway. As bioethanol at present can only be used in mixtures with traditional gasoline, its potential as a long-term renewable fossil fuel alternative is limited. Bioethanol is currently produced in large quantities from grains such as corn, which is a major concern, since this directly competes with the food supply.2,3 Also, bioethanol suffers from several limitations, including low energy density, high volatility and contamination by the absorption of water from the atmosphere. Unless cellulosic bioethanol production technology4,5 develops to the next improved level for economic production, researchers will look to lignocellulosic biomass feedstock as a promising alternative.The global amount of terrestrial plant biomass produced annually has been estimated to be about 56.8 × 109 tonnes of elemental carbon.6 Lignocellulose is estimated to make up about 70–95% of this amount.7 The components of lignocelluloses (40–50% cellulose, 16–33% hemicelluloses and 15–30% lignin),8 are available in several industrial waste streams, especially from the paper and agricultural industries. Since cellulose is the main component of lignocellulosic biomass, it has attracted most attention. For successful chemical methods to be developed, detailed knowledge of the recalcitrant chemical structure of celluloses is indispensable to fully tackle the difficulties associated with the chemical transformations.9 Recently, several solvents and catalysts including supercritical water, ionic liquids, mesoporous carbon functionalized with ruthenium metal or SO3H groups, and sulfonated ion exchange resins have been investigated for the hydrolysis of cellulose.10,11 Extensive reviews on cellulose conversions to sugar alcohols and also on cellulose conversion in ionic liquids have been published.12–15 To overcome resistance to degradation through breaking of hydrogen bond networks and β-1,4-glycosidic bonds, ionic liquids have been employed to form homogeneous solutions of cellulose prior to hydrolysis.15–19 Although homogeneous hydrolysis can be carried out under mild conditions with high glucose yield, the workup for separation of sugars, dehydrated products, and unreacted cellulose from the ionic liquid is normally complicated.20 It is difficult even for enzymes to access glycosidic bonds of the cellulose network due to the complex H-bonded structure of lignocellulosic materials, which must be pretreated before hydrolysis, and the cost of such processes is another bottleneck.
In order to overcome the problems encountered in conventional processes with more environmentally sustainable approaches,21 Fukuoka and Dhepe demonstrated the direct conversion of cellulose into sugar alcohols by use of the solid bifunctional catalyst Pt/γ-Al2O3, unfortunately in this process a considerable degradation of cellulose is restricted.22 Hara et al. intensely studied a sulfonated activated carbon which nearly completely hydrolyzed cellulose into β-1,4-glucans at 100 °C.23,24 Onda et al. further demonstrated that sulfonated activated carbon can convert ball-milled cellulose (amorphous) into glucose with a maximum 41% yield.25 Sulfonated silica/carbon nanocomposites of varying acid density (0.15–0.93 mmol g−1) were developed for cellulose hydrolysis, however, the catalysts undergo deactivation by leaching of polycyclic aromatic hydrocarbon-containing –SO3H groups.26 Cellobiose and cellulose hydrolysis using nanosheets of niobium molybdate (HNbMoO6) have demonstrated the accessibility of saccharides into the strong acidic interlayer gallery of the solid, but only a low yield of glucose was achieved from cellulose, probably due to the low surface area (5 m2 g−1).27 Later, Zhang et al. employed a sulfonated mesoporous carbon for cellulose hydrolysis that results in a maximum 75% yield of glucose.28
It is understood from the outcomes of above referenced studies that a high cellulose-to-liquid ratio is important for large scale applications. However, to achieve a high yield of glucose, the process is usually conducted at a low cellulose-to-liquid ratio which means the process is energy-consuming, requiring the concentration of the product glucose solution for the production of ethanol. As for the practical processes for glucose production from renewable biomass, solid catalysts can be a viable alternative with which cellulose can be nearly completely converted except the residual lignin components and humin side-product formation.29,30 Furthermore, an analysis of the spent catalyst and solid residues may be of great importance for mechanistic development. Thus researchers have initiated biomass degradation/selective transformation using engineered solid catalysts with substantial surface modification with acidic functional groups to enhance selectivity and efficiency. Meso/nanoporous materials with uniform pore size distribution and functionalized pores with acidic functional groups (–SO3H, –COOH etc.) are the most promising candidates. For example, aluminosilicate MCM-41 (Mobil Composition of Matter) with pore diameter of 2–10 nm (Fig. 1), renders the processing of large organic molecules especially polymeric cellulose and starch present in wood-biomass feedstocks.31 Mesoporous materials can be applied, however, attempts have been made to improve the hydrothermal stability of these catalysts by stream treatment, as these systems must tolerate aqueous phase biomass conversion at high temperature by introducing noble and/or transition metals to enhance the oxygen removal capacity and/or promote decarboxylation reactions. Pyrolysis of wood-biomass in the presence of zeolite H-ZSM-5 (Fig. 1) has been studied in which acidic sites of the zeolite operate by a carbonium ion mechanism.31 H-ZSM-5 activated at 500 °C has predominantly Brønsted acid sites, however at higher temperatures, Lewis acid sites form. Mesoporous silica has been regarded as an ideal support for heterogeneous catalysts because of its high surface area and tunable pore size. Functionalization of mesoporous silica can be accomplished by using either post-synthesis grafting or co-condensation.32,33
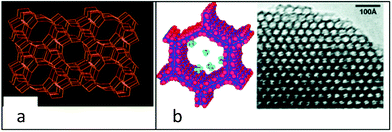 | ||
| Fig. 1 (a) Microporous H-ZSM-5 and (b) mesoporous aluminosilicate MCM-41. Small molecules are shown schematically in the pores. 2D array of hexagonal units of MCM-41 from HRTEM (reproduced from ref. 31 with permission, Copyright 2008, Wiley-VCH). | ||
Mesoporous materials with large surface area and pore volume, and narrowly distributed and tunable pore diameters of 2–10 nm have received great attention for their potential applications in adsorption,31,34 catalysis,35–37 microelectronics,38,39 and optical devices.40 Among the mesoporous silica materials, SBA-15 of 2D-hexagonal p6mm pore structure is particularly desirable because of its relatively large pore diameter and higher hydrothermal stability in comparison with that of MCM-41.41 Sulfonic acid-functionalized mesoporous silica materials have served as solid acid catalysts for fine chemical and biodiesel syntheses.42,43 However, the pore diameter of conventional SBA-15 is 5–8 nm, while the pore length is in the micrometer scale. Molecular diffusion and pore blockage in the long-channeling pores are the main concerns when applying these materials as catalysts for the conversion of bulky molecules.44,45
In this Minireview, the scopes of many surface modified solid catalyst and their potential applications for biomass transformations to platform chemicals have been discussed. Discussion is separated in different sections such as cellulose to sugar conversion, production of ethylene glycol, HMF and furfural by recently explored various materials as catalysts, including meso/nanoporous functionalized silica, porous metallo-organic frameworks, aluminosilicates (zeolites), functionalized zeolites, immobilized ionic liquids, and surface decorated carbon materials. The effect of meso/nanoporous channels on the diffusion of cellulosic materials while undergoing hydrolysis has been addressed with suitable examples. Particular emphasis is expended to explain how the biomass conversion depends on various factors related to catalysts, including the effect of the size of pore windows, Brønsted/Lewis acidic site ratios, composition of the solid, surface area and surface functionalization. Results obtained from application of these solid materials have been summarized such that a pre-requisite knowledge can be extracted for the rational design of new materials for selective biomass transformations, particularly, cellulose to glucose and platform chemicals. For the sake of length and cohesion, it is opted to highlight the factors involved in most of the recently reported solid acid catalyzed biomass transformation process, instead of covering a large number of materials that are already the subject of numerous reviews.
2. Cellulose hydrolysis
Cellulose is an important renewable feedstock for the production of biocomposites and biofuels.46 However, cellulose consists of linear glucose polymer chains that form a very tight hydrogen-bonded supramolecular network, and as such strongly resists chemical and enzymatic hydrolyses; further, access of reactants and catalysts to the biopolymer chain is restricted, and makes the substrate insoluble in conventional solvents. Disassembling of the supramolecular cellulose structure has been developed with ionic liquids which enhances the reactivity of cellulose by breaking of the glycosidic bonds, however, the recovery of these solvents poses a barrier without a practical solution.47 Therefore, there has been a growing interest to modify the cellulose structure in such a way that it can be transformed to valuable end-products via degradation. Replacement of ionic-liquid assisted cellulose degradation process with alternative methods is also a major target.48 Leading work by Schüth and co-workers16 has emphasized the efficient synergy between ionic liquids and acidic solid catalysts (e.g. Amberlyst®15) under mild conditions to hydrolyze cellulose to mono- and oligosaccharides. Success of this strategy lies on intrinsic Brønsted acid properties of sulfonic acid decorated Amberlyst®15. The optimum concentration of glucose is also an important factor especially for bioethanol production because concentration of the glucose solution is energy consuming and so effective treatment of high cellulose loadings with a suitable catalyst is desired. High cellulose/liquid ratio was successfully demonstrated by Jacobs et al. for hexitol production with heteropoly acid and Ru/C as catalyst by using concentrated cellulose.49 It was also demonstrated that depolymerization of cellulose into polyols takes place on noble50 and transition metals51 on carbon supports by combination of hydrolysis in hot water and hydrogenation under compressed hydrogen gas. The role of heterogeneous acids for the hydrolysis of cellulose has been intensely described in the past.10,52 However, significant development in this area has been witnessed recently using materials with mesoporous morphology along with large pore size and volume which offers controlled reaction and improved selectivity. It was demonstrated that ordered mesoporous materials upon functionalization with sulfonic acid groups act as excellent catalysts for hydrolysis.53 To further improve the strategy, if such solid acids can be made responsive to an external magnetic force, mechanical separation of catalyst would be an added advantage. Indeed, such materials already find wide applications in catalysis, biosensors and drug delivery.54 Fu and co-workers first demonstrated that high yields of glucose can be obtained from cellulose hydrolysis in aqueous medium by using a magnetic solid acid catalyst comprising of sulfonated mesoporous silica (SBA-15) and magnetic iron oxide (Fe3O4) nanoparticles (MNPs) that operates at a remarkably high cellulose/liquid ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 or 1:15).55 Template synthesis protocol of the mesoporous silica with its pores functionalized with sulfonic acid groups involves block copolymer P123 (P = pluronic) for co-condensation of tetraethoxysilane (TEOS) followed by oxidation of marcapto groups with hydrogen peroxide (Scheme 1).
:10 or 1:15).55 Template synthesis protocol of the mesoporous silica with its pores functionalized with sulfonic acid groups involves block copolymer P123 (P = pluronic) for co-condensation of tetraethoxysilane (TEOS) followed by oxidation of marcapto groups with hydrogen peroxide (Scheme 1).
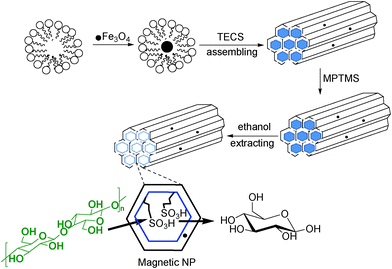 | ||
| Scheme 1 Design and structure of magnetic solid acid. | ||
By combining magnetic Fe3O4 nanoparticles (MNPs) and mesoporous SBA-15, the solid acids are not only easily separable under a magnetic field but also gain hydrothermal stability and perform even better than concentrated sulfuric acid. It is postulated that in this process, protons diffusing from the SBA-15 channels on the surface of cellulose undergo hydrolysis.56 Results of comparative studies revealed that sulfonated activated carbon (AC-SO3H) and macroporous resin Amberlyst-15 are less effective catalysts than Fe3O4-SBA-SO3H presumably due to the higher surface area of the latter. The superior catalytic activity of Fe3O4-SBA-SO3H has been further demonstrated for hydrolysis of cellobiose resulting in 96% glucose yield and, in the case of lignocellulosic biomass corncob, with a total reducing sugar yield of 45%.
Porous coordination polymers decorated with sulfonic acid groups can serve as effective catalysts for cellulose hydrolysis as recently demonstrated by Akiyama et al.57 The pore surface of a chromium oxide solid, composed of a terephthalate unit (MIL-101) framework, was fabricated with Brønsted acidic –SO3H groups and exhibited a large surface area (1915 m2 g−1) (Fig. 2).
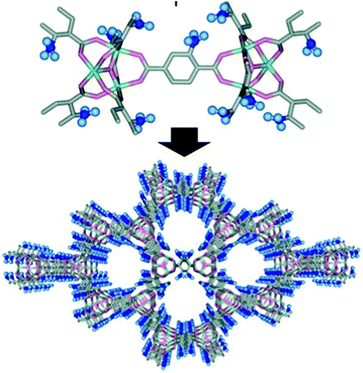 | ||
| Fig. 2 Structure of sulfonic acid functionalized porous coordination polymer for cellulose hydrolysis (reproduced from ref. 57, copyright 2011, Wiley-VCH). | ||
Cellulose hydrolysis using this –SO3H functionalized coordination polymer as catalyst produces xylose, glucose and cellobiose with increase in yield with reaction time. As realized, the advantage of using acid functionalized porous coordination polymer as catalyst is two-fold: (1) prevention of the formation of unfavourable side products such as 5-hydroxymethylfurfural, levulinic acid and formic acid, and (2) a water resistant and thermally stable catalyst that can perform at high temperature. Generally it should be disfavored for cellulose to diffuse deep inside the porous solid since the dimensions of cellulosic microfibrils are some hundred times larger than the average inner diameter of the pores of the framework solid (≈ 3 nm). Because most of the cellulose is crystallized and insoluble in water, it is assumed that only one end of the cellulose chain is accommodated in the pores and that the catalytic reaction takes place in the constricted conditions in the nanospace, resulting in a clean conversion. It is also arguable that the reaction efficiency depends on the contact time and frequency between substrates and catalytic sites when the micropores trap the substrate.15
Compared to resin-type or oxide-type solid acids, sulfonated carbons are more robust in hot water besides having preferable textural properties. By sulfonation at appropriate conditions, surface acidity and hydrophilicity/hydrophobicity may be tuned to facilitate the hydrolysis of cellulose. It was recently demonstrated that hydrolysis of cellulose strongly depends on the sulfonation temperature of the carbon material (activated carbon AC).58 A study demonstrated that sulfonation at 150–250 °C generates a high acid density (0.88 mmol g−1) without causing a decrease in specific surface area (412 m2 g−1). The correlation of acid density and catalytic performance can thus be made. When C-sulfonated CMK-3 (ordered mesoporous carbon) was employed as the catalyst, a glucose yield as high as 74.5% was attained. Comparative cellulose hydrolysis activity of sulfonated carbon materials can be arranged in the following order in terms of glucose yield: ACB < MWCNT < cell-carbon < CSAC < resin-carbon < CMK-3. Acid density and hydrophobicity of the carbon materials are the two important factors to be considered in order to explain the difference in activity for cellulose hydrolysis. Kinetic study of cellulose hydrolysis over the AC-N-SO3H-250 catalyst revealed a fast hydrolysis of cellulose into glucose in the initial stage, followed by a slow progress which supports that degradation may be inhomogeneous.
An excellent comparative study of the hydrolysis of cellulose over the solid acid catalysts including H-form zeolites with different Si/Al ratios, sulfated zirconia, ion-exchange resin and sulfated activated-carbon under hydrothermal conditions (423 K in water) revealed that zeolite catalysts with high Si/Al ratio, rSi:Al (H-beta (rSi:Al = 75), H-ZSM-5 (rSi:Al = 45)) are more highly active for glucose selectivity than zeolites with relatively low Si/Al ratio (H-beta (rSi:Al = 13) and H-mordenite (rSi:Al = 10)).59 This indicates the zeolite catalysts with high Si/Al ratios have high hydrophobic character, which may be a cause of the relatively high glucose yield. However, sulfated zirconia and Amberlyst-15 (high density of SO42− ions) exhibit higher activity than the H-form zeolites.
It is realized that very limited attention has been paid to solid acid catalysis for the depolymerization of cellulose in water under realistic conditions, i.e. temperatures below about 200 °C (to avoid secondary transformation of the sugars), with high cellulose to catalyst ratio and avoiding the use of energy-intensive pretreatment methods for cellulose, such as long-time ball-milling.58 Crystalline cellulose conversion is dependent on the efficient solid–solid interaction between the external surface of the catalyst and the crystalline cellulose, along with limitation of secondary reactions of the formed products.
When screening the performance of micro- and mesoporous solid acid catalysts (H-BEA, H-MOR, sulfated zirconia supported over mesoporous silica (SZ-SBA-15) and Amberlyst-15), it was revealed that the conversion of cellulose to glucose in aqueous medium at 190 °C is driven by the shape-selectivity effects of the macropores which also limits the polymerization of glucose to humin-type species.60 The comparative activity indicates inside pores acidity play a minor role for activity, but a more relevant role regarding the selectivity is played by the secondary reactions of glucose. To deconstruct cellulose selectively, a proper contact is required between the cellulose chains and surface acidic sites of the material, followed by diffusion of the depolymerization products inside the pores of the catalyst, leading to the formation of glucose from oligomers and its subsequent dehydration to HMF. Even with large channels as those present in mesoporous SBA-15, the internal accessibility is limited. In sulfated zirconia (SZ-SBA15) acidic sites are mainly located inside the channels of the mesoporous support but the bulky oligomers can hardly diffuse inside. Consequently this catalyst demonstrated weaker activity in terms of glucose yield. Sulfated zirconia over mesoporous SBA-15 can enhance the surface area and act as a potentially bifunctional catalyst with combined Brønsted and Lewis acidity. The strong Brønsted acidity created from sulfonation of supported zirconia is responsible for breaking the β-1,4-glycosidic bonds in cellobiose. However, with crystalline cellulose, poor performance indicates low degree of solid–solid interaction between the catalyst and the crystalline cellulose. In this case most of the acid sites are located inside the channels, where glucose may diffuse, but secondary polymerization is inhibited from shape-selectivity effects. This is probably the reason why SZ-SBA-15 shows a poor glucose yield. The larger channels in SBA-15 (≈6 nm) with respect to zeolites (H-BEA and H-MOR) do not limit these secondary reactions on glucose, leading to humin-type side-products.
In a recent study, CoFe2O4-embedded silica nanoparticles containing sulfonic acid groups were fabricated and found as highly active catalysts for hydrolysis of disaccharides (sucrose and cellobiose) and polysaccharides (starch and cellulose) with facile magnetic separation.61 The use of nanoparticles as solid catalysts may well overcome the above difficulties of solid-solid reactions. The nanoparticulate acid catalysts are expected to be dispersed in aqueous medium, resulting in facile interaction with cellulose. However, the use of nanoparticulate materials simultaneously suffers from the difficulty of catalyst recovery from the solution by sedimentation or filtration, though this could be solved by incorporation of magnetic particles into the catalyst. A 30% yield of total reducing sugars (TRS) with 7% glucose was obtained by using MNPs@SiO2SO3H as catalyst for a cellulose hydrolysis. Even though Amberlyst-15 afforded comparable results, MNPs@SiO2SO3H were found to be more effective in terms of turnover (3.8 vs. 0.4).
Amorphous carbon is a solid Brønsted acid containing flexible polycyclic carbon sheets with SO3H, COOH and phenolic hydroxyl (OH) groups in a 3D network that can be readily sulfonated to result in a strong acid capable of providing synergistic effect of both –SO3H and –COOH groups during the acid catalyzed hydrolysis of cellulose. Of note here is the –SO3H-bearing amorphous carbon incorporates a large amount of hydrophilic molecules, including water, into the bulk, which plays a major role in solid–solid interaction in the interface (Fig. 3).62 As a result, many parameters will influence the hydrolysis of cellulose and it is therefore difficult to optimize such a complicated reaction system. Artificial neural network (ANN) and a response surface methodology (RSM) approaches revealed that the amount of water is a dominant factor and that the hydrolysis rate increases with an increase in the amount of cellulose.63 Crystalline pure cellulose is not hydrolyzed by conventional strong solid Brønsted acid catalysts such as niobic acid, H-mordenite, Nafion and Amberlyst-15, and instead amorphous carbon bearing SO3H, COOH and OH proved more efficient. The apparent activation energy for the hydrolysis of cellulose into glucose using the carbon catalyst is estimated to be 110 kJ mol−1 much improved than that for sulfuric acid under optimal conditions (170 kJ mol−1).63 The catalytic performance of the carbon catalyst is attributed to the ability of the material to adsorb β-1,4-glucan. The interfacial hydrolysis of pure crystalline cellulose when in contact with surface functionalized carbon material lead to an increase in the amount of cellulose and decrease in the amount of water requiring distillation. Hydrolysis of cellulose using Brønsted acidic carbon material involves two stages: H+ attack of hydrogen and β-1,4-glycosidic bonds in crystalline cellulose to form water-soluble β-1,4-glucan, followed by hydrolysis of the β-1,4-glycosidic bonds in the β-1,4-glucan to form glucose. Efficient conversion of cellulose into glucose using a solid acid catalyst therefore requires a strong interaction between the solid acid and β-1,4-glucan because the Brønsted acid sites of the solid acid cannot approach the cellulose surface without such an interaction. This suggests that the glycosidic bond in β-1,4-glucan participates in the adsorption on the carbon material. The ability to adsorb β-1,4-glucan may be attributable to the high densities of hydrophilic groups (phenolic –OH) bonded to the graphene sheets.22 The effective surface area of the carbon material during hydrolysis is estimated to exceed 560 m2 g−1, much larger than the BET surface area after hydrolysis (only 2 m2 g−1) at water vapor pressure, suggesting that the large amounts of water can be incorporated into the carbon bulk due to the high density of the hydrophilic groups (–OH) bound to the flexible graphene domains. This incorporation provides good accessibility for cellulose chains in solution to the –SO3H groups giving rise to improved performance.64
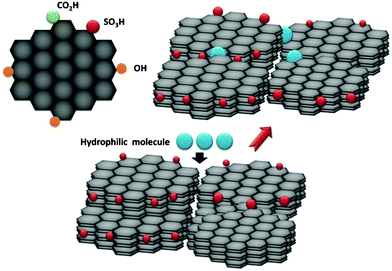 | ||
| Fig. 3 Schematic structure of carbon-based solid acid and incorporation of hydrophilic molecules into the carbon bulk. | ||
In another case, –SO3H-bearing amorphous carbon, prepared by partial carbonization of cellulose followed by sulfonation in fuming H2SO4, was applied for the acid-catalyzed hydrolysis of β-1,4-glucan, including cellobiose and crystalline cellulose.65 Structural analyses revealed that the resulting carbon material consists of graphene sheets with 1.5 mmol g−1 of SO3H groups, 0.4 mmol g−1 of COOH, and 5.6 mmol g−1 of phenolic OH groups. The catalytic performance of the carbon catalyst is attributed to the large adsorption capacity for hydrophilic reactants and the adsorption ability of β-1,4-glucan. This is distinct from hydrolysis in the homogeneous sulfuric acid system. The –SO3H-bearing carbon material, which is capable of adsorbing β-1,4-glucan, can thus be expected to exhibit higher activity for hydrolysis than other solid acids.
The strategy of applying cellulose derived acid was realized by our group; cellulose was hydrothermally treated with p-toluene sulfonic acid (TsOH) at 180 °C for aging and resulted in a sulfonic acid decorated carbon material (Cellu-TsOH) (Fig. 4) with surface area of 47.5 m2 g−1.66 This was found to be a promising candidate for catalyzing cellulose hydrolysis as found from an initial study.
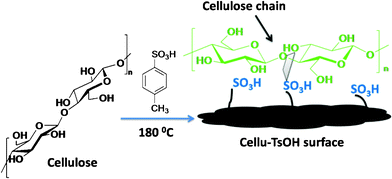 | ||
| Fig. 4 Cellu-TsOH obtained from cellulose and application in cellulose hydrolysis. | ||
A strategy to adsorb/attract cellulose on the catalyst surface and subsequent hydrolysis would be an ideal mimic to the enzymatic cellulase catalyzed cellulose hydrolysis. A solid catalyst with both cellulose-binding sites (–Cl) and catalytic sites (–SO3H) (Fig. 5) is a promising candidate which has been successful in lowering the apparent activation energy of the process (83 kJ mol−1) in comparison to that using sulfuric acid (170 kJ mol−1) as revealed in a recent study.67 Installation of an active site for an effective adsorption of cellulose chains on the surface of the solid catalyst was possible because of selecting a sulfonated polystyrene resin (CP-SO3H) containing chloromethyl groups even when containing a low density of acid sites (0.0033 mmol g−1). The chloride group (–Cl) acts as a cellulose-binding site because of its ability to form stronger hydrogen bonds with hydroxyl groups of cellulose and disrupt intra- and inter-hydrogen bonds of cellulose, thus enhancing the dissolution of cellulose.
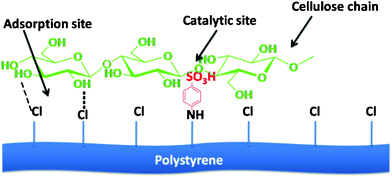 | ||
| Fig. 5 Cellulase-mimetic solid acid catalyst for cellulose hydrolysis via cleavage of glycosidic linkages. | ||
Cellulose adsorption on to the solid surface is an interesting phenomenon to study by tuning the surface with other hydrogen bond forming functional groups. Analysis of the microstructure of such materials would also be interesting. Moreover, the strategy of installation of combined H-bond forming sites and Brønsted acidic sites for improved hydrolysis rate of cellulose can be extended to nanomaterials with hierarchical structure, MOFs etc.
3. Bifunctional materials for cellulose conversion
Selective conversion of cellulose into high-value chemicals or fuels is of significant importance.68,69 Cellulose conversion to a desired polyol is challenging, however several studies on hydrolytic hydrogenation of cellulose to mixtures of sugar alcohols using a wide range of heterogeneous catalysts have mainly been reported, most of which lack selectivity.11,15 Mechanistically, selective cellulose conversion to sugar alcohols must include hydrolysis of cellulose into glucose, which is proposed to be catalyzed by an acid catalyst, followed by a metal catalyzed hydrogenation of glucose to a sugar alcohol (sorbitol, ethylene glycol etc.). Currently ethylene glycol (EG) is produced mainly from ethylene via the intermediate ethylene oxide in the petrochemical industry. The global production of EG in 2007 was estimated to be 17.8 million tonnes, up by 5.4% from 2006.70 In view of the importance and increasing demand of EG in the plastics industry for the manufacture of polyester fibers and resins, and in the automotive industry as an antifreeze, its production directly from cellulose will open a new way for reducing dependence on petroleum.Carbon-supported Ni-promoted W2C has been an efficient catalyst for the conversion of cellulose with a high selectivity towards ethylene glycol (EG) due to the synergistic effect between Ni and W2C.71 The introduction of Ni however causes severe sintering of W2C particles due to the methanation of the carbon support catalyzed by Ni during carburization, which would be detrimental to the conversion of cellulose. Addition of Ni to the W2C/AC facilitates the formation of the W2C phase at a lower temperature. On the other hand, it also induces significant sintering of W2C particles.72 Surface analysis indicate that the enhanced EG yield is at least partially due to weaker bonding between EG and Ni-promoted W2Cs. The problem of sintering of the effective W2C component of bimetallic Ni–W2C catalyst has been solved by improving the dispersion (number of atoms exposed to the surface) of active components of the catalyst by post-introduction of Ni which induces sintering resistance of the W2C crystallites.73 This result proved that partial dissolution of W in water or aqueous solutions of Ni-nitrate indeed occurred during the impregnation step. It was this dissolution of W that resulted in redispersion of particles. The originally large W2C particles became smaller due to dissolution starting from the outer surfaces of the particles. High CO uptake (chemisorption) by Ni–W2C/AC prepared by post-impregnation method led to considerably higher dispersion of the active components than that prepared by co-impregnation.
Zhang's group has developed a series of Ni–W bimetallic catalysts supported by mesoporous silica (SBA-15) which exhibited high activity and selectivity for EG (maximum 76.1%).74 In this process, W is found to be the key component for degradation of cellulose, i.e. C–C cracking. The advantage of this bimetallic catalyst resides on the ability to change the weight ratio of Ni and W to tune the competition between the hydrogenation and C–C cracking capabilities, so tuning the competition between hydrogenation and C–C cracking, and accordingly selectivity towards EG. In view of the importance of EG in the petrochemical industry, this approach may open a new avenue for the production of valuable chemicals from renewable resources.
Bifunctional materials based on nickel as a less expensive alternative metal catalyst for the transformation of cellulose into C6 sugar alcohols has been recently developed. Bifunctional nickel phosphide catalyst (16% Ni2P/AC (activated carbon), Ni2P/SiO2) was found to produce a maximum of 48% sorbitol under optimized reaction conditions (498 K, 6 MPa H2 at RT).75 Speciality of this catalyst lies in the feature of tuning the bifunctionality (number of acid sites of excess P vs. number of metallic sites of Ni2P) by varying the initial Ni/P ratio. This result also suggests that for obtaining maximum yield of sorbitol from cellulose, the relative number of acid sites and hydrogenation sites of the catalyst must match.
In comparison with conventional carbon materials (carbon nanotubes) and porous silica,76 aggregates of carbon nanofibers (CNFs) exhibit interesting properties, such as enhanced mechanical strength and filterability, high hydrothermal stability, controlled surface chemistry, and reduced mass transfer limitations associated with the presence of mesopores and a high surface area. Limited accessibility of active catalytic sites within the restricted space inside traditional pore systems, which prevents the polymeric biomolecules from penetrating to the metal sites has been a major barrier when using porous solid based catalysts due to the incompatibility between substrate and catalyst. Such obstacles can be overcome by using carbon nanofibers instead of porous solids as demonstrated for the conversion of cellulose over as-synthesized carbon nanofibers (CNFs) with nickel particles77 which contain Ni at the tips of the carbon filaments when formed by catalytic vapor deposition (CVD) of methane over nickel nanoclusters supported by γ-alumina (Fig. 6a). Fine tuning of nickel loading on the carbon nanofibers during the growth process leads to an improved yield of sugar alcohol which is comparable to that of Ni2P/AC materials for cellulose conversion (Fig. 6b and c). Acid-catalyzed hydrolysis by in situ produced H+ ions followed by rapid hydrogenation of glucose over supported Ni nanoclusters is a possible route for this conversion. A difference in function of the porous material and nanofibers for cellulose conversion lies in the fact that entangled threadlike carbon nanofibers around the waster-insoluble cellulose matrix promotes immediate hydrogenation of released glucose units due to the efficient accessibility of Ni particles attached at the tip of the nanofibers. It is still a matter of debate whether Brønsted acidity formed in water at high temperature (503 K) or intrinsic acid sites on the catalyst support material (γ-Al2O3) plays the major role in cellulose hydrolysis. It is the dispersion of Ni nanoparticles on the support (γ-Al2O3) that drives the growth of carbon nanofibers. Another interesting part of this study resides in understanding the growth mechanism of carbon nanofibers which unveils a larger proportion of the Ni(111) surfaces via crystallographic reconstruction of Ni nanoclusters.
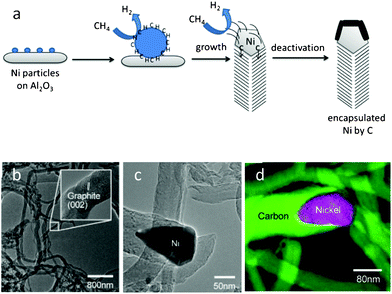 | ||
| Fig. 6 (a) Schematic representation of CNF growth by precipitation of angled graphene layers from a faceted catalyst particle to form a nanofiber. TEM images of CNF growth over Ni nanoclusters, (b) details of graphene layers in the nanofibers, (c) and (d) Ni particles at the tip of a CNF (reproduced from ref. 77, copyright 2010, Wiley-VCH). | ||
It is also interesting that sorbitol selectivity decreases significantly when CNF was used as support for a Ni/CNF for the conversion of cellulose under similar reaction conditions,77 however, effective anchoring of well-dispersed Ni particles also opens perspectives on tailoring the acid strength/concentration for improved catalytic activity during hydrolytic hydrogenation of cellulose. By using a Ni/CNF catalyst under optimal conditions a highest 76% yield of hexitol is obtained and catalytic results indicate that the balance between the hydrogenation (Ni loading) and the acid functions (density of Brønsted acid sites) is crucial for the desired selectivity in cellulose conversion, however, better selectivity can be achieved with Ni/CNF with low density of Brønsted acid sites.78 The advantage of using Ni/CNF is in the tuning of the dispersion of Ni particles by varied Ni loading and number of oxygen containing surface groups.
In order to perform effective cellulose hydrolysis under mild conditions (low activation energy), it is highly important that the bifunctional materials contain cellulose binding sites which enhance the substrate-adsorbing ability. To reduce the catalytic domain of a bifunctional material for the hydrolysis step, increase in Brønsted acid density might be an important factor, however, a low density of Brønsted acid sites is more effective in certain cases which further support for a balance between hydrolytic and hydrogenation sites of a bifunctional catalyst.
4. HMF production
HMF is one of the most important platform chemical for a range of polymer building blocks79–81 and biofuels82–85 because it contains two different functional groups, hydroxyl and aldehyde, which allows its chemical transformation to a variety of products via oxidation, hydrogenolysis and condensation.86,87 Several solid acids containing Lewis acidic sites88–92 and mesoporous materials93,94 are known to catalyze the dehydration of carbohydrates to HMF. Production of HMF from glucose faces several disadvantages because it involves expensive enzyme catalysis, numerous reactors and toxic ionic liquids. One-pot production of HMF from glucose or cellulose remains a challenge and towards a possible solution, several new catalytically active solid materials have been synthesized and investigated. Glucose is the most abundant monosaccharide to be used as feedstock for the large scale production of HMF.95 Conversion of glucose to HMF occurs via isomerisation of glucose to fructofuranose in the presence of enzymes or base catalyst followed by dehydration of fructose to HMF in the presence of acid catalyst (Scheme 2).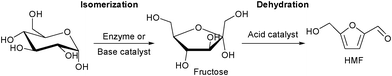 | ||
| Scheme 2 Reaction pathway for glucose conversion to HMF. | ||
Among recent approaches, Takagaki et al. have demonstrated that a combination of Amberlyst-15 (solid acid) and Mg–Al hydrotalcite (solid base) in N,N-dimethylformamide medium can be used to produce HMF from glucose with maximum 73% conversion and 58% HMF selectivity.96 To overcome the problems including separation costs, sensitivity to reaction conditions, and undesirable side-product formation, advanced methods have been developed. Recently, Nikolla et al. has reported a tin-containing high-silica molecular sieve with the zeolite beta topology (Sn-beta) which efficiently catalyzes the isomerisation of glucose to fructose in aqueous medium at low pH (≈1). The process involved a combination of Sn-beta with acid catalyst in a biphasic water/tetrahydrofuran reactor to produce HMF from glucose, cellobiose and starch.97 Long term recyclability of the robust Sn-beta zeolite catalyst was evident from morphology analysis (SEM) before and after the reaction which revealed that the crystal structure and pore volume of the catalyst is unaffected under the reaction conditions.
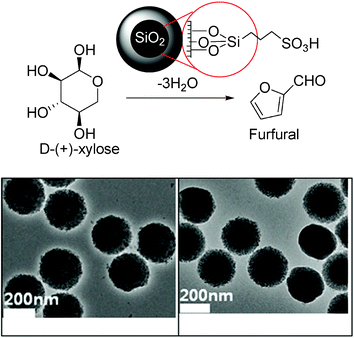
Scott and Dumesic have reported a bifunctional material 3-((3-(trimethoxysilylpropyl)thio)propane-1-sulfonic acid (TESAS) consisting of silane, incorporated into mesoporous SBA-15 type silica by co-condensation.98 Because of the better hydrophobicity, thioether-containing TESAS-SBA-15 showed improved activity (71% HMF selectivity at 84% conversion) than the sulfone derivative in the aqueous phase dehydration of fructose. Controlled hexagonal pore structure of TESAS-SBA-15 (Fig. 7) and particle morphology with minimal defects can result in better interfacial activity and impressive selectivity as revealed in this study.
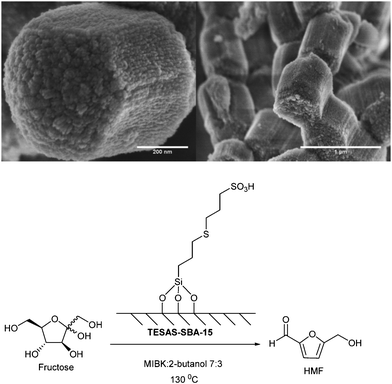 | ||
| Fig. 7 SEM images of TESAS-SBA-15 showing hexagonally shaped particles (reproduced from ref. 94, copyright 2011, American Chemical Society). | ||
Since metallorganic frameworks (MOFs) contain much larger pore diameter than zeolites and unprecedented surface areas, that allows inclusion of large molecules including drugs, dyes etc. in the pores.99–101 MIL-101 with cubic structure constructed from chromium carboxylate units in a corner-sharing fashion, is an ideal material for catalysis application because the pore windows are large enough (29 and 34 Å; pore volumes: 12700 and 20600 Å3) to allow access to large reactant molecules by diffusion into the pores and large product molecules can also easily move out of the pore network.101a Phosphotungstic acid (PTA, H3PW12O40) encapsulated MIL-101, a successful catalyst for the esterification of n-butanol with acetic acid in liquid phase, has been employed for the dehydration of carbohydrates.102 High crystallinity and sharp reflection of edges of the (PTA)x/MIL-101 materials (Fig. 8) reveals a homogeneous distribution of all elements. N2 adsorption–desorption analysis showed that total pore volume (1.47 cm3 g−1) and the BET surface area (2772 m2 g−1) decreases with increased loading of PTA in PTAx/MIL-101 and even at high PTA loading, the structure of the MOF is maintained. Of note, HMF selectivity for fructose dehydration improved consistently upon increasing the PTA loading in the MIL-101. This strategy demonstrates the utilization of free PTA confined in the nanospace of MIL-101 rather than using a metal salt of PTA.
 | ||
| Fig. 8 SEM images of MIL-101 and PTA (3.0)/MIL-101 (reproduced from ref. 102, copyright 2011, Wiley-VCH). | ||
A solid heteropolyacid Cs2.5H0.5PW12O40 catalyst has been employed in a biphasic (MIBK/water) medium for fructose dehydration to HMF with maximum 94.7% selectivity and the catalyst was tolerant to high concentration of feedstock (50 wt% fructose).103 The TOF range of a number of solid catalysts from a comparative study shows the order: Amberlyst-15 > sulfated-zirconia > Cs2.5H0.5PW12O40 >zeolite; higher HMF selectivity and fructose conversion were observed with Cs-PTA under relatively mild conditions at 30 wt% feedstock.
A combination of acid groups including sulfonic, carboxylic and phenolic contained in Glu-TsOH, derived from glucose and p-toluenesulfonic acid (TsOH), exhibited superior catalytic performance than Amberlyst-15 in spite of lower concentration of sulfonic groups and low surface area.104 It is possible that the presence of a variety of surface groups on Glu-TsOH can enhance the adsorption of fructose and at the same time does not influence desorption of the product HMF, resulting in a significantly higher yield of HMF (91.2%) in dimethyl sulfoxide (DMSO) at 130 °C after only 1.5 h and good reusability.
Among the recently developed strategies for the high yield production of HMF from carbohydrates supported ionic liquid catalysts have been employed. Nanoparticles of size in the range 290–630 nm have been prepared by immobilization of ionic liquid, 1-(triethoxysilylpropyl)-3-methylimidazolium hydrogen sulfate (IL-HSO4) on the surface of silica via covalent grafting through the reaction of the ethoxy groups with SiO2 surface silanols (Scheme 3).105 Investigation of fructose dehydration with nanoparticulate catalyst (SILnPs, d ≈ 610 nm) in DMSO showed a maximum conversion of 99.9% with 63% HMF yield.
 | ||
| Scheme 3 IL-HSO4 immobilization on silica nanoparticles. | ||
SEM and TEM images for SiO2 nanoparticles before and after IL immobilization (Fig. 9) revealed that morphologic homogeneity was maintained after grafting. However, particle coalescence may occur through the formation of interparticle necks possibly due to the presence of the IL covering the silica surface.
 | ||
| Fig. 9 SEM and TEM images of Si-2-IL-HSO4, respectively (reproduced from ref. 105, copyright 2011, RSC). | ||
The simultaneous effects of reaction temperature and catalyst amount on HMF yield over Si-IL-HSO4 NPs revealed that this catalyst is more effective than conventional solid acids such as the H-form of ZSM-5 and mordenite zeolites as well as Amberlyst-15, commercially available Dowex 50wx8-100 ion exchange resin, sulfonyl chloride (SiO2–SO2Cl) and sulfuric acid (SiO2–SO3H) modified silica gel.
As found recently, the reactivity of CrCl2 in [EMIM]Cl is related to the dynamic nature of the catalytic system.106 In such an environment, transient self-organization of CrII complexes into dimers promotes the glucose to fructose isomerization that determines the overall HMF selectivity. In this case, the mobility of the Cr complexes and the basic Cl sites are thought to be pivotal to explain the high HMF selectivity. Molecular level understanding of this successful homogeneous system forms the basis for the rational design of a solid catalyst for glucose dehydration. A supported Cr containing catalyst was reported by Degirmenci et al. which consists of molecular grafting of the ionic liquid propyl-3-methylimidazolium chloride (PMim+Cl−) moieties on the hydroxyl group on mesoporous silica SBA-15 resulting in Im-SBA-15 covered with (PMim+Cl−) moieties (Scheme 4).107 The CrCl2-Im-SBA-15 material shows coordination of CrCl2 to (PMim+Cl−), which is covalently grafted on the mesoporous silica support by creating a reaction environment on the surface that locally resembles the characteristics of the bulk ionic liquid and isolates the catalytic species (Cr2+) from excessive interaction with the solvent.
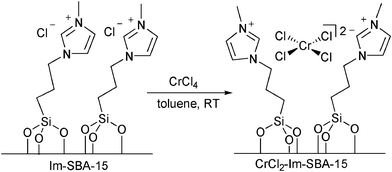 | ||
| Scheme 4 Construction of CrCl2-Im-SBA-15 shows coordination of CrCl2 to (PMim+Cl−). | ||
Results of this study suggests that introduction of CrCl2 into PMIm+Cl− immobilized on the surface of SBA-15 leads to the formation of loosely bound, catalytically active Cr2+ species. The resulting high mobility of these complexes is beneficial for selective dehydration. At the same time, the weak electrostatic binding of the reactive species results in the decomposition of the active phase of the catalyst in the presence of coordinating solvents such as water. In the presence of a biphasic 2-BnOH/MIBK (7:3) extraction layer, glucose dehydration by using CrCl2-Im-SBA-15 leads to a 70% HMF selectivity at 50% glucose conversion. The effectiveness of the IL-immobilized catalyst can be rationalised as the results are comparable with various homogeneous ionic liquid-mediated glucose conversions.104 A combination of Amberlyst-15 and hydrotalcite (anionic layered clay) afforded high HMF selectivity under moderate reaction temperature (Scheme 5).109 In this case, Amberlyst-15, a strongly acidic ion-exchange resin, functioned as acid catalyst with high selectivity and in the presence of a solid base hydrotalcite, isomerisation of glucose to fructose preferentially occurred, resulting in successive dehydration of fructose to HMF.
The successful strategy of immobilization of ionic liquid on a mesoporous silica SBA-15 or similar supports could be an excellent route for cellulose depolymerisation when the ionic liquid moiety is functionalized with Brønsted acidic sites.
As found recently, HMF yield can be enhanced significantly by the addition of organic solvent (MIBK) in dehydration of fructose over zeolites. However the HMF selectivity during the addition of MIBK is significantly increased in the order: H-MOR > ZSM-5 > BEA > amorphous aluminosilicate; which corresponds to the order of decrease in acid strength.110 In effect, the hydrophobic nature of zeolites should promote the adsorption of organic solvent (MIBK 0.24 g g−1) in the pores and thus results in formation of strong hydrogen bonds when the Brønsted acidic sites of zeolites strongly interact with the ketone group of adsorbed MIBK (Fig. 10a). It is expected that MIBK molecules inside of the pores should suppress oligomerization of HMF by displacement of HMF from the acid sites in the reaction medium and also by dilution of fructose and HMF inside the pores by organic solvent molecules. MIBK basically prevents the return of HMF into the pores of zeolite. The concept lies in using an inert compound that just adsorbs product HMF and prevents its transformation to oligomeric compounds. This modification was introduced to reduce the activity of the heterogeneous catalysts during fructose dehydration for the deposition of insoluble humins on the catalyst pores. Addition of organic solvent (MIBK) during the dehydration of fructose in the presence of solid acid promotes the deactivation of the external surface acid sites and catalytic testing of such samples with deactivated external acidity indicates that the main effect of organic solvent at high fructose conversion is due to the suppression of the external acidity of the zeolites. Upon addition of a large amount of MIBK, true selectivity of HMF formation inside the zeolite pores is expected. The HMF selectivity over H-MOR, H-ZSM-5, H-BEA and aluminosilicate Al2O3–SiO2vs. peak maximum temperature of TPD ammonia curves (Fig. 10(b)) describes the performance of the solid catalysts in the presence of MIBK.
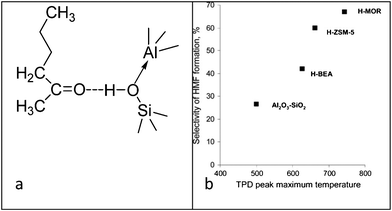 | ||
| Fig. 10 (a) Interaction of MIBK at the acidic sites of zeolite. (b) Selectivity of HMF formation over H-MOR, H-ZSM-5, H-BEA and aluminosilicate Al2O3–SiO2vs. peak maximum temperature of TPD ammonia curves (reproduced from ref. 110, copyright 2012, Elsevier). | ||
Lewis acidity of mesoporous TiO2 nanoparticles have been explored previously for glucose and fructose conversions to HMF by importing mesopores on the surface of the material when prepared by a soft-templating route using salicyclic93 and aspartic acid94 as templates. Recently, biopolymer templated porous TiO2 was found to be excellent catalyst for the unutilized hemicellulosic sugars (mannose, galactose, lactose) to HMF conversions in organic and water–organic biphasic solvents with maximum 45% HMF yield111 which is higher than the previously reported mannose conversion using chromium catalysts.112 High activity of porous TiO2 for the conversion of glucose epimer (mannose) is probably due to the biopolymer (alginate) templating that creates large mesopores in the sample with pore volumes of 0.44 cm3 g−1 and hydrothermally prepared particles contain larger pores of ca. 11.68 nm (Fig. 11a).113 Textural mesoporosity of this kind is responsible for the exceptionally high Lewis acidity of the spherical TiO2 particles (3.11 mmol g−1) (Fig. 11b) that is reflected in the catalytic activity.112
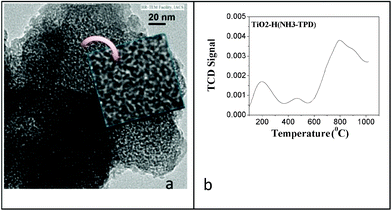 | ||
| Fig. 11 (a) HR-TEM images of typical nanostructure of TiO2-H showing the extensive porous particles (inset photo). (b) Temperature programmed desorption (TPD) of ammonia over mesoporous TiO2-H nanoparticles. | ||
A hierarchically macro/mesoporous titanium phosphate synthesized through slow evaporation using P123 as template has been found to be effective for the biomass conversion to HMF, however, the remarkable influence of hierarchical structure and framework acid sites on the biomass conversion was not studied previously.114 A wide pore aperture of this titanium phosphate material (∼7 nm) was very suitable for cellulose and lignocellulosic biomass conversion perhaps by accommodating glucan (shorter length cellulose) strands inside the active catalytic sites (Fig. 12).
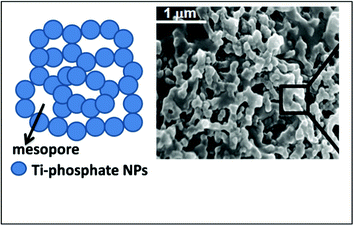 | ||
| Fig. 12 Hierarchically porous titanium phosphate nanoparticles and SEM image displaying the mesopores. | ||
The question that arises on the catalytic activity of hierarchically porous TiPO4 is whether macropores in the catalyst are truly favorable for improving the selectivity for HMF. Zeolites exhibit a low furfural selectivity due to the strong irreversible adsorption of the furfural in the micropores (0.028 cm−3g−1) which causes an increase in rate of humin formation in a pore confinement effect.115 Arguably, macropores of TiPO4 might also favor humin formation, however, this possibility has not been addressed yet, so as to perhaps evaluate the true effectiveness of the hierarchically porous catalysts containing macro- and mesopores. It can be speculated that in the presence of organic solvents, displacement of substrate and HMF product from the acid sites is highly probable which would prevent the return of HMF into the pores and subsequent oligomerization.
Brønsted and Lewis acidities of HPAs and related salts were found to be effective in catalyzing hydrolysis of polysaccharides including cellobiose and cellulose to glucose.116,117 However, by using HPA as catalyst, one-pot conversion of cellulose was not successful prior to a Brønsted–Lewis–surfactant-combined HPA catalyst Cr[(DS)H2PW12O40]3 (DS = OSO3C12H25 dodecyl sulfate) that enabled cellulose depolymerization and glucose conversion to HMF in a single process under mild conditions.118 Cr[(DS)H2PW12O40]3 is an amphiphilic material consisting of a hydrophilic head group (CrH2PW12O40) and a hydrophobic tail (OSO3C12H25) therefore, Cr[(DS)H2PW12O40]3 exhibits amphiphilic properties to assemble micelles in aqueous solution and accumulation of cellulose on HPA micelles. A remarkable HMF yield (52.7%) was obtained by using the HPA–micellar catalyst in purely aqueous phase cellulose conversion. This doubly acidic (Brønsted and Lewis) catalyst practically overcomes the diffusion problem in the solid–solid interaction and enhances the reaction rate by providing a hydrophobic environment for protecting HMF from decomposition, and prevents humin formation.
5. Furfural production
Furfural is a common lignocellulosic biomass derived compound with an annual production of more than 200000 tons.119 Furfural can be used as a feedstock to produce gasoline, diesel or jet quality fuel.120–122 Of particular interest is the selective conversion of furfural to a potential biofuel, 2-methylfuran (MF) in the gas or liquid phase which is related to its efficient production from xylose.121 Furfural is generally derived from C5 sugars, mainly xylose and arabinose, that are contained in the hemicelluloses of lignocellulosic materials. Lignocellulosic xylose is mainly present as glucuronoxylan, in addition to some xyloglucan, in hardwoods and as glucoronoarabinoxylan (GAX) in grasses. Xylose is generally produced as an aqueous stream. It would, therefore, be preferable to convert furfural in water, ideally at the concentration level delivered by the pretreatment. The dehydration of xylose to furfural in water typically proceeds at 150–220 °C with up to 60–70 mol% furfural yield under stripping conditions,123 due to the activation barrier of ∼30 kcal mol−1.124 Conventional furfural production relies on hemicellulose rich feedstocks such as corncobs or sugarcane bagasse using sulfuric acid catalyst and the process produces several co-products such as acetic acid, acetone and methanol in significant amounts.122 In addition, numerous studies have been reported on dehydration of xylose to furfural in various media using Lewis or solid acids as catalysts. For example, Moreau et al. reported xylose to furfural conversion using HY and H-MOR zeolites at 170 °C in biphasic water/toluene or water/MIBK.125 Under these conditions, the furfural yield is ∼30% when carried out in a single-phase system.126 Recently, alkylphenols were identified as effective extractants for furfural even at low extractant/oil ratio.127 Pure organic solvents, such as dimethylsulfoxide, also allow higher furfural yields without the benefit of the solvent extraction of furfural.128 It was found that addition of a solid base (hydrotalcite) to a solid acid (Amberlyst-15) brought down the xylose to furfural conversion temperature at <100 °C, although at the cost of loss of yield at temperatures >100 °C.129 Furfural and xylose can react together to form solid humins, which are polymerized into insoluble carbonaceous species. Furfural can also self-polymerize to form solid humins. In a biphasic regime, the furfural can be extracted into an organic solvent, and this step is governed by mass transfer. It would be desirable to obtain solid catalysts that exhibit high activity and selectivity comparable to homogeneous catalysts for aqueous-phase dehydration.
Many acid catalysts have been explored for dehydration of xylose,88 however, processes that are more in line with sustainable chemistry principles and of higher furfural selectivity need to be developed. The conversion of xylose to furfural proceeds in two steps by the isomerisation of xylose to xylulose, followed by xylulose dehydration to furfural.129 These steps parallel the conversion of hexoses, for example, the isomerization of glucose to fructofuranose followed by dehydration to 5-hydroxymethylfurfural (HMF). From the perspectives of biorefining, for solid materials, catalyzed furfural production in aqueous or aqueous/organic biphasic medium, different types of materials have been employed which can be categorized as solid acids with shape selectivity (e.g. aluminosilicates), supported strong acids (HPA on mesoporous silica), inorganic oxides or mixed oxides with tailored acid strength (surface decorated ZrO2), ion-exchange resins and layered structured materials (metal phosphates).130,131
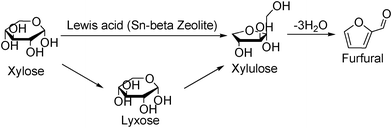
Earlier, Sn-beta zeolite was employed for the isomerization of glucose to fructose via 1,2-intrahydride transfer132 in which case the efficiency was comparable to biological catalysts.133,134 Sn-beta zeolite was employed for various reactions including reduction of carbonyl compounds,135 and production of lactate derivatives,136,137 with the active site containing partially hydrolyzed Sn–OH groups.138 When Sn-beta zeolite with micropores (0.16 cm3 g−1) with Sn/Si ratio 0.01 is combined with a Brønsted acid catalyst (HCl and Amberlyst-15), furfural was obtained from xylose in aqueous medium at much lower temperature (100 °C) and the analysis of the product mixture (xylulose 27%, lyxose 11% at ∼60% xylose conversion) suggests the Lewis acid catalyzes the isomerization of xylose to xylulose (Scheme 6).139 It was found that xylose does not react with Amberlyst-15 or HCl at low temperatures; however, when xylulose is the reactant, conversion is ∼66%, and furfural yield is 24%. This result supports a reaction network in which xylulose dehydrates rapidly to furfural via Brønsted-acid catalysis and xylose isomerizes to xylulose with a Lewis-acid catalyst, advocating for dual acidic sites in a catalyst. Formation of xylulose is a key step to furfural production and requires either functional group rearrangement or a configurational change around the C1 and C2 carbon atoms. Structural studies using X-ray absorption fine structure (EXAFS) technique reveals that Sn is substituted in pairs on opposite sides of six-membered rings, i.e. uniform crystallographic location of Sn in the β crystal structure, that leads to sites with uniform catalytic activity and high chemical selectivity (Fig. 13). The mechanistic evidence indicates that the active site of the catalyst interacts with the carbonyl group of C1 and the adjacent hydroxyl group on C2. Kinetic studies of isomerization reactions indicate that certain acids and metals are able to transfer the hydrogen directly through a hydride shift between C-2 and C-1.87 Lewis acidity of catalyst is essential to polarize the carbonyl group in the ketone while also coordinating both the alcohol and the ketone to facilitate a hydride shift between them.88 It is therefore plausible that Sn in zeolite Beta performs the isomerization reaction via an intramolecular hydride shift between the carbonyl-containing C-1 and the hydroxyl-bearing C-2 of glucose by way of a five-membered complex. Important factors in the Sn-Beta isomerization of glucose in aqueous media include the role of the solvent, the confinement and polarity effects within the micropores of the zeolite, and the impact of the coordination state of the Sn atom in the framework as either partially hydrolyzed framework Sn centers (–Si–O–)3Sn–OH or fully framework coordinated Sn atoms Sn(–Si–O–)4.
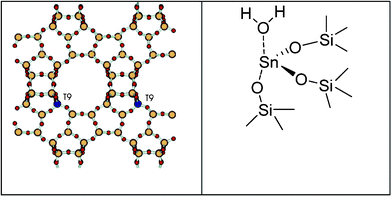 | ||
| Fig. 13 Topology of Sn-beta zeolite showing T9 positions (reproduced from Fig. 1 of ref. 138 with permission, copyright 2006, American Chemical Society) and (b) proposed active site structure of the material for isomerization of xylose. | ||
The use of conventional microporous zeolites as catalysts for xylose dehydration is compromised by diffusion limitations inherent in the relatively narrow pore dimensions. One possible solution could be delamination of layered precursors of zeolites. Valente and co-workers demonstrated that a delaminated zeolite with Si/Al = 29 is effective as a catalyst in a water/toluene biphasic reactor system at 170 °C. While comparing the results with that H-MOR (Si/Al ∼6), a higher yield of furfural (47%) was obtained with aluminosilicate as catalyst prepared from lamellar precursor (Nu-6(1)) compared to that with the H-MOR, possibly due to the six to seven times higher specific surface area of the material derived from the layered zeolite (Nu-6(1)) and a greater number of acid sites.140 A similar correlation was observed for the cyclodehydration of xylose in the presence of layered titanate, niobates and titanoniobates, and the corresponding exfoliated-aggregated materials with higher SBET.141 The material, designated as del-Nu-6(1), is a promising alternative to conventional zeolites or mesoporous materials for the production of furfural. Moreover, del-Nu-6(1) is considerably more selective to furfural at 40–90% xylose conversion. The catalytic efficiency of del-Nu-6(1) is mainly attributed to the easier accessibility of its active sites. The catalytic performance may be further improved by optimizing the Si/Al ratio and the delamination procedure. However, an important drawback is the formation of coke, which may lead to a drop in furfural selectivity at high conversions. Reducing the crystallite size of the catalyst to the nanoscale would increase the surface area to micropore volume ratio and decrease the diffusion path lengths, which may avoid severe coking. On the other hand, the reduction in crystallite size may alter the Lewis/Brønsted acid sites ratio on which stability of intermediate species and furfural selectivity would depend.142
Aqueous phase cyclodehydration of xylose at 170 °C was carried out with a composite material consisting of zeolite beta (BEA) nanocrystals (Si/Al = 12) embedded in a pure siliceous TUD-1 mesoporous matrix (BEATUD).143 Furfural yields at high xylose conversion (98%) was higher with BEATUD (74%) than with bulk BEA (54%) as the former contains a lower amount of carbonaceous matter and favorable adsorption effect caused by the surrounding silica matrix. Fine tuning of Si/Al ratio, might change the total amount of acid sites and catalyst surface polarity, and variation in zeolite loading may influence the dispersion and the number of accessible acid sites of the zeolite. In this way, catalytic performance for composites of the type BEATUD can be controlled.
Carbonaceous materials are promising catalysts due to their high surface area which may provide adequate catalytic active sites. An excellent candidate of this class would be sulfonated graphene oxide (SGO) that has been demonstrated as a rapid and water-tolerant carbocatalyst that performs at a low catalyst loading at 200 °C with an average yield of 61% of furfural from xylose.144 Surface area analysis and reaction results suggests that the aryl sulfonic acid groups are the key active sites for high temperature production of furfural in water. The material SGO exhibited a sheet-like appearance, despite the presence of oxygen-bearing and –SO3H functional groups that might disrupt the sp2 carbon network in SGO.
Ion-exchange membrane Nafion 117, a sulfonated tetrafluoroethylene based fluoropolymer-copolymer, as a robust and reusable catalyst is promising in terms of economical furfural production as this possesses excellent chemical and thermal stability under the xylose dehydration conditions.145 Deprotonation of the sulfonic acid groups of Nafion 117 would deactivate the catalyst by reducing the number of available acid sites for xylose dehydration, and organic residue deposition on the surface of the Naflon is also responsible for deactivation. Porous siliceous materials with solid silica core and porous silica shell can enhance the hydrothermal stability of mesoporous silica and were investigated for the dehydration of D-xylose in water. Modified mesoporous shell silica (MSHS) (Fig. 14) beads when modified with sulfonic acid performs as an efficient catalyst for dehydration of xylose with higher furfural selectivity than the unmodified aluminosilicate.146
 | ||
| Fig. 14 Mesoporous silica beads with solid core and mesoporous shell for catalytic cyclodehydration of xylose to furfural (reproduced from ref. 146, copyright 2011, Elsevier). | ||
The result of a recent study showed sulfated tin oxide (SO42−/SnO2) exhibited superior catalytic activity. The density of SO42− groups on SnO2 plays an important role in producing furfural from xylose by governing the catalyst performance.147 Furthermore, gas-phase characterization of acid sites can be used to predict catalytic activity and it was revealed from the comparative study of catalytic activity of acid catalysts, including Zr–P, SiO2–Al2O3, WOX/ZrO2, γ-Al2O3 and HY zeolite, that catalyst selectivity is a function of the Brønsted to Lewis acid sites.148 Comparative study showed that the Lewis acid sites decrease furfural selectivity by catalyzing a side reaction between xylose and furfural to form humins (insoluble degradation products). At 20% xylose conversion, catalysts with high Brønsted to Lewis acid ratios, such as Zr–P, exhibit furfural selectivity as much as 30 times higher than catalysts with higher Lewis acid sites. Dehydration reactions using ion-exchange polymer resins with high Brønsted acid sites showed similar selectivities to that of Zr–P and HCl. HY zeolite showed low furfural selectivity due to strong irreversible adsorption of the furfural in the pores, causing an increase in the rate of humin formation. Crystalline microporous silico-aluminophosphates (SAPO), with Brønsted and Lewis acidity are effective catalysts, and result in 58–65% furfural yield along with 100% xylose conversion.149 Considering that the “catalytic pore sizes” of zeolites are often found to exceed the crystallographic ones by as much as 2 Å in the case of H-MOR, the xylose molecules may be able to diffuse into the channels of all three framework types, under the reaction conditions used for catalysis. Indeed, according to the literature, the 8.6 Å glucose molecule is able to diffuse into the water filled 7.4 Å Y-zeolite pore.150 In the liquid phase, the solute diffuses as a solute–solvent assemblage and catalyst–solvent interactions may reduce the effective diffusivity of xylose within the liquid-filled pores of the SAPO materials. Diffusion may also be facilitated at higher temperatures. Smaller crystallite/particle sizes may enhance the overall reaction rate by increasing the number of accessible acid sites and decreasing the intracrystalline diffusion path lengths. To probe the existence of such effects, the catalytic activities of two SAPO-11 materials possessing different particle sizes have been compared, and the differences in furfural selectivities may be partly due to differences in the Lewis/Brønsted ratio. The SAPO systems are promising and comparable with H-MOR zeolite (Si/Al ∼6); further for a specific SAPO system the catalytic performance may be fine-tuned, based on the effects of the preparation method, crystallinity, morphology, silicon content and acidity on the catalytic performances. Such studies may also provide insights into the factors that influence the target vs. undesired reaction pathways. In semi-batch conditions, sulfonic ion-exchange resin Amberlyst-70 was used as catalyst for the xylose dehydration to furfural with furfural stripping using nitrogen.151 Constant liquid–vapor equilibrium along with stripping could be maintained for different xylose loadings. In this semi-continuous stripping, nitrogen as stripping agent led to the formation of two visible water–furfural phases. Indeed, increased furfural efficiency at 200 °C could be due to the reason that when temperature is increased that leads to (ΔG ~ 0) resulting negligible polymerization and fragmentation which increases the furfural selectivity.
The effectiveness of the Lewis acidic Sn4+ center for the isomerization of xylose to xylulose in furfural production indicates in future more efficient Sn-containing materials may be employed. A feature that needs to be studied in such reaction catalyzed by Sn-materials is the dependence of humin formation on the morphology of the material. Systematic study on this is absent in the literature, except for the report by Ordomsky et al.,110 which discusses the order of the decrease in the strength in the acid sites and its influence on the HMF selectivity with different zeolitic materials.
In order to understand the humin formation mechanism more clearly, it could be useful to employ a single catalytic material of different morphology depending on the preparation methods, and compare how the humin formation depends on the textural property of the material by carrying out analytical studies of the product mixture of the dehydration process. Despite the many new developments described above, the cost and energy expense of furfural production and recovery still requires significant improvement by the use of efficient solid catalysts and superior extracting media.
6. Humin formation
The catalyst selectivity is a function of the Brønsted to Lewis acid site ratio for both the heterogeneous and homogeneous reactions. Lewis acid sites decrease furfural selectivity by catalyzing a side reaction between xylose and furfural to form humins (insoluble degradation products). Similarly, loss of product yields in cellulosic biomass transformation to HMF is associated with the formation of dark solid humins. In the case of acid-catalyzed hydrolysis of cellulose, humins that form have not been characterized nearly as thoroughly, probably because the focus has been on improving the selectivity to desired products such as HMF, LA etc. Chemistry of humin formation is poorly understood and polymerization of furanic compounds were thought to be the reason for humin formation.152,153 Elemental composition of humins (55–65% C, 4–5% H and 30–40% O)154,155 and scanning electron microscopy (SEM) images (Fig. 15) reveals agglomerated spherical particles with a broad distribution of diameters (5–10 μm) from gas-phase components due to the degradation of reactants/products in a batch process.146 | ||
| Fig. 15 SEM image of insoluble humins (reproduced with permission from ref. 155, copyright RSC, 2006). | ||
Sievers et al.156,157 found strong similarities in a cross-polarization magic-angle spinning (CP MAS) 13C NMR spectral pattern between the starting cellulose and the humins derived via ionic liquid catalyzed hydrolysis, supporting that monosaccharides are a primary components of humins and also other species must be involved in their formation. Earlier, it was suggested that polymerization of HMF and furfural is responsible in the formation of humins. In aqueous phase fructose dehydration catalyzed by AlCl3·6H2O, the brown materials obtained were found to be partially soluble in DMSO-d6, and the 1H NMR spectrum obtained was similar to that of HMF.158 Sievers et al. has found that the particle morphology of the humins changes with the HMF conversion in a acid catalyzed and hydrothermal conversion (Fig. 16).159
 | ||
| Fig. 16 SEM image of humins recovered after 2 h (90% HMF conversion) catalyzed by 0.1 M sulfuric acid (a), and after hydrothermal conversion at 135 °C for 49 h (45% HMF conversion) (b) (reproduced with permission from ref. 156, copyright 2009, American Chemical Society). | ||
From the experimental results Patil and Lund have postulated that humin growth involves reaction between 2,5-dioxo-6-hydroxyhexanal and HMF including acid-catalyzed aldol condensation.29 Two extreme cases of humin formation mechanism have been postulated based on the IR spectral and DFT results that showed humin retains the furan ring and hydroxymethyl group of HMF but that the carbonyl group of HMF is not present supporting humin growth via aldol addition/condensation of HMF and 2,5-dioxo-6-hydroxyhexanal. In order to explain humin generation during acid-catalyzed conversion of carbohydrates, important points to consider would be (a) possible aldol addition/condensation of 2,5-dioxo-6-hydroxyhexanal with HMF resulting in humin without carbonyl groups and (b) 2,5-dioxo-6-hydroxyhexanal condensing with HMF leading to products which are less reactive toward further addition/condensation of another HMF. Equilibrium geometry and vibrational frequency calculation of IR spectral results proved that the carbonyl group of HMF was eliminated in the aldol addition/condensation in the product and the actual humin growth process could fall anywhere between the two extremes.160 Experimental findings suggest that humin formation occurred through condensation polymerization of sugars (glucose) with the products of their dehydration (HMF) (Fig. 17)159 which propagates with the increase in water concentration of the medium leading to inhibition by water solvation of protons, making free protons less available for the protonation of the oxygen atoms of the carbonyl groups of HMF.28
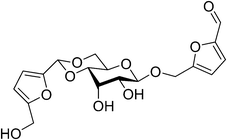 | ||
| Fig. 17 Proposed humin formation via condensation polymerization of glucose and HMF. | ||
The humin formation mechanism might be an interesting topic for further exploration as it is rather important to investigate the factors responsible for detrimental humin formation in the case of solid material catalyzed biomass conversions. Whether the crystalline nature of the material suppresses the humin formation is an important fundamental question which needs to be addressed by a systematic study. It is also realized that the presence of Lewis and Brønsted acidic sites are necessary for the xylose conversion to furfural via xylulose pathway. For example, materials with mesoscopic void spaces (interparticle voids) and substantial textural mesoporosity may be ideal candidates to explore their potential for sugar conversion to platform chemicals and associated humin formation. Although the humin formation mechanism may be easier to study in the homogeneous phase using mineral acid as catalyst, studies should be extended to the more relevant solid acid catalysts for which the formation mechanism of humin has not yet been explored experimentally nor any theoretical studies reported.
7. Factors influencing catalytic effectiveness
Diffusion limitations
For a porous solid catalyzed transformation in aqueous medium, multilayers of water are incorporated in pores via sequential diffusion. Results suggest contributions of hydrogen bonds between the adsorbate–substrate and adsorbate–adsorbate interactions, and the predominance of the former might be responsible for single-molecule migration on the surface. These molecules are entrapped at higher coordination sites in pores when compared the activity with nonporous materials as catalysts.161 It was found that accumulation of solvents, e.g. phenol inside the pores of ZSM-5 crystals, is considered the major cause of deactivation due to strong adsorption and slow diffusion. However, the sole presence of micropores can impose diffusion limitations due to the restricted access and slow mass transport to and from active sites located within the micropores. This phenomenon represents the major drawback in cellulose conversions catalyzed by acid functionalized zeolites. To improve this situation, formation of “hierarchical” structures in acid catalysts could be interesting for further study. For example, hierarchically structured zeolites integrate at two levels of porosity, i.e. micro- and mesopores. Design aspects of such hierarchically porous materials would be the focus for future study.161 The extent to which these ordered pores of different widths influence the selectivity of biomass conversions requires further evaluation.Hydrophobic/hydrophilic environment
Surface hydrophobicity and hydrophilicity of the solid are two important factors that determine the potential of the material as a catalyst for an aqueous phase reaction. Cellulose degradation is a solid–solid interactions at the initial stages of the reaction and key catalytic steps of adsorption of feed cellulose and desorption of product depend strongly on these properties. Biomass transformation involves hydrophilic and hydrophobic molecules. Therefore, tuning of the surface properties is also necessary to optimize the adsorption and desorption processes and thus the overall catalytic transformation. Cellulose contains hydrophilic components and during conversion to intermediates or to liquid fuels, the oxygen content will decrease in successive steps and the hydrophobicity of the substrate will increase after a number of steps. These transformations should take advantage of hydrophilic surfaces, as they can facilitate the approach of the substrates and desorption of the less hydrophilic products. For example, interaction between catalyst and cellulose was facilitated in the case of micellar catalyst Cr[(DS)H2PW12O40]3 containing an anionic surfactant compared to its non-micellar form Cr[H2PW12O40]3.119 The Lewis acidity of Cr[(DS)H2PW12O40]3 originates from the metal cation as an electron pair acceptor, and Brønsted acidity is generated from protons of HPAs. The concentration of cellulose is accumulated compared to the surrounding water phase through interactions with the micelle surface or through insertion into the micelle itself containing a hydrophilic head group (CrH2PW12O40) and hydrophobic tail (OSO3C12H25) by overcoming the insolubility of cellulose in water as described in Fig. 18.![Micellar activity of Cr[(DS)H2PW12O40]3.](/image/article/2012/RA/c2ra20922e/c2ra20922e-f18.gif) | ||
| Fig. 18 Micellar activity of Cr[(DS)H2PW12O40]3. | ||
Development of new water-tolerant solid acids with tailored porosity and adjusted surface polarity could impact on biomass transformation at an industrial scale, however, there is high probability of poisoning of acid sites by water.162 Results also suggest that zeolite catalysts H-beta (rSi:Al = 75) and H-ZSM-5 (rSi:Al = 45) with high Si/Al ratios have high hydrophobic character, which may be a cause of the relatively high glucose yield.
Solvent effect
Effect of organic solvent addition in aqueous dehydration of hexose and pentose to platform chemicals is closely associated with the acid strength of the material employed as catalyst. It has been demonstrated that selectivity of HMF formation over zeolites significantly increases upon MIBK addition which is attributed to the suppression of humin formation due to the filling of the pores of zeolite with MIBK and resulting desorption of the product HMF.107 Dehydration of fructose proceeds through cyclic intermediates, which might also be very reactive and contribute in the transformation into humins as well. In the case of fructose dehydration in aqueous media, with the formation and desorption of HMF and its subsequent transformation into oligomeric products, the selectivity should continually decrease from the beginning of the experiment. Considering the diffusion limitation of fructose molecules into the pores of H-ZSM-5 (5 Å) and H-MOR (6.5–7 Å),163 HMF selectivity will not differ to a considerable extent due to the very small difference of the pore size. Since the partition ratio for HMF between water and MIBK at reaction temperature is about 0.8 (volume ratio of MIBK to water = 3:1), the amount of HMF present in the organic phase is 2.5 times higher than the amount in water. This should lead to a decrease in the rate of HMF condensation in the aqueous phase. The hydrophobic properties of the zeolites in comparison with amorphous aluminosilicate should promote adsorption of organic solvent in the pores.164 Ordomsky's study showed amorphous aluminosilicate adsorbs just a small amount of MIBK, whereas that in zeolites BEA, MOR and ZSM-5 corresponds to the total pore volume of that in zeolites.110 Molecules of MIBK inside of the pores should suppress oligomerization of HMF by displacement of HMF from the acid sites into the reaction medium and by dilution of fructose and HMF inside of the pores by organic solvent molecules. Absorption of HMF by MIBK prevents the return of HMF molecules in the pores of zeolite and thus prevents oligomerization, resulting in an increase in selectivity of the process. Interaction of organic solvent with hydrophilic amorphous aluminosilicate without micropores does not allow the removal of HMF from the surface. The decrease in the conversion of fructose over H-MOR with the addition of MIBK not only indicates a strong interaction of this solvent with the acid sites of the catalyst but also an increase in the residence time of a zeolite crystal in the organic solvent when amount of organic solvent is increased. The maximal effect of MIBK addition over H-MOR should be attributed to the highest strength of acid sites in H-MOR, which also leads to the highest selectivity without the addition of MIBK. Interaction of organic solvent with the strong acid sites of H-MOR should be more intense and lead to a higher retention time of solvent in the pores in comparison with H-BEA and H-ZSM-5. Ordomsky's observations indicate that MIBK continues to interact with catalyst. Such an interaction should be especially intense with acid sites with a higher accessibility such as on the surface of the zeolite crystal. The effect of other organic extracting solvents (2-sec-butylphenol, 4-propylguaiacol) can also be examined for mesoporous solid catalysts. However, the extracting agent should be an inert compound that simply absorbs formed HMF and prevents it transformation into oligomeric side-products. Extensive studies with other organic solvent in biphasic media will be interesting to reveal many yet unknown facts. In addition, it would be highly interesting to investigate surface decorated metal organic frameworks with acidic sites for such studies, especially MOFs with narrow pore windows.
Lewis and Brønsted acid sites
To design more efficient aqueous-phase dehydration catalysts, a high ratio of Brønsted to Lewis acid sites is desirable as revealed from the comparative aqueous phase dehydration of xylose using a series of solid catalyst with varied Brønsted to Lewis ratio.111 It is important to examine the role of Lewis and Brønsted sites of solid acid catalysis for aqueous dehydration of carbohydrates for the optimal design of solid catalysts. In the study of Huber et al. it was found that the catalysts with the highest number of Lewis acid sites were found to be the most active and on a per site basis, the catalyst activity decreases as follows, γ-Al2O3 ∼ WOx/ZrO2 > SiO2–Al2O3 ∼ HY > Zr–P ∼ HCl.115 Furfural selectivity is increased in the same order, indicating Brønsted sites are responsible for controlled reaction and suppression of by-product formation. A higher rate of xylose disappearance and lower furfural selectivity has been observed for catalysts with increased Lewis sites and in addition, furfural selectivity is a direct function of the Brønsted acid sites concentration, and not necessarily the nature of the acid sites. This hypothesis is further supported by the fact that dehydration reactions with acidic ion-exchange polymer resins which contain only Brønsted acidic sites show comparable furfural selectivity with Zr–P and HCl. Similarly, NH3-TPD studies showed exceptionally highly acidic sites for a TiO2 material obtained via biopolymer templating. However, using the same material as catalyst, only moderate yields of HMF from sugar derivatives (glucose epimers) has been obtained.108The significance of the joint action of Lewis and weak Brønsted acid sites was further demonstrated by a carbon-mesoporous silica MCM-41 based composite material (CSM) containing Lewis acidity provided by grafting Sn(IV) with the silica component and weak Brønsted acidity originating from oxygen containing functional groups (CO2H, CO, OH) in the polyaromatic to graphite-like carbon network inside the pores of silica (Fig. 19).165 The advantage of using this material in cascade reactions for sugars lies in independently tunable Brønsted (B) and Lewis acid (L) sites (B/L ratio) and readily tunable pore texture. Composite materials with well-balanced Lewis/Brønsted sites and tunable Brønsted acidity have been successful in producing alkyl lactates and lactic acid selectively from sugars in organic and in aqueous media, respectively. In this material intraporous carbon is decorated with various surface functional groups based on the loadings and synthetic techniques which exhibit mild Brønsted acidity. Selective production of alkyl lactate from hexoses is achieved by tailoring the pore size of CSMs by controlling the number of Brønsted acid sites (ratio of B/L).
 | ||
| Fig. 19 Bifunctional catalyst containing tunable Lewis and Brønsted acid sites; B = Brønsted site, L = Lewis site. | ||
Dispersion of the catalyst
Because of the improved dispersion of active components, selective conversion of cellulose to EG can be achieved with considerable yield improvement, as has been demonstrated for Ni promoted W2C catalysts.73 This depends on modifications in the preparation method of the catalyst. In this case, a controlled Ni loading has been achieved. Redispersion of W2C crystallites took place at the impregnation step, undergoing a partial dissolution during impregnation. and the originally large W2C particles became smaller due to dissolution starting from the outer surfaces of the particles.8. Summary and outlook
This review has highlighted materials which are efficient for selective conversion of biomass to platform chemicals. The key technologies using solid catalysts for the large scale production of chemicals and fuels from biorenewable substrates can be constructed by understanding of the structure and reactivity pattern of catalysts which have had their own share of success towards this goal. Study of their reactivity pattern from a mechanistic point of view will certainly improve our understanding to develop better catalysts that operate in aqueous medium and at low temperature. We would like to list a number of investigations yet to be done in the field of biomass conversion using solid materials. To this list, first must be the control of solid humin formation by using highly porous materials with pores decorated with Brønsted acidic sites (e.g. –SO3H groups) and when using such materials with different pore widths varied systematically, how the humin growth rate varies and what factors dominate the selectivity of the end-products. The second aspect is the influence of the particle morphology of the catalytic materials on the selectivity. For example, what factor will decide selectivity between materials with mesoporous or macro/mesoporous combined texture for cellulose conversion, i.e. which performs better for a selective conversion. This requires systematic study of a specific material with different texture or particle morphology.Groundwork for the production of cellulose constituent sugars via selective hydrolysis in aqueous medium with chemocatalysts is an active area of research and the field might benefit most from systematic studies to further elucidate the fundamental factors that govern the performance of meso/nanoporous solid catalysts. Conceptually, it is clear that a high acid density, β-glucan affinity, good accessibility of strong acid sites and a high surface area should increase the performance of hydrolysis catalysts, and at the same time selectivity of hydrolysis is a primary issue to obtain glucose as the major product from cellulose, and this depends on the pore size and pore volume and functional groups of the catalyst. Systematic studies to investigate the individual importance of different factors are thus highly important for creating a complete picture. Detailed study on the diffusion phenomena of certain biomolecules in the porous structures and the rational design of porous catalysts should help in this endeavor.
Selective hydrolysis of cellulose for glucose production can still be rather enigmatic; less water slows hydrolysis, but too much water also slows the rate of hydrolysis, supposedly because of hydration of the acid sites, though this remains to be proven. Examples showed that the role of water in cellulose hydrolysis is also not fully understood. This should be further investigated and if possible exploited to develop highly active and selective catalysts for large-scale industrial applications. Hybrid surface structures constituted by interpenetrated silica and carbon components, facilitating the adsorption of β-1,4-glucan on the solid catalyst as observed in the case of sulfonated silica/carbon nanocomposites, should be investigated further. Heteropoly acids have been found as active and selective for cellulose conversions,113,166,167 but their water solubility prevents them from being industrially feasible catalysts. Several authors reported the incorporation of HPAs into materials such as zirconia, activated carbon, MCM-41, silica or MOFs.168 In water however; these materials exhibit significant leaching of the HPA, even at modest temperatures. Another approach might be the use of insoluble salts of HPAs.169 If these issues can be solved, heteropoly acids could be very powerful catalysts for cellulose hydrolysis.
Efforts to convert cellulosic biomass to base and intermediate chemicals for biofuels synthesis have been successful to a considerable extent in recent years. In the next decade, we expect to see more processes that employ highly advanced catalytic systems and pretreatment methods to convert lignocellulosic biomass from a wide range of sources into fuels, base chemicals and fine chemicals. We are moving to a renewable and more sustainable economy but further effective development is necessary. Furfural upgrade technologies still require improvement to produce cost-competitive biofuels, as conventional hydrolysis of lignocellulose still proceeds with low yields (∼10 wt% or 50–60% of the theoretical value), and high demand for energy. More investigations toward the one-pot conversion of xylose to furfural and 2-methylfuran is necessary for which only limited developments have thus far been possible using conventional catalysts. Towards this, we have recently started our program on production of DMF and 2-MF from algal and lignocellulosic biomass using supported catalysts in a “one-pot” process,83,170 and we hope to see promising results in the near future.
Promising development on heterogeneously catalyzed conversion of cellulose into polyols and platform chemicals has received intensive attention over the past few years and breakthroughs have been achieved therein to a limited extent. It might be appropriate to conclude that heterogeneously catalyzed conversion of cellulose and lignocellulose to hydrolyzed products with selectivity and further conversion to platform chemicals remains in its infancy until there are significant developments in bifunctional or multifunctional material catalysis. Recent advances in carbon nanofiber based materials, surface functionalized MOF and tunable Lewis–Brønsted acid grafted mesoporous materials etc. hold promise for further possible advances. The introduction of interesting multifunctional materials for direct conversion of cellulose into promising liquid fuels in the near future would certainly lead to advancement of research at the interface of materials and biomass conversion towards a new level.
Acknowledgements
The author gratefully acknowledges all researchers whose excellent contributions have enriched the field of heterogeneously catalyzed biomass conversions chemistry discussed in this review. The author gratefully thanks to United States–India Educational Foundation (USIEF), NewDelhi and Council for International Exchange of Scholars (CIES), Washington, DC for financial support through a Fulbright–Nehru Postdoctoral Fellowship.References
- M. Balat and H. Balat, Appl. Energy, 2009, 86, 2273–2282 CrossRef CAS.
- U. Chakravartya, B. Magne and M. Moreaux, Journal of Economic Dynamics and Control, 2008, 32, 1181–1203 CrossRef.
- T. W. Simpson, A. N. Sharpley, R. W. Howrath, H. W. Parel and K. R. Mankin, J. Environ. Qual., 2008, 37, 318–324 CrossRef CAS.
- O. R. Inderwildi and D. A. King, Energy Environ. Sci., 2009, 2, 343–346 CAS.
- K. Nakashima, K. yamaguchi, N. Taniguchi, S. Arai, R. yamada, S. Katahira, N. Ishida, H. Takahashi, C. Ogino and A. Kondo, Green Chem., 2011, 13, 2948–2953 RSC.
- M. Imhoff, L. Bounoua, T. Ricketts, C. Loucks, R. Harriss and W. Lawrence, Nature, 2004, 429, 870–873 CrossRef CAS.
- G. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- P. Mäki-Arvela, B. Holmbom, T. Salmi and D. Murzin, Catal. Rev. Sci. Eng., 2007, 49, 197–340 Search PubMed.
- M. E. Himmel, S. Y. Ding, D. K. Johnson, W. S. Adney, M. R. Nimlos, J. W. Brady and T. D. Foust, Science, 2007, 315, 804–807 CrossRef CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS.
- S. Van de Vyver, P. A. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef CAS.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- P. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS.
- D. Alonso, J. Bond and J. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- J. A. Gedoers, S. Van De Vyver, R. Ooms, B. O. de Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 Search PubMed.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS.
- C. Z. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177–182 RSC.
- C. Z. Li and Z. K. B. Zhao, Adv. Synth. Catal., 2007, 349, 1847–1850 CrossRef CAS.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516–4521 CrossRef CAS.
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC.
- Y. C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 CAS.
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS.
- M. Kitano, D. Yamaguchi, S. Suganuma, K. Nakajima, H. Kato, S. Hayashi and M. Hara, Langmuir, 2009, 25, 5068–5075 CrossRef CAS.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- (a) S. Van de Vyver, L. Peng, J. Geboers, H. Schepers, F. de Clippel, C. J. Gommes, B. Goderis, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1560–1563 RSC; (b) Y. Y. Wu, Z. H. Fu, D. L. Yin, Q. Xu, F. L. Liu, C. L. Lu and L. Q. Mao, Green Chem., 2010, 12, 696–700 RSC.
- (a) A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363–5365 RSC; (b) A. Takagaki, C. Tagusagawa, S. Hayashi, M. Hara and K. Domen, Energy Environ. Sci., 2010, 3, 82–93 RSC.
- F. Pang, A. Q. Wang, M. Y. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- S. J. Dee and A. T. Bell, ChemSusChem, 2011, 4, 1166–1173 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- M. Stöcker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef.
- X. Fang, G. E. Frywell, L. Q. Wang, A. Y. Kim, J. Liu and K. M. Kemner, Science, 1997, 276, 923–926 CrossRef.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- J. M. Sun, H. Zhang, R. J. Tian, D. Ma, X. H. Bao, D. S. Su and H. F. Zou, Chem. Commun., 2006, 1322–1324 RSC.
- A. Corma, Chem. Rev., 1997, 97, 2373–245 CrossRef CAS.
- S. Y. Chen, J. F. Lee and S. Cheng, J. Catal., 2010, 270, 196–205 CrossRef CAS.
- S. Y. Chen, C. Y. Tang, J. F. Lee, L. Y. Jang, T. Tatsumi and S. Cheng, J. Mater. Chem., 2011, 21, 2255–2265 RSC.
- S. Baskaran, J. Liu, K. Domansky, N. Kohler, X. Li, C. Coyle, G. E. Frywell, S. Thevuthasan and R. E. Williford, Adv. Mater., 2000, 12, 291–294 CrossRef CAS.
- C. M. Yang, A. T. Cho, F. M. Pan, T. G. Tsai and K. J. Chao, Adv. Mater., 2001, 13, 1099–1102 CrossRef CAS.
- M. H. Bartl, S. P. Puls, J. Tang, H. C. Lichtenegger and G. D. Stucky, Angew. Chem., Int. Ed., 2004, 43, 3037–3040 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- J. Melero, R. V. Grieken and G. Morales, Chem. Rev., 2006, 106, 3790–3812 CrossRef CAS.
- I. K. Mbaraka, D. R. Radu, V. S. Y. Lin and B. H. Shanks, J. Catal., 2003, 219, 329–336 CrossRef CAS.
- G. D. Cremer, M. B. J. Roeffaers, E. Bartholomeeusen, K. Lin, P. Dedecker, P. P. Pescarmona, P. A. Jacobs, D. E. D. Vos, J. Hofkens and B. F. Sels, Angew. Chem., Int. Ed., 2010, 49, 908–911 CrossRef.
- K. Lin, P. P. Pescarmona, K. Houthoofd, D. Liang, G. V. Tendeloo and P. A. Jacobs, J. Catal., 2009, 263, 75–82 CrossRef CAS.
- T. Arioli, L. Peng, A. S. Betzner, J. Burn, W. Wittke, W. Herth, C. Camilleri, H. Hofte, J. Plazinski, R. Birch, A. Cork, J. Glover, J. Redmond and R. E. Williamson, Science, 1998, 279, 717–720 CrossRef CAS.
- (a) N. Sun, H. Rodr_guez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405–1421 RSC; (b) A. Stark, Energy Environ. Sci., 2011, 4, 19–32 RSC.
- A. El Seoud, A. Koschella, L. C. Fidale, S. Dorn and T. Heinze, Biomacromolecules, 2007, 8, 2629–2647 CrossRef.
- J. Geboers, S. Van de Vyver, K. Carpentier, K. de Blochouse, P. Jacobs and B. F. Sels, Chem. Commun., 2010, 46, 3577–3579 RSC.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef CAS.
- R. Palkovits, Chem. Ing. Tech., 2011, 83, 411–419 CrossRef CAS.
- W. M. Van Rhijn, D. E. De Vos, B. F. Sels, W. D. Bossaert and P. A. Jacobs, Chem. Commun., 1998, 317–318 RSC.
- S. M. Zhu, Z. Y. Zhou and D. Zhang, ChemPhysChem, 2007, 8, 2478–2483 CrossRef CAS.
- D.-M. Lai, L. Deng, Q.-X. Guo and Y. Fu, Energy Environ. Sci., 2011, 4, 3552–3557 CAS.
- D. Lai, L. Deng, J. Li, B. Liao, Q. Guo and Y. Fu, ChemSusChem, 2011, 4, 55–58 CrossRef CAS.
- G. Akiyama, R. Matsuda, H. Sato, M. Takata and S. Kitagawa, Adv. Mater., 2011, 23, 3294–3297 CrossRef CAS.
- J. Peng, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC.
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC.
- P. lanzafame, D. M. Temi, S. Perathoner, A. N. Spadaro and G. Centi, Catal. Today, 2012, 179, 178–184 CrossRef CAS.
- A. Takagaki, M. Nishimura, S. Nishimura and K. Ebitani, Chem. Lett., 2011, 40, 1195–1197 CrossRef CAS.
- M. Hara, Energy Environ. Sci., 2010, 3, 601–607 CAS.
- D. Yamaguchi, M. Kitano, S. Suganuma, K. Nakajima, H. Kato and M. Hara, J. Phys. Chem. C, 2009, 113, 3181–3188 CAS.
- M. Okamura, A. Takagaki, M. Toda, J. N. Kondo, T. Tatsumi, K. Domen, M. Hara and S. Hayashi, Chem. Mater., 2006, 18, 3039–3045 CrossRef CAS.
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, Solid State Sci., 2010, 12, 1029–1034 CrossRef CAS.
- S. De, S. Dutta and B. Saha, unpublished results.
- L. Shuai and X. Pan, Energy Environ. Sci., 2012, 5, 6889–6894 CAS.
- B. Kamm, Angew. Chem., Int. Ed., 2007, 46, 5056–5058 CrossRef CAS.
- M. R. Klaas and H. Schone, ChemSusChem, 2009, 2, 127–128 CrossRef CAS.
- ICIS Chemical Business (Weekly), 352 (2008) July 28.
- (a) N. Ji, T. Zhang, M. Y. Zheng, A. Q. Wang, H. Wang, X. D. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang, Y. Shu, A. L. Stottlemyer and J. G. Chen, Catal. Today, 2009, 147, 77–85 CrossRef CAS.
- N. Ji, M. Zheng, A. Wang, T. Zhang and J. G. Chen, ChemSusChem, 2012, 5, 939, DOI:10.1002/cssc.201100575.
- M.-Y. Zheng, A.-Q. Wang, N. Ji, J.-F. Pang, X.-D. Wang and T. Zhang, ChemSusChem, 2010, 3, 63–66 CrossRef CAS.
- L.-N. Ding, A.-Q. Wang, M.-Y. Zheng and T. Zhang, ChemSusChem, 2010, 3, 818–821 CrossRef CAS.
- (a) W. Deng, X. Tan, W. Fang, Q. Zhang and Y. Wang, Catal. Lett., 2009, 133, 167–174 CrossRef CAS; (b) J. K. Chinthaginjala, K. Seshan and L. Lefferts, Ind. Eng. Chem. Res., 2007, 46, 3968–3978 CrossRef CAS.
- S. V. D. Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. V. Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef.
- S. V. D. Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549 CrossRef.
- O. Casanova, S. Iborra and A. Corma, ChemSusChem, 2009, 2, 1138–1144 CrossRef CAS.
- B. Saha, S. Dutta and M. M. Abu-Omar, Catal. Sci. Technol., 2012, 2, 79–81 CAS.
- S. Dutta, S. De and B. Saha, ChemPlusChem, 2012, 77, 259–72 CrossRef CAS , and references therein.
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- S. De, S. Dutta and B. Saha, ChemSusChem, 2012 DOI:10.1002/cssc.201200031.
- M. I. Alam, S. Dutta, S. De and B. Saha, RSC Adv., 2012, 2, 6890 RSC.
- S. Saravanamurugan, O. N. Van Buu and A. Riisager, ChemSusChem, 2011, 4, 723–726 CrossRef CAS.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2009, 11, 1327–1331 RSC.
- X. H. Qi, M. Watanabe, T. M. Aida and R. L. Smith, ChemSusChem, 2009, 2, 944–946 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith Jr., Catal. Commun., 2009, 10, 1771–1775 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 6276–6278 RSC.
- S. De, S. Dutta, A. K. Patra, A. Bhaumik and B. Saha, J. Mater. Chem., 2011, 21, 17505–17510 RSC.
- S. Dutta, S. De, A. K. Patra, M. Sasidharan, A. Bhaumik and B. Saha, Appl. Catal., A, 2011, 409–410, 133–139 CrossRef CAS.
- A. I. Torres, P. Daoutidis and M. Tsapatsis, Energy Environ. Sci., 2010, 3, 1560–1572 CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Etibani, Chem. Commun., 2009, 6225–6227 Search PubMed.
- E. Nikolla, Y. Román-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- A. J. Crisci, M. H. Tucker, M.-Y. Lee, S. G. Jang, J. A. Dumesic and S. L. Scott, ACS Catal., 2011, 1, 719–728 CrossRef CAS.
- (a) P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS; (b) P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS.
- (a) Z. Wang, G. Chen and K. L. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS; (b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC; (c) L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- (a) J. Kim, S. Bhattacharjee, K. E. Jeong, S. Y. Jeong and W. S. Ahn, Chem. Commun., 2009, 3904–3906 RSC; (b) A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC; (c) C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883–1888 CrossRef CAS; (d) N. V. Maksimchuk, M. N. Timofeeva, M. S. Melgunov, A. N. Shmakov, Y. A. Chesalov, D. N. Dybtsev, V. P. Fedin and O. A. Kholdeeva, J. Catal., 2008, 257, 315–323 CrossRef CAS.
- Y. Zhang, V. Degirmenci, C. Li and E. J. M. Hensen, ChemSusChem, 2011, 4, 59–64 CrossRef CAS.
- Q. Zhao, L. Wang, S. Zhao, X. Wang and S. Wang, Fuel, 2011, 90, 2289–2293 CrossRef CAS.
- J. Wang, W. Xu, J. Ren, X. Liu, G. Lu and Y. Wang, Green Chem., 2011, 13, 2678–2681 RSC.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- E. A. Pidko, V. Deirmenci, R. A. van Santen and E. J. M. Hensen, Angew. Chem., Int. Ed., 2010, 49, 2530–2534 CrossRef CAS.
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef CAS.
- M. E. Zakrzewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Chem. Rev., 2011, 111, 397–417 CrossRef CAS.
- M. Ohara, A. takagaki, S. Nishimura and K. Ebitani, Appl. Catal., A, 2010, 383, 149–155 CrossRef CAS.
- V. V. Ordomsky, J. Van der Schaaf, J. C. Schouten and T. A. Nijhuis, J. Catal., 2012, 287, 68–75 CrossRef CAS.
- S. De, S. Dutta, A. K. Patra, B. S. Rana, A. K. Sinha, B. Saha and A. Bhaumik, Appl. Catal., A, 2012, 435–436, 197, DOI:10.1016/j.apcata.2012.06.002.
- J. P. Binder, A. V. Cefali, J. J. Blank and R. T. Raines, Energy Environ. Sci., 2010, 3, 765–771 CAS.
- S. Dutta, A. K. Patra, S. De, A. Bhaumik and B. Saha, ACS Appl. Mater. Interfaces, 2012, 4, 1560–1564 CAS.
- A. Dutta, A. K. Patra, S. Dutta, B. Saha and A. Bhaumik, J. Mater. Chem., 2012, 22, 14094, 10.1039/C2JM30623A.
- R. Weingarten, G. A. Tompsett, W. C. Conner Jr. and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itayab and A. Satsumaa, Green Chem., 2009, 11, 1627–1632 RSC.
- J. Tian, J. H. Wang, S. Zhao, C. Jiang, X. Zhang and X. H. Wang, Cellulose, 2010, 17, 587–594 CrossRef CAS.
- S. Zhao, M. Cheng, J. Li, J. Tian and X. Wang, Chem. Commun., 2011, 47, 2176–2178 RSC.
- J. B. Binder, J. J. Blank, A. V. Cefali and R. T. Raines, ChemSusChem, 2010, 3, 1268–1272 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- J.-P. Lange, E. Van der Heide, J. Van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- A. Corma, O. De La Torre, M. Renz and N. Villandier, Angew. Chem, Int. Ed., 2011, 50, 2375–2378 CAS.
- K. J. Zeitsch, The Chemistry and Technology of Furfural and its many By-Products, Elsevier, Amsterdam, 2000. Search PubMed.
- I. Agirrezabal-Telleria, A. Larreategui, J. Requies, M. B. Guemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS.
- C. Moreau, R. Durand, S. Razigade, J. Duhamet, P. Faugeras, P. Rivalier, P. Ros and G. Avignon, Appl. Catal., A, 1996, 145, 211–224 CrossRef CAS.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- E. Gürbuz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef.
- A. S. Dias, M. Pillinger and A. A. Valente, J. Catal., 2005, 229, 414–423 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Lett., 2010, 39, 838–840 CrossRef CAS.
- R. Karinen, K. Vilonen and M. Niemela, ChemSusChem, 2011, 4, 1002–1016 CrossRef CAS.
- S. Dutta, S. De, B. Saha and M. I. Alam, Catal. Sci. Technol., 2012 10.1039/C2CY20235B.
- Y. Roman-Leshkov, M. Moliner, J. A. Labinger and M. E. Davis, Angew. Chem., Int. Ed., 2010, 49, 8954–8957 CrossRef CAS.
- M. Moliner, Y. Roman-Leshkov and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6164–6168 CrossRef CAS.
- E. N. E. Nikolla, Y. Roman-Leshkov, M. Moliner and M. E. Davis, ACS Catal., 2011, 1, 408–410 CrossRef CAS.
- A. Corma, M. E. Domine, L. Nemeth and S. Valencia, J. Am. Chem. Soc., 2002, 124, 3194–3195 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS.
- E. Taarning, S. Saravanamurugan, M. S. Holm, J. M. Xiong, R. M. West and C. H. Christensen, ChemSusChem, 2009, 2, 625–627 CrossRef CAS.
- M. Boronat, A. Corma and M. Renz, J. Phys. Chem. B, 2006, 110, 21168–21174 CrossRef CAS.
- V. Choudhary, A. B. Pinar, S. I. Sandler and D. G. Vlachos, ACS Catal., 2011, 1, 1724–1728 CrossRef CAS.
- S. Lima, M. Pillinger and A. A. Valente, Catal. Commun., 2008, 9, 2144–2148 CrossRef CAS.
- A. S. Dias, S. Lima, D. Carrazo, V. Rives, M. Pillinger and A. A. Valente, J. Catal., 2006, 244, 230–237 CrossRef CAS.
- H. D. Mansilla, J. Baeza, S. Urzua, G. Maturana, J. Villasenor and N. Duran, Bioresour. Technol., 1998, 66, 189–193 CrossRef CAS.
- S. Lima, M. M. Antunes, A. Fernandes, M. Pillinger, M. F. Ribeiro and A. A. Valente, Appl. Catal., A, 2010, 388, 141–148 CrossRef CAS.
- E. Lam, J. H. Chong, E. Majid, Y. Liu, S. Hrapovic, A. C. W. Leung and J. H. T. Luong, Carbon, 2012, 50, 1033–1043 CrossRef CAS.
- E. Lam, E. Majid, A. C. W. Leung, J. H. Cong, K. A. Mahmoud and J. H. T. Luong, ChemSusChem, 2011, 4, 535–541 CrossRef CAS.
- G. H. Jeong, E. G. Kim, S. B. Kim, E. D. Park and S. W. Kim, Microporous Mesoporous Mater., 2011, 144, 134–139 CrossRef CAS.
- T. Suzuki, T. Yokoi, R. Otomo, J. N. Kondo and T. Tatsumi, Appl. Catal., A, 2011, 408, 117–124 CrossRef CAS.
- R. Weingarten, G. A. Tompsett, W. C. Conner Jr. and G. W. Huber, J. Catal., 2011, 279, 174–182 CrossRef CAS.
- S. Lima, A. Fernandes, M. M. Antunes, M. Pillinger, F. Ribeiro and A. A. Valente, Catal. Lett., 2010, 135, 41–47 CrossRef CAS.
- R. Netrabukkana, K. Lourvanij and G. L. Rorrer, Ind. Eng. Chem. Res., 1996, 35, 458–464 CrossRef CAS.
- I. Agirrezabal-Telleria, A. Lerreategui, J. Requies, M. B. Guemez and P. L. Arias, Bioresour. Technol., 2011, 102, 7478–7485 CrossRef CAS.
- F. S. Asghari and H. Yoshida, Ind. Eng. Chem. Res., 2007, 46, 7703–7710 CrossRef CAS.
- R. Hashaikeh, I. S. Butler and J. A. Kozinski, Energy Fuels, 2006, 20, 2743–2747 CrossRef CAS.
- B. Girsuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- C. Sievers, M. B. Valenzuela-Olarte, T. marzialetti, I. Musin, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2009, 48, 1277–1286 CrossRef CAS.
- C. Sievers, I. Musin, T. Marzialetti, M. B. V. Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2, 665–671 CrossRef CAS.
- S. De, S. Dutta and B. Saha, Green Chem., 2011, 13, 2859–2868 RSC.
- R. Weingarten, J. Cho, W. C. Conner and G. W. Huber, Green Chem., 2010, 12, 1423–1429 RSC.
- R. Souda, Surf. Sci., 2011, 605, 1257–1262 CrossRef CAS.
- D. Verboekend and J. Perez-Ramirez, Catal. Sci. Technol., 2011, 1, 879–890 CAS.
- Y. Ogihara, R. L. Smith Jr., H. Inomata and K. Ara, Cellulose, 2005, 12, 595–606 CrossRef CAS.
- C. Moreau, R. Durand, C. Pourcheron and S. Razigade, Ind. Crops Prod., 1994, 3, 85–90 CrossRef CAS.
- G. S. Haegh, Stud. Surf. Sci. Catal., 1985, 24, 605–609 CrossRef CAS.
- F. D. Clippel, M. Dusselier, R. V. Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 120608112610009, DOI:10.1021/ja301678w.
- K. Shimizu, H. Furukawa, N. Kobayashi, Y. Itaya and A. Satsuma, Green Chem., 2009, 11, 1627–1632 RSC.
- R. Palkovits, K. Tajvidi, A. Ruppert and K. Procelewska, Chem. Commun., 2011, 47, 576–578 RSC.
- B. Devassy and S. Halligudi, J. Catal., 2005, 236, 313–323 CrossRef CAS.
- J. Geboers, S. Van de Vyver, K. Carpentier, P. Jacobs and B. Sels, Green Chem., 2011, 13, 2167–2174 RSC.
- S. Dutta, S. De and B. Saha, unpublished results.
| This journal is © The Royal Society of Chemistry 2012 |