A facile synthesis of oxyavicine†
Ravi
Jangir
and
Narshinha P.
Argade
*
Division of Organic Chemistry, National Chemical Laboratory (CSIR), Pune 411 008, India. E-mail: np.argade@ncl.res.in
First published on 5th July 2012
Abstract
Starting from dimethyl 3,4-methylenedioxyhomophthalate, a concise total synthesis of oxyavicine has been described via the base catalyzed monoalkylation, homophthalimide formation, oxidative selective iodination, regioselective reductive dehydration and an intramolecular Heck coupling reaction followed by the oxidation pathway with 16% overall yield in six steps.
A large number of bioactive benzo[c]phenanthridine alkaloids are known in the literature and this important class of natural products possesses various practical biological activities (Fig. 1).1 To date, several novel synthetic approaches to these imperative alkaloids have been known in the literature.2 Oxyavicine has been isolated from Zanthoxylum avicennae, Broussonetia papyrifera and Zanthoxylum nitidum plant species.3 Oxyavicine is known to exhibit analgesic and anti-inflammatory activity, and it is also useful for the treatment of ophthalmic disorders.3 Oxyavicine has been synthesized by several research groups utilizing an array of elegant synthetic methodologies.4 In continuance of our cyclic anhydrides/imides to bioactive natural products studies; we reasoned that the homophthalimides would be the potential building blocks for the synthesis of the benzophenanthridine class of alkaloids.5 In this context we herein report our results on the synthesis of oxyavicine (Scheme 1).
![Representative benzo[c]phenanthridine alkaloids.1,2](/image/article/2012/RA/c2ra20915b/c2ra20915b-f1.gif)
 | ||
| Scheme 1 Convergent access to oxyavicine via an intramolecular Heck coupling reaction. | ||
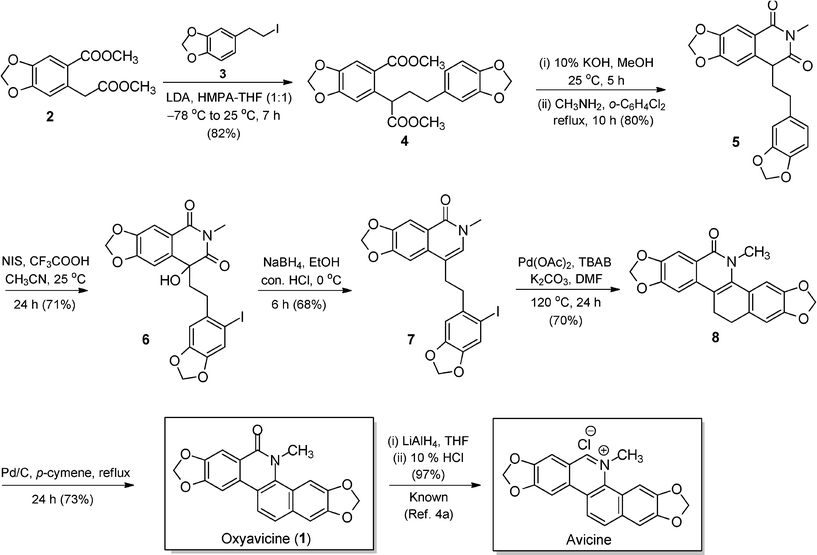
Methyl 6-(2-methoxy-2-oxoethyl)benzo[d][1,3]dioxole-5-carboxylate (2) on base catalyzed alkylation with 5-(2-iodoethyl)benzo[d][1,3]dioxole (3) exclusively furnished the desired mono-alkylated coupling product 4 in 82% yield.6 The potassium hydroxide induced hydrolysis of diester 4 provided the corresponding dicarboxylic acid. The obtained dicarboxylic acid was treated with an aqueous solution of methyl amine to form the corresponding salt. Then the excess of amine and water were removed under vacuum and the obtained salt was dried using vacuum pump. The thermal intramolecular cyclization of the above specified salt in refluxing o-dichlorobenzene provided the N-methylhomophthalimide 5 in 80% yield. At this stage we planned to introduce the iodine at an appropriate position in an aryl part of an alkyl unit in imide 5 for the Heck coupling reaction.7 We avoided the direct coupling of diester 2 with the corresponding o-iododerivative of compound 3, foreseeing the plausibility of formation of an undesired aryne intermediate. The selective electrophilic iodination of the activated aryl moiety in compound 5 using N-iodosuccinimide (NIS, 1.10 equiv) in the presence of a catalytic amount of trifluoroacetic acid yielded the required iodo-compound. In the above specified iodination reaction of compound 5, an air-oxidation of the activated methine carbon also took place simultaneously to actually form the hydroxylated iodo-compound 6 in 71% yield. The activated methine/methylene positions of the homophthalimides are known to be prone to facile air-oxidation and respectively form the corresponding tertiary alcohols/ketones.8 The presence of a tertiary hydroxyl unit in compound 6 was confirmed on the basis of IR (stretching frequency at 3460 cm−1), 1H NMR (broad singlet at δ 3.92), 13C NMR (quaternary carbon at 74.6 ppm) and mass spectral data (M+Na+ peak at 532). At this stage the NaBH4 reduction of compound 6 was studied in detail to obtain the desired lactam 7. The regioselective NaBH4 reduction of the more reactive unconjugated carbonyl group of the homophthlimide 6 followed by the acid-catalyzed dehydration took place in one-pot to provide the desired pivotal building block 7 in 68% yield. An intramolecular palladium-catalyzed Heck coupling reaction of thus formed unsaturated lactam 7 under the standard set of reaction conditions delivered the desired oxyavicine architecture 8 in 70% yield. Finally, the palladium on charcoal induced dehydrogenation of compound 8 in refluxing p-cymene furnished the natural product oxyavicine (1) in 73% yield.4g The oxyavicine was obtained in six steps with 16% overall yield and the analytical and spectral data obtained for the synthesized natural product were in complete agreement with the reported data.3,4 The conversion of oxyavicine to the avicine alkaloid with 97% efficiency is known in the literature.4a
In summary, we have accomplished the convergent synthesis of oxyavicine with the construction of ring B and C utilizing an intramolecular Heck-coupling reaction as the key step. We feel that our present approach is general in nature and will be useful to design several naturally occurring benzophenanthridine alkaloids, their unnatural derivatives and congeners for SAR studies.
Experimental section
The 1H NMR spectra were recorded on 200 MHz NMR and 400 MHz NMR spectrometer using TMS as an internal standard. The 13C NMR spectra were recorded on 200 NMR spectrometer (50 MHz), 400 NMR spectrometer (100 MHz) and 500 NMR spectrometer (125 MHz). Mass spectra were taken on a MS-TOF mass spectrometer. HRMS (ESI) were taken on an Orbitrap (quadrupole plus ion trap) and TOF mass analyzer. The IR spectra were recorded on an FT-IR spectrometer. Elemental analysis was taken at NCL. Column chromatographic separations were carried out on silica gel (60–120 mesh). Commercially available n-BuLi, o-dichlorobenzene, NIS, NaBH4, Pd(OAc)2, Pd/C and p-cymene were used. The starting materials methyl 6-(2-methoxy-2-oxoethyl)benzo[d][1,3]dioxole-5-carboxylate (2) and 5-(2-iodoethyl)benzo[d][1,3]dioxole (3) were prepared using the known procedures.9,10Methyl 6-(4-(benzo[d][1,3]dioxol-5-yl]-1-methoxy-1-oxobutan-2-yl)benzo[d][1,3]dioxole-5-carboxylate (4)
A freshly prepared solution of LDA from diisopropylamine (1.67 mL, 12.00 mmol) and n-BuLi (1.60 M in hexane, 8.12 mL, 13.00 mmol) in THF (5 mL) under argon atmosphere at 0 °C was added to solution of compound 2 (2.52 g, 10.00 mmol) in THF (15 mL) and HMPA (15 mL) mixture at −78 °C under argon atmosphere and the reaction mixture was further stirred at the same temperature for 30 min. To the above reaction mixture was added solution of compound 3 (3.04 g, 11.00 mmol) in THF (5 mL) in a dropwise fashion. The reaction mixture was allowed to gradually attain the room temperature for 7 h. The reaction was quenched with saturated NH4Cl solution. The solvent was removed in vacuo and the obtained residue was dissolved in ethyl acetate (50 mL), washed with water (20 mL), brine (25 mL) and dried over Na2SO4. The concentration of the organic layer in vacuo followed by silica gel column chromatographic purification of the resulting residue using 25% ethyl acetate–petroleum ether as an eluent gave pure product 4 as a thick oil (3.28 g, 82%). 1H NMR (CDCl3, 200 MHz) δ 1.90–2.10 (m, 1H), 2.25–2.60 (m, 3H), 3.66 (s, 3H), 3.84 (s, 3H), 4.75 (t, J = 8 Hz, 1H), 5.91 (s, 2H), 6.02 (d, J = 2 Hz, 2H), 6.53–6.75 (m, 3H), 6.91 (s, 1H), 7.38 (s, 1H); 13C NMR (CDCl3, 50 MHz) δ 33.4, 35.5, 45.9, 52.0 (2 carbons), 100.7, 101.9, 108.0, 108.4, 108.8, 110.4, 121.1, 123.0, 135.3, 136.6, 145.6, 146.4, 147.5, 150.9, 167.0, 174.4; ESIMS (m/z) 423 [M+Na]+; HRMS (ESI) calcd for C21H20O8 [M + H]+ 401.1236, found 401.1240; IR (CHCl3) νmax 1716, 1666, 1620 cm−1. Anal. Calcd for C21H20O8: C, 63.00; H, 5.03. Found: C, 62.82; H, 4.77.8-(2-(Benzo[d][1,3]dioxol-5-yl)ethyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinoline-5,7(6H,8H)-dione (5)
The compound 4 (2.80 g, 7.00 mmol) was added to a stirred solution of 10% aqueous KOH (25 mL) and MeOH (25 mL) at 0 °C. The reaction mixture was allowed to reach room temperature and further stirred for 12 h and then it was acidified with 2 N HCl. The reaction mixture was concentrated in vacuo and the obtained residue was dissolved in ethyl acetate (50 mL), washed with water (20 mL), brine (25 mL) and dried over Na2SO4. The organic layer was concentrated in vacuo and to the obtained residue was added 40% aqueous solution of methyl amine (3 mL) and it was stirred for 5 min. The above reaction mixture was again concentrated in vacuo and dried. To the obtained salt was added o-dichlorobenzene (20 mL) and it was refluxed for 10 h. The reaction mixture was allowed to gradually attain room temperature. The direct silica gel column chromatographic purification of the resulting solution using 25% ethyl acetate–petroleum ether as an eluent gave pure product 5 as a yellow thick oil (2.05 g, 80%). 1H NMR (CDCl3, 400 MHz) δ 2.13–2.25 (m, 1H), 2.31–2.43 (m, 2H), 2.43–2.53 (m, 1H), 3.29 (s, 3H), 3.87 (t, J = 8 Hz, 1H), 5.90 (s, 2H), 6.09 (d, J = 4 Hz, 2H), 6.51 (d, J = 8 Hz, 1H), 6.57 (s, 1H), 6.67 (d, J = 8 Hz, 1H), 6.68 (s, 1H), 7.59 (s, 1H); 13C NMR (CDCl3, 100 MHz) δ 26.8, 30.9, 38.3, 45.3, 100.8, 102.1, 106.0, 107.7, 108.1, 108.8, 119.8, 121.3, 134.0, 135.0, 145.8, 147.55, 147.61, 152.6, 164.1, 173.5; ESIMS (m/z) 389 [M+Na]+; HRMS (ESI) calcd for C20H17NO6 [M + H]+ 368.1134, found 368.1125; IR (CHCl3) νmax 1709, 1663, 1623 cm−1.8-Hydroxy-8-(2-(6-iodobenzo[d][1,3]dioxol-5-yl)ethyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinoline-5,7(6H,8H)-dione (6)
To a stirred solution of compound 5 (1.50 g, 4.08 mmol) in acetonitrile (30 mL) along with CF3COOH (0.50 mL) was added N-iodosuccinimide (1.01 g, 4.50 mmol). The reaction mixture was stirred at room temperature for 24 h and then it was concentrated in vacuo. The obtained residue was dissolved in ethyl acetate (30 mL), washed with 5% aq. Na2S2O3 (10 mL), water (20 mL), brine (25 mL) and dried over Na2SO4. The concentration of the organic layer in vacuo followed by silica gel column chromatographic purification of the resulting residue using 25% ethyl acetate–petroleum ether as an eluent gave pure product 6 as yellow crystalline solid (1.47 g, 71%). Mp 128–130 °C; 1H NMR (CDCl3, 200 MHz) δ 1.83–2.10 (m, 2H), 2.35–2.63 (m, 2H), 3.38 (s, 3H), 3.92 (br s, 1H), 5.90 (s, 2H), 6.09 (s, 2H), 6.58 (s, 1H), 7.12 (s, 1H), 7.16 (s, 1H), 7.54 (s, 1H); 13C NMR (CDCl3, 50 MHz) δ 27.6, 35.0, 47.2, 74.6, 87.4, 101.5, 102.2, 105.2, 107.6, 109.0, 118.46, 118.53, 135.6, 137.3, 146.9, 148.2, 148.5, 152.8, 163.3, 176.9; ESIMS (m/z) 532 [M+Na]+; HRMS (ESI) calcd for C20H16INO7 [M+Na]+ 531.9869, found 531.9865; IR (CHCl3) νmax 3460, 1715, 1668, 1621 cm−1.8-(2-(6-Iodobenzo[d][1,3]dioxol-5-yl)ethyl)-6-methyl-[1,3]dioxolo[4,5-g]isoquinolin-5(6H)-one (7)
To a stirred solution of compound 6 (1.20 g, 2.35 mmol) in EtOH (20 mL) was portion wise added NaBH4 (717 mg, 18.86 mmol) at 0 °C. The reaction mixture was further stirred for 6 h with the periodic addition of 2 drops solution of EtOH (8 mL) along with 2 N HCl (2 mL) at the interval of 15 min. The excess of NaBH4 was quenched at 0 °C by using 2 N HCl in EtOH until the reaction mixture became acidic. The reaction mixture was concentrated in vacuo and the obtained residue was extracted with ethyl acetate (25 mL × 3). The combined organic layer was washed with water (20 mL), brine (25 mL) and dried over Na2SO4. The concentration of the organic layer in vacuo followed by silica gel column chromatographic purification of the resulting residue using 40% ethyl acetate–petroleum ether as an eluent gave pure product 7 as crystalline solid (764 mg, 68%). Mp 189–192 °C; 1H NMR (CDCl3, 200 MHz) δ 2.70–3.00 (m, 4H), 3.55 (s, 3H), 5.95 (s, 2H), 6.09 (s, 2H), 6.67 (s, 1H), 6.78 (s, 1H), 7.14 (s, 1H), 7.24 (s, 1H) 7.84 (s, 1H); 13C NMR (CDCl3, 50 MHz) δ 30.7, 36.9, 40.8, 87.5, 101.0, 101.6, 101.8, 106.2, 109.4, 114.8, 118.6, 121.8, 129.5, 133.9, 137.0, 147.0, 147.5, 148.5, 151.9, 161.4; ESIMS (m/z) 500 [M+Na]+; HRMS (ESI) calcd for C20H16INO5 [M+Na]+ 499.9971, found 499.9955; IR (CHCl3) νmax 1655, 1611 cm−1.5-Methyl-12,13-dihydro-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-c][1,3]dioxolo[4,5-j]phenanthridin-6(5H)-one (8)
To a stirred solution of compound 7 (600 mg, 1.26 mmol) in DMF (15 mL) was added K2CO3 (1.04 g, 7.56 mmol), TBAB (812 mg, 2.52 mmol) and Pd(OAc)2 (30 mg, 10 mol%) at 25 °C under argon atmosphere and the reaction mixture was heated at 120 °C for 24 h. The reaction mixture was allowed to gradually attain the room temperature and then it was diluted with ethyl acetate (50 mL), washed with water (20 mL), brine (25 mL) and dried over Na2SO4. The concentration of the organic layer in vacuo followed by silica gel column chromatographic purification of the resulting residue using 40% ethyl acetate–petroleum ether as an eluent gave pure product 8 as crystalline solid (307 mg, 70%). Mp 230–232 °C (lit.4g 237–241 °C); 1H NMR (CDCl3, 200 MHz) δ 2.60–2.85 (m, 4H), 3.72 (s, 3H), 5.99 (s, 2H), 6.07 (s, 2H), 6.80 (s, 1H), 6.93 (s, 1H), 7.06 (s, 1H), 7.84 (s, 1H); 13C NMR (CDCl3, 125 MHz) δ 24.2, 29.1, 37.9, 100.3, 101.2, 101.7, 106.2, 106.7, 108.4, 115.4, 120.4, 124.0, 132.9 (2 carbons), 136.9, 146.1, 146.7, 147.4, 152.1, 163.4; ESIMS (m/z) 372 [M+Na]+; IR (CHCl3) νmax 1650, 1615 cm−1.5- Methyl-[1,3]dioxolo[4′,5′:4,5]benzo[1,2-c][1,3]dioxolo[4,5-j]phenanthridin-6(5H)-one (oxyavicine, 1)
A mixture of compound 8 (200 mg, 0.57 mmol) and 10% Pd–charcoal (180 mg) in p-cymene (5 mL) in a sealed tube was heated at 200 °C under argon atmosphere for 24 h. The reaction mixture was allowed to attain room temperature, diluted with CHCl3 (25 mL) and the catalyst was filtered off. The concentration of the organic layer in vacuo followed by silica gel column chromatographic purification of the resulting residue using 30% ethyl acetate–petroleum ether as an eluent gave pure oxyavicine (1) as a crystalline solid (145 mg, 73%). Mp 278–279 °C (lit.4g 278–283 °C); 1H NMR (CDCl3, 400 MHz) δ 3.98 (s, 3H), 6.11 (s, 2H), 6.14 (s, 2H), 7.19 (s, 1H), 7.57 (d, J = 8 Hz, 1H), 7.63 (s, 1H), 7.64 (s, 1H), 7.91 (s, 1H), 7.94 (d, J = 8 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 41.2, 100.7, 101.5, 101.9, 102.7, 104.8, 106.6, 116.8, 118.5, 120.7, 120.9, 123.3, 131.0, 131.9, 135.8, 147.0, 147.6, 148.2, 152.4, 164.1; ESIMS (m/z) 370 [M+Na]+; IR (CHCl3) νmax 1646 cm−1.Acknowledgements
R. J. thanks CSIR, New Delhi, for the award of research fellowship. N. P. A. thanks the Department of Science and Technology, New Delhi, for financial support.References
- (a) V. Simanek, in The alkaloids, ed. A. Brossi, Academic Press, New York, 1985, vol. 26, pp. 185–229. Search PubMed; (b) B. D. Krane, M. O. Fagbule and M. Shamma, J. Nat. Prod., 1984, 47, 1 CrossRef CAS; (c) S.-D. Fang, L.-K. Wang and S. M. Hecht, J. Org. Chem., 1993, 58, 5025 CrossRef CAS; (d) C.-T. Chang, S.-C. Doong, I.-L. Tsai and I.-S. Chen, Phytochemistry, 1997, 45, 1419 CrossRef CAS; (e) T. Ishikawa, Med. Res. Rev., 2001, 21, 61 CrossRef CAS , and references cited therein 1a–e.
- (a) B. Clement, M. Weide, U. Wolschendorf and I. Kock, Angew. Chem., Int. Ed., 2005, 44, 635 CrossRef CAS; (b) K. Orito, Y. Satoh, H. Nishizawa, R. Harada and M. Tokuda, Org. Lett., 2000, 2, 2535 CrossRef CAS; (c) M. Cushman and J.-K. Chen, J. Org. Chem., 1987, 52, 1517 CrossRef CAS; (d) M. Shamma and H. H. Tomlinson, J. Org. Chem., 1978, 43, 2852 CrossRef CAS , and references cited therein 2a–d.
- (a) H. R. Arthur, W. H. Hui and Y. L. Ng, J. Chem. Soc., 1959, 4007 RSC; (b) S.-Q. Wang, G.-Q. Wang, B.-K. Huang, Q.-Y. Zhang and L.-P. Qin, Chem. Nat. Compd., 2007, 43, 100 CrossRef; (c) J. Hu, W.-D. Zhang, R.-H. Liu, C. Zhang, Y.-H. Shen, H.-L. Li, M.-J. Liang and X.-K. Xu, Chem. Biodiversity, 2006, 3, 990 CrossRef CAS.
- (a) P. Lv, K. Huang, L. Xie and X. Xu, Org. Biomol. Chem., 2011, 9, 3133 RSC; (b) C.-C. Liu, K. Parthasarathy and C.-H. Cheng, Org. Lett., 2010, 12, 3518 CrossRef CAS; (c) R. P. Korivi and C.-H. Cheng, Chem.–Eur. J., 2010, 16, 282 CrossRef CAS; (d) T. N. Le and W.-J. Cho, Chem. Pharm. Bull., 2006, 54, 476 CrossRef CAS; (e) T. N. Le, S. G. Gang and W.-J. Cho, J. Org. Chem., 2004, 69, 2768 CrossRef CAS; (f) H. Ishii, T. Ishikawa, Y.-I. Ichikawa, M. Sakamoto, M. Ishikawa and T. Takahashi, Chem. Pharm. Bull., 1984, 32, 2984 CrossRef CAS; (g) I. Ninomiya, T. Naito, H. Ishii, T. Ishida, M. Ueda and K. Harada, J. Chem. Soc., Perkin Trans. 1, 1975, 762 RSC; (h) S. F. Dyke, M. Sainsbury and B. J. Moon, Tetrahedron, 1968, 24, 1467 CrossRef CAS; (i) K. W. Gopinath, T. R. Govindachari and N. Viswanathan, Tetrahedron, 1961, 14, 322 CrossRef CAS.
- (a) P. S. Deore and N. P. Argade, J. Org. Chem., 2012, 77, 739 CrossRef CAS; (b) R. M. Patel, V. G. Puranik and N. P. Argade, Org. Biomol. Chem., 2011, 9, 6312 RSC; (c) P. B. Wakchaure and N. P. Argade, Synthesis, 2011, 2838 CAS; (d) U. A. Kshirsagar, V. G. Puranik and N. P. Argade, J. Org. Chem., 2010, 75, 2702 CrossRef CAS; (e) P. B. Wakchaure, S. Easwar and N. P. Argade, Synthesis, 2009, 1667 CAS; (f) P. B. Wakchaure, V. G. Puranik and N. P. Argade, Tetrahedron: Asymmetry, 2009, 20, 220 CrossRef CAS; (g) K. P. Haval and N. P. Argade, J. Org. Chem., 2008, 73, 6936 CrossRef CAS.
- R. Okunaka, T. Honda, M. Kondo, Y. Tamura and Y. Kita, Chem. Pharm. Bull., 1991, 39, 1298 CrossRef CAS.
- (a) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320 CrossRef CAS; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009 CrossRef CAS , and references cited therein.
- (a) H. Heaney, M. O. Taha and A. M. Z. Slawin, Tetrahedron Lett., 1997, 38, 3051 CrossRef CAS; (b) P. B. Wakchaure, S. Easwar, V. G. Puranik and N. P. Argade, Tetrahedron, 2008, 64, 1786 CrossRef CAS.
- M. Wu, L. Li, A.-Z. Feng, B. Su, D.-M. Liang, Y.-X. Liu and Q.-M. Wang, Org. Biomol. Chem., 2011, 9, 2539 CAS.
- S. Shahane, F. Louafi, J. Moreau, J.-P. Hurvois, J.-L. Renaud, P. V. D. Weghe and T. Roisnel, Eur. J. Org. Chem., 2008, 4622 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20915b/ |
| This journal is © The Royal Society of Chemistry 2012 |