Chemoenzymatic preparation of optically active 3-(1H-imidazol-1-yl)cyclohexanol-based ionic liquids: application in organocatalysis and toxicity studies†
Caroline E.
Paul
a,
Vicente
Gotor-Fernández
a,
Iván
Lavandera
a,
Jose
Montejo-Bernardo
b,
Santiago
García-Granda
b and
Vicente
Gotor
*a
aDepartamento de Química Orgánica e Inorgánica, Instituto Universitario de Biotecnología, Universidad de Oviedo, Calle Julián Clavería 8, Oviedo (Asturias), Spain. Fax: +34 985103448; Tel: +34 985103448E-mail: vgs@uniovi.es
bDepartamento de Química Física y Analítica, Universidad de Oviedo-CINN, Calle Julián Clavería 8, Oviedo (Asturias), Spain
First published on 18th June 2012
Abstract
A straightforward and robust modular synthetic approach was developed for the asymmetric synthesis of imidazolium salts, in which several engineered molecular vectors were considered to evaluate their toxicological profile. The diastereoselective synthesis of cis-3-(1H-imidazol-1-yl)cyclohexanol was achieved by the Michael addition of imidazole to cyclohex-2-en-1-one under microwave conditions followed by reduction of the ketone. The racemic cis-alcohol obtained was successfully resolved through a lipase-catalysed kinetic resolution, Pseudomonas cepacia lipase proved to be a good biocatalyst for the exclusive acetylation of the (1R,3S)-cis enantiomer. Using the remaining (1S,3R)-cis alcohol enantiomer as a synthetic precursor, the optically active (1R,3R)-trans alcohol enantiomer was also prepared. The corresponding chiral salts and ionic liquids were obtained via quaternisation with alkyl halides, followed by anion exchange with inorganic salts. In this manner, a family of novel imidazolium-based ionic liquids was prepared, and their properties as phase-transfer catalysts in the Michael addition of diethyl malonate to trans-chalcone were analysed. The toxicity of these compounds against E. coli cells was also evaluated to understand their structural implications through the presented systematic synthetic approach.
Introduction
Ionic liquids (ILs) are considered as valuable materials due to their synthetic applications as “greener” alternatives to volatile organic solvents1 or organocatalysts.2 In recent years, extensive studies have been performed on their use in synthetic transformations, their role as stationary phases in gas chromatography,3 and their applications in gas–liquid or liquid–liquid extractions,4 electrochemistry,5 mass spectrometry,6 and infrared, Raman and fluorescence spectroscopy.7 The asymmetric synthesis of their chiral versions continues to be an attractive and challenging aim for organic chemists since chiral ionic liquids have shown a great ability to produce significant chirality transference in organocatalytic reactions,8 also presenting different physico-chemical properties when compared to the corresponding racemates. Organic cations (e.g. imidazolium, pyridinium, ammonium, phosphonium) can be easily combined with a variety of organic or inorganic anions (e.g. halides, PF6−, BF4−, SbF6−), leading to a myriad of possibilities to modify or finely tune the properties of the ionic liquid.9 Imidazolium salts are the most used common cores because of their accessibility and easy manipulation and modification from raw materials. Nonetheless, toxicity measurements of ionic liquids have shown that imidazolium-based structures may not be the most benign.10Herein, a synthetic modular approach was developed for the synthesis of various chiral imidazolium structures to further study their possible applications in catalytic processes and to obtain information on the influence of defined motifs with regards to their toxicity. The data thus obtained can be further used for preparing more ecological ionic systems. In this context, for the synthesis of a variety of chiral imidazolium salts and ionic liquids, several key points were considered as tuneable vectors (Fig. 1): (1) the imidazole N-3 substitution (R); (2) the relative position of the cyclohexyl substituents and the absolute configuration of the stereogenic carbon atoms; and finally (3) the anion present in the molecular structure (X−).
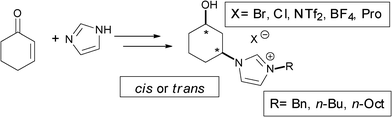 | ||
| Fig. 1 The engineered molecular vectors considered for the asymmetric synthesis of imidazolium salts. | ||
Results and discussion
Chemoenzymatic asymmetric synthesis of imidazole derivatives
The synthesis of racemic 3-(1H-imidazol-1-yl)cyclohexanone (3) was previously reported from commercially available cyclohex-2-en-1-one (1) and imidazole (2) using a Brønsted or Lewis acid, lanthanides, or an inorganic solid support such as cellulose, with moderate to high yields (63–85% yield).11 To develop a more efficient synthesis, the reaction was first carried out in the absence of catalyst using 1,4-dioxane or acetonitrile (MeCN) as solvent under reflux. Only 80% conversion was attained even after several days of reaction and changing the reagent ratio. Subsequently, the reaction was performed under microwave conditions (Scheme 1),12 and after optimisation of the power, temperature and reaction time, a preparative scale (300 mg) of imidazole (2) with an excess of ketone 1 (2 equiv.) in MeCN produced the racemic ketone 3 in 25 min with 98% yield after purification through various washes with Et2O. An up-scale of the reaction to 3 g was made, affording 96% of the pure product after only 2 h 30 min of microwave reaction under the same conditions.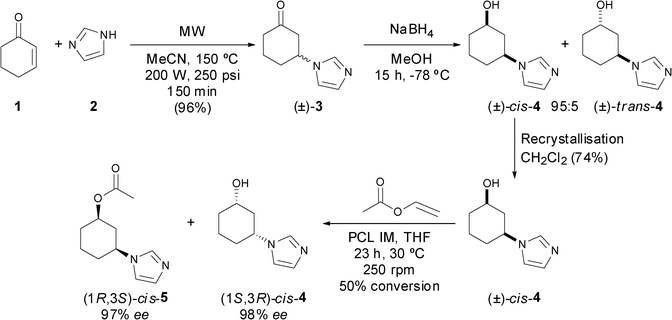 | ||
| Scheme 1 Chemical synthesis and lipase-catalysed kinetic resolution of racemic alcohol cis-4. | ||
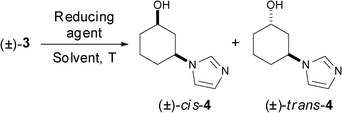
As a first approach to selectively obtain either one of the cis- or trans-alcohol 4 enantiomers, a screening of the alcohol dehydrogenases (ADHs)13 from Candida parapsilosis, Lactobacillus brevis, Lactobacillus kefir, Thermoanaerobacter sp., Ralstonia sp. and Rhodococcus ruber was carried out for the bioreduction of ketone 3 under mild reaction conditions in aqueous buffer. However, the products obtained were mixtures of cis- and trans-alcohol 4, along with traces of 1 and 2 due to the retro-Michael reaction of ketone 3 occurring in water. This reversibility was corroborated by dissolving ketone 3 in solvents such as methanol or chloroform, recovering a mixture of 1, 2 and 3 after several hours. Consequently, the chemical reduction of 3 was explored using various reducing agents to obtain only one of the racemic isomers as detailed in Table 1.
| Entry | Reducing agent | Solvent | T (°C) | t (h) | cis/trans4a |
|---|---|---|---|---|---|
| a Ratio calculated by 1H NMR of the crude reaction product. All reactions led to the full reduction of the ketone. | |||||
| 1 | TiCl4/BH3·Py | CH2Cl2 | −78 | 2 | 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33 :33 |
| 2 | CeCl3/LiEt3BH | THF | −78 | 15 | 89:11 |
| 3 | LiEt3BH | THF | −78 | 15 | 95:5 |
| 4 | L-Selectride | THF | −78 | 15 | 50:50 |
| 5 | NaBH4 | MeOH | 0 | 2 | 85:15 |
| 6 | NaBH4 | MeOH | −20 | 5 | 89:11 |
| 7 | NaBH4 | MeOH | −78 | 15 | 95:5 |
| 8 | NaBH4 | EtOH | −78 | 15 | 95:5 |
First, two different complexes formed by titanium chloride (IV) and pyridine borane (entry 1) or cerium chloride (III) and lithium triethylborohydride (entry 2) were used at −78 °C,14 affording a good induction towards the formation of the cis-4 alcohol isomer. A considerable improvement was achieved by simply using LiEt3BH in THF (entry 3), obtaining a mixture of 95:5 (90% diastereomeric excess) after 15 h favouring the cis-isomer. A more sterically hindered reducing agent, L-Selectride, was also used at −78 °C, however no stereopreference was observed (entry 4). Sodium borohydride (NaBH4) was used in MeOH at different temperatures (0 to −78 °C, entries 5–7), and as expected, higher cis/trans ratios were observed at lower temperatures, affording alcohol 4 in moderate to excellent diastereomeric excess (70% at 0 °C, 78% at −20 °C and 90% at −78 °C). The best selectivity was thus reached at lower temperatures, with no difference observed with ethanol as solvent (entry 8). An up-scale (5 g) of the reaction was performed with NaBH4, because of its easier handling, higher stability and lower cost with respect to LiEt3BH, without any loss of selectivity, affording alcohol 4 with 98% yield and 90% diastereomeric ratio (dr). Separation of cis- and trans-4 isomers was not possible by column chromatography, so the mixture was later recrystallised in dichloromethane yielding the racemic cis-alcohol in 74% yield and >99% dr (Scheme 1).
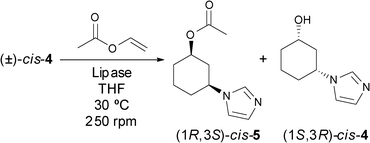
Biocatalytic asymmetric methodologies such as kinetic resolutions (KRs), dynamic kinetic resolutions (DKRs) or enantioselective desymmetrisations (EDs) are very solicited processes for academic and industrial purposes.15 Among all the known enzymes, hydrolases and mainly lipases have attracted considerable attention due to their commercial availability and multiple applications in chemo-, regio- or stereoselective processes.16 With poor selectivities obtained in the bioreduction of racemic 3-(1H-imidazol-1-yl)cyclohexanone 3 and because of the retro-Michael reaction occurring in solution, the lipase-catalysed kinetic resolution of the racemic cis-alcohol 4 was explored to obtain enantiomerically pure products. Employing anhydrous THF as solvent, vinyl acetate as acyl donor (VinOAc, 3 equiv.) and a temperature of 30 °C,17 the influence of the lipase in the KR process was studied by screening the enzymes Candida antarctica lipase B (CAL-B) and different preparations of Pseudomonas cepacia lipase (PCL, also currently known as Burkholderia cepacia lipase). While the reaction with CAL-B was extremely fast, yielding the (1S,3R)-alcohol 4 in the enantiopure form after only 2 h and 54% conversion (entries 1 and 2, Table 2), all PCL preparations led to the stereoselective formation of the (1R,3S)-acetate 5 in high ee. Both lipases supported on ceramic (PCL-C I, entry 3) or immobilised on diatomaceous earth (PCL IM, entry 4) reached 50% conversion after 23 h leading to almost enantiopure substrate (99% ee) and product (97% ee). Not surprisingly, PCL SD attained a lower conversion (8%, entry 5), as this preparation is more suited to an aqueous medium. Therefore, the PCL IM was identified as the best biocatalyst for the lipase-catalysed resolution of the racemic cis-alcohol 4 (Scheme 1). Furthermore, after chemical hydrolysis of the acetyl group of (1R,3S)-5 with K2CO3 in MeOH, the alcohol enantiomer (1R,3S)-4 was obtained without any loss of its optical purity (97% ee).
| Entry | Enzyme | t (h) | ee S (%)a | ee P (%)a | c (%)b | E c |
|---|---|---|---|---|---|---|
| a Enantiomeric excess (eeS for alcohol; eeP for acetate) determined by HPLC. Isolated yields in parenthesis. b Conversion: c = eeS/(eeS + eeP). c Enantiomeric ratio: E = ln[(1 − c) × (1 − eeS)]/ln[(1 − c) × (1 + eeS)]. | ||||||
| 1 | CAL-B | 1 | 82 | 88 | 48 | 40 |
| 2 | CAL-B | 2 | >99 | 84 | 54 | 87 |
| 3 | PCL-C I | 23 | 98 (83) | 96 (95) | 50 | >200 |
| 4 | PCL IM | 23 | 99 (98) | 97 (98) | 50 | >200 |
| 5 | PCL SD | 23 | 8 | >99 | 8 | >200 |
For trans-alcohol 4, since only the cis-alcohol 4 could be obtained with the explored methods, the Mitsunobu reaction18 was performed on the racemic cis-alcohol 4. Thus, p-nitrobenzoic acid, diethyl azadicarboxylate (DEAD) and triphenylphosphine in anhydrous THF were employed, and the formed ester was deprotected with sodium methoxide at room temperature yielding the racemic trans-alcohol 4 in 51% overall yield (Scheme 2).
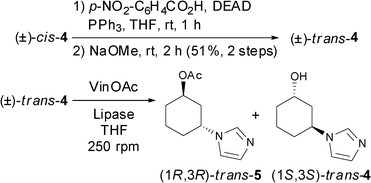 | ||
| Scheme 2 Synthesis and lipase-catalysed KR of (±)-trans-alcohol 4. | ||
The biocatalysed kinetic resolution of the racemate was attempted to give access to both enantiomers (1S,3S)-trans-alcohol 4 and (1R,3R)-trans-acetate 5 (Table 3). Unfortunately, low selectivity and reactivity were obtained with CAL-B (entries 1 and 2) while PCL-C I required 60 °C to attain 23% conversion of the acetate in enantiopure form (entry 3). However, longer reaction times did not lead to an increased conversion. In addition, more extreme conditions such as using the acyl donor (VinOAc) as the reaction medium provoked a dramatic loss in the stereoselectivity of the process (entry 4). Therefore, an alternative chemoenzymatic approach for the synthesis of (1R,3R)-trans-4 was developed. Hence, starting from enantioenriched (1S,3R)-cis-alcohol 4, the optically active (1R,3R)-trans-alcohol 4 was obtained following a Mitsunobu inversion–deprotection sequence, as previously described for the racemic trans-4 in Scheme 2.
| Entry | Enzyme | Solvent | VinOAc | T (°C) | t (h) | ee S (%)a | ee P (%)a | c (%)b | E c |
|---|---|---|---|---|---|---|---|---|---|
| a Enantiomeric excess (eeS for alcohol; eeP for acetate) determined by HPLC. b Conversion: c = eeS/(eeS + eeP). c Enantiomeric ratio: E = ln[(1 − c) × (1 − eeS)]/ln[(1 − c) × (1 + eeS)]. | |||||||||
| 1 | CAL-B | THF | 3 equiv. | 30 | 169 | 17 | 63 | 21 | 5 |
| 2 | CAL-B | THF | 3 equiv. | 60 | 23 | 23 | 31 | 43 | 3 |
| 3 | PCL-C I | THF | 3 equiv. | 60 | 23 | 29 | >99 | 23 | >200 |
| 4 | PCL-C I | VinOAc | 0.1 M | 60 | 23 | 37 | 28 | 57 | 2 |
Preparation of chiral ionic imidazolium salts and ionic liquids
Having in hand the adequate optically active pure precursors for the synthesis of the imidazolium salts, a general synthetic sequence based on the quaternisation of the N-3 imidazole nitrogen atom and the subsequent anion exchange to modulate the physico-chemical properties of the ionic salt was developed in a similar manner as previously reported for the synthesis of 1,2-derivatives.19 The fine tuning of ionic liquid properties through structural design not only opens new possibilities for their use in different applications, but also enables the study of the critical factors applied to Sustainable Chemistry that can affect their toxicity. Herein, structural diversity (Fig. 1) was considered based on the counter-anion (Br−, Cl−, BF4−, NTf2− or Pro−), the imidazolium alkylating chain (Bn, n-Bu or n-Oct) and the configuration of the two stereogenic centres including their relative disposition (cis or trans). Hence, the nomenclature of the salts was defined by the following descriptors: the first one includes the absolute configuration of C1 and C3 at the cyclohexyl ring [i.e., (R,S)-]. The relative configuration at these carbon atoms is reported by the second descriptor trans or cis [i.e., (R,S)-cis-]. The term IM refers to an imidazole derivative [i.e., (R,S)-cis-IM-]. The fourth descriptor indicates the presence of OH at position C1 of the ring [i.e., (R,S)-cis-IM-OH-]. The nature of the alkyl chain at N-3 of the imidazolium moiety is indicated by the use of Bn (benzyl), Bu (n-butyl) or Oct (n-octyl) [i.e., (R,S)-cis-IM-OH-Bn-]. Finally the last descriptor refers to the anion [i.e., (R,S)-cis-IM-OH-Bn-Br].The synthesis of the chiral imidazolium salts and ionic liquids was carried out starting from both the racemic and optically active cis- and trans-alcohol 4, by reaction with alkyl or benzyl halides at 70 or 100 °C for the mono-quaternisation of the N-3 position (Scheme 3). Brominated salts were prepared using a small excess of the alkylating agent (1.2 equiv.), with a larger excess (10 equiv.) and longer reaction time for chloride salts, yielding the desired halide salts in good to excellent yields (82–98%) after successive washes with Et2O. In addition, the quaternisation reaction time could be shortened under microwave conditions (0.5–1 h).
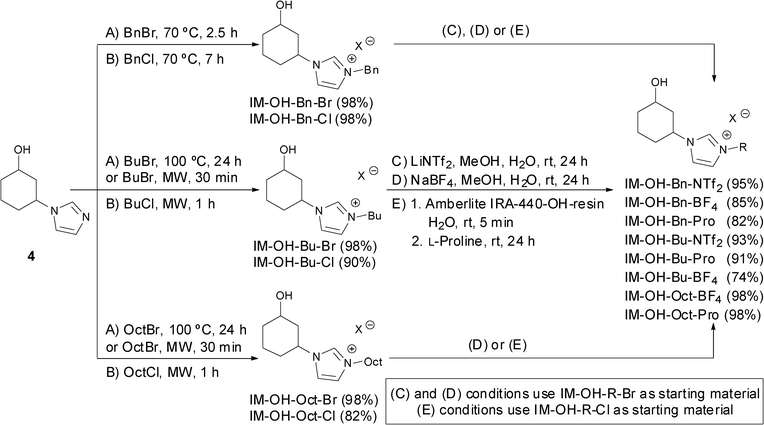 | ||
| Scheme 3 The synthesis of imidazolium salts and ionic liquids through the quaternisation and anion exchange chemical sequence. | ||
Exchange of the halide anion was possible using inorganic salts such as lithium bis(trifluoromethane sulfonimide) (LiNTf2) or sodium tetrafluoroborate (NaBF4), obtaining yields between 74 to 98% after 24 h at room temperature. In an attempt to prepare proline-derived organocatalysts, reactions of halide salts with potassium prolinate generated in situ from proline and potassium hydroxide were carried out. The corresponding prolinate salts were smoothly formed, however their poor solubility in organic solvents and the formation of potassium bromide during the anion exchange in water made their purification impossible. An alternative strategy was envisaged using the chloride salts as starting material. Anion exchange of IM-OH-R-Cl with basic Amberlite IRA-440-OH resin led to the corresponding IM-OH-R-OH intermediates,20 to which L-proline was subsequently added, affording IM-OH-R-Pro in good to excellent yields (82–98%).
Moreover, the optically active compound (1S,3R)-cis-IM-OH-Bn-Br was successfully crystallised and its structure was solved by single crystal X-ray diffraction, revealing the absolute configuration of the stereogenic centres and confirming the selective quaternisation at the N-3 atom suggested by the NMR studies. The solid-state structure (Fig. 2) corroborated the lipase-catalysed acetylation at the (1R)-position of the cis-alcohol 4,21 in accordance with the stereopreference observed for CAL-B and PCL in the lipase-catalysed acetylation towards other 1,3-amino alcohols.22 Consequently, after a Mitsunobu inversion–deprotection reaction, the configuration of (1R,3R)-trans-alcohol 4 was also confirmed. Once both the racemic and optically active salts were obtained, their possible use in organocatalysis was explored by using them as phase transfer catalysts in the model reaction of the Michael addition of diethyl malonate to trans-chalcone, and their inhibition properties in the growth of E. coli cells were also evaluated as a test of their biocompatibility.
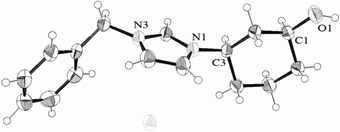 | ||
| Fig. 2 The solid-state structure of the imidazolium salt (1S,3R)-cis-IM-OH-Bn-Br determined through X-ray diffraction analysis. | ||
Application of imidazolium salts and ionic liquids in a Michael-type addition reaction
Asymmetric phase-transfer catalysts (APTCs) are recognised as powerful tools due to their chirality transfer abilities, encouraging the development of different systems over the past few years.23 Recently, enantiopure triazolium salts were employed as APTC in the Michael addition between diethyl malonate (6) and trans-chalcone (7),24 so the potential use of the above synthesised 1H-imidazol-3-ium salts in this catalytic process was envisioned. The reactions were performed using dry potassium carbonate, observing only trace amounts of the final adduct (±)-8 in the absence of the catalyst. The results using the salts and ILs as catalysts in this reaction are summarised in Table 4. As can be seen, generally good to excellent conversions were achieved, although with low stereoselectivities, by varying the absolute configuration of the final product depending on the disposition of the substituents in the cyclohexyl ring.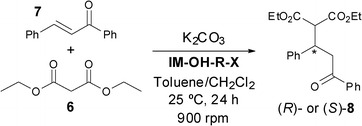
| Entry | Catalyst | c (%)a | ee P (%)b |
|---|---|---|---|
| a Determined by 1H NMR analysis of the crude reaction product. b Determined by HPLC. | |||
| 1 | (+)-(1R,3S)-cis-IM-OH-Bn-Br | 89 | <3 |
| 2 | (+)-(1R,3S)-cis-IM-OH-Bu-Br | 89 | <3 |
| 3 | (+)-(1R,3S)-cis-IM-OH-Oct-Br | 98 | <3 |
| 4 | (−)-(1R,3R)-trans-IM-OH-Bn-Br | 20 | 5 (S) |
| 5 | (−)-(1S,3R)-cis-IM-OH-Bn-Cl | 95 | 6 (R) |
| 6 | (−)-(1S,3R)-cis-IM-OH-Bu-Cl | >99 | 6 (R) |
| 7 | (−)-(1S,3R)-cis-IM-OH-Oct-Cl | >99 | 6 (R) |
| 8 | (+)-(1R,3S)-cis-IM-OH-Bn-Cl | 90 | <3 |
| 9 | (+)-(1R,3S)-cis-IM-OH-Bu-Cl | 98 | <3 |
| 10 | (+)-(1R,3S)-cis-IM-OH-Oct-Cl | 98 | <3 |
| 11 | (±)-cis-IM-OH-Bn-BF4 | 76 | 0 |
| 12 | (−)-(1S,3R)-cis-IM-OH-Bn-BF4 | 97 | 7 (R) |
| 13 | (−)-(1R,3R)-trans-IM-OH-Bn-BF4 | 99 | <3 |
| 14 | (−)-(1S,3R)-cis-IM-OH-Bn-NTf2 | 28 | <3 |
| 15 | (−)-(1R,3R)-trans-IM-OH-Bn-NTf2 | 13 | <3 |
| 16 | (±)-cis-IM-OH-Bn-Pro | 67 | 0 |
| 17 | (−)-(1S,3R)-cis-IM-OH-Bn-Pro | 78 | 8 (R) |
| 18 | (−)-(1S,3R)-cis-IM-OH-Bu-Pro | 60 | 6 (R) |
| 19 | (−)-(1S,3R)-cis-IM-OH-Oct-Pro | 83 | 4 (R) |
| 20 | (−)-(1R,3R)-trans-IM-OH-Bn-Pro | 87 | 3 (S) |
Clear trends could be observed, for instance octyl derivatives led to higher activity values compared to benzyl or butyl ones, and all derivatives afforded conversions over 89% (entries 1–3, 5–7 and 8–10) except when prolinate was the anion (entries 17–19). While racemates led to adduct 8 with no enantiopreference (entries 11 and 16), optically active catalysts gave low, albeit appreciable, selectivities (entries 12 and 17), usually favouring the (R)-product for cis-catalysts, while (S)-8 was preferentially obtained when employing trans-derivatives (entries 4 and 20). Bistrifluoromethane sulfonimides showed poorer activity values (13–28% conversion, entries 14 and 15), while chlorides and tetrafluoroborates led to excellent levels of conversion (entries 5–10 and 12–13). Finally, the effect of proline-derived catalysts is remarkable, because while adding L-proline to the reaction no conversion was observed, pleasingly all the prolinate salts catalysed the formation of the final product in moderate to good conversions (entries 17–20).
In summary, the structural features of the organocatalyst employed influenced the outcome of the Michael addition reaction, highlighting the importance of modular synthetic approaches for the synthesis of chiral catalysts. In general, excellent levels of activity were achieved with halogenated salts and tetrafluoroborate ionic liquids, and the highest stereoselectivities were obtained with the prolinate and chloride imidazolium compounds. Finally, racemates obviously did not induce any chirality and most remarkably, a reversed stereospecificity was achieved when changing the absolute configuration of the stereogenic centres.
Biological evaluation of imidazolium salts and ionic liquids through the agar diffusion test
The choice of reactants and solvents is of particular interest, especially when considering an up-scale of a reaction. In this respect, a realistic view of the eco-toxicity parameters must be considered.25 The agar diffusion test is a simple and effective method to screen the toxicity caused by ionic liquids based on their ability to inhibit the growth of a determined microorganism.26 The absence of sophisticated equipment and the use of an inexpensive medium appear as clear advantages for the development of this method. In order to further employ the imidazolium salts for biocatalytic purposes in the near future, and because nowadays many bacterial enzymes can be easily cloned and over-expressed on E. coli as the host microorganism, this bacterium was chosen as a suitable model to study their biocompatibility. Therefore, E. coli cells were spread onto LB agar plates, and a filter paper disc was soaked with the corresponding imidazolium alcohol, salt or ionic liquid and placed on the lawn of cells. All plates were incubated for 24 h at 30 °C, and the inhibition zone around the filter paper was measured. Data are compiled in Table 5.| Entry | Compound | Amount of compound (μmol) | Inhibition zone (mm) | Normalised inhibition zone (mm μmol−1) |
|---|---|---|---|---|
| 1 | Blank | — | 0 | 0 |
| 2 | (±)-IM-cis-OH | 0.0126 | 0 | 0 |
| 3 | (±)-trans-IM-OH-Bn-Br | 0.0152 | 4.5 | 296 |
| 4 | (±)-cis-IM-OH-Bn-Br | 0.0170 | 3 | 176 |
| 5 | (−)-cis-(1S,3R)-IM-OH-Bn-Br | 0.0246 | 8 | 325 |
| 6 | (+)-cis-(1R,3S)-IM-OH-Bn-Br | 0.0187 | 14 | 749 |
| 7 | (±)-cis-IM-OH-Bu-Br | 0.0300 | 2 | 67 |
| 8 | (−)-cis-(1S,3R)-IM-OH-Bu-Br | 0.0284 | 2 | 71 |
| 9 | (+)-cis-(1R,3S)-IM-OH-Bu-Br | 0.0330 | 2 | 61 |
| 10 | (±)-cis-IM-OH-Oct-Br | 0.0171 | 11.5 | 673 |
| 11 | (−)-cis-(1S,3R)-IM-OH-Oct-Br | 0.0053 | 4 | 757 |
| 12 | (+)-cis-(1R,3S)-IM-OH-Oct-Br | 0.0206 | 5.5 | 267 |
| 13 | (±)-trans-IM-OH-Bn-Cl | 0.0114 | 4.5 | 395 |
| 14 | (±)-cis-IM-OH-Bn-Cl | 0.0256 | 5 | 195 |
| 15 | (±)-cis-IM-OH-Bu-Cl | 0.0213 | 2 | 94 |
| 16 | (±)-cis-IM-OH-Oct-Cl | 0.0130 | 12.25 | 938 |
| 17 | (±)-trans-IM-OH-Bn-BF4 | 0.0257 | 6.5 | 253 |
| 18 | (±)-cis-IM-OH-Bn-BF4 | 0.0081 | 1.25 | 154 |
| 19 | (±)-cis-IM-OH-Bu-BF4 | 0.0190 | 1.75 | 92 |
| 20 | (±)-cis-IM-OH-Oct-BF4 | 0.0131 | 12.5 | 954 |
| 21 | (±)-trans-IM-OH-Bn-NTf2 | 0.0067 | 5 | 746 |
| 22 | (±)-cis-IM-OH-Bn-NTf2 | 0.0085 | 2 | 235 |
| 23 | (±)-cis-IM-OH-Bu-NTf2 | 0.0117 | 2.5 | 214 |
| 24 | (±)-trans-IM-OH-Bn-Pro | 0.0051 | 1 | 196 |
| 25 | (±)-cis-IM-OH-Bn-Pro | 0.0142 | 2.25 | 158 |
| 26 | (±)-cis-M-OH-Bu-Pro | 0.0226 | 2 | 88 |
| 27 | (±)-cis-IM-OH-Oct-Pro | 0.0133 | 10.5 | 798 |
The toxicity measurements were performed taking into account the μmoles used for each substrate in order to compare the effect of the imidazole derivatives on the inhibition of the E. coli cell growth. In all cases, the perfect water-miscibility of the salts and ILs tested allowed a good diffusion through the agar plate. Furthermore, no precipitate or chemical reaction in the paper disc was observed as previously mentioned for other ionic compounds.26 In the case of the blank experiment without IL, no inhibition of growth was observed. Representative photos are shown in Fig. 3.
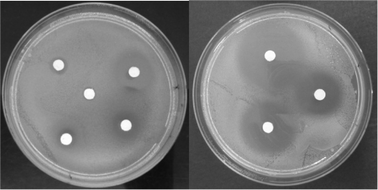 | ||
| Fig. 3 The agar diffusion tests for a series of imidazolium compounds. Left plate: top from left (±)-cis-IM-OH-Bn-Pro; (±)-trans-IM-OH-Bn-BF4; centre (±)-cis-IM-OH-Bn-BF4; bottom from left (±)-cis-IM-OH-Bn-NTf2; (±)-trans-IM-OH-Bn-NTf2. Right plate: top from left (±)-cis-OH-Oct-Br; (+)-cis-OH-Oct-Br; bottom (−)-cis-OH-Oct-Br. | ||
Defined trends were clearly observed in Table 5 and are more detailed in Tables S1–S4 in the ESI†. While the bistrifluoromethane sulfonimide-derived imidazolium compounds behaved as the more toxic ILs (Table S1†), the bromide and proline derivatives generally were the ones that led to lower toxicity values. The longer the cation alkyl chain length was, the lower the biocompatibility was, as previously observed by Frade et al. (n-Bu < Bn < n-Oct).27 Thus, the octyl derivatives clearly reached the highest values for the inhibition of E. coli cell growth (Table S2†).
Studying the effect of the relative disposition of the stereogenic centres was of special interest as there are few reports that evaluate this feature for ionic liquids.28trans-Isomers led in all cases to higher inhibition values rather than cis-isomers (Table S3†). Finally, an appreciable difference was observed when comparing the racemic and optically active compounds for the cis-IM-OH-R-Br family (Table S4†), however a clear tendency could not be identified, although it should be noted that the enantiomer toxicity effects were not cumulative.
Conclusions
In summary, a robust and straightforward approach was developed for the synthesis of enantiopure salts and ILs, combining both chemical conventional methods and biocatalytic reactions. Two key steps were clearly identified: (1) the selective reduction of ketone 3 to the racemic cis-alcohol 4 using NaBH4 at low temperature; and (2) its subsequent kinetic resolution using PCL IM as biocatalyst and vinyl acetate as acyl donor, thus enabling the preparation of the adequate building blocks for the synthesis of imidazolium-based ionic liquids after quaternisation with the corresponding alkyl halide and anion exchange with the tetrafluoroborate or bis(trifluoromethane sulfonimide) salts. Furthermore, an easy protocol to obtain the L-prolinate salts was conceived starting from the corresponding chloride salts.Exploring the catalytic possibilities of the synthesised compounds, the Michael addition between diethyl malonate and trans-chalcone was studied with a representative range of imidazolium derivatives, achieving in general high activities although low stereopreferences. The best activities were reached with halogenated salts and tetrafluoroborate ionic liquids, and although stereoselectivities were very low for synthetic purposes, prolinate and chloride imidazolium compounds presented the highest values. Remarkably, a reversed stereospecificity was noted when changing the absolute configuration of the stereogenic centres.
Finally, a study of the inhibited growth of E. coli cells in the presence of these derivatives enabled the identification of some correlations between the structural motifs and the toxicity properties of the ionic liquids prepared, a larger cation alkyl chain length such as octyl, bistrifluoromethane sulfonimide anion and trans-isomers being the less benign components. Synthesis of (chiral) ionic liquids is an important and active field nowadays, and researchers should think not only of the practical but also of the ecological implications that these derivatives show. Robust and flexible synthetic methods plus the performance of toxicity tests for newly obtained ionic liquids are highly advisable for generating useful structures, rather than only collecting a series of compounds created in many cases without additional advantages. The development of simple, fast and cost-effective tests such as the agar diffusion test should accompany these synthetic routes.29 This test has proven to be a quick, inexpensive and reliable method that gives a clear idea about the ecological implications of ionic systems.
Experimental section
Candida antarctica lipase type B (CAL-B, Novozyme 435, 7300 PLU/g) was obtained from Novozymes and PCL-C I (1638 U/g) was purchased from Sigma-Aldrich. PCL SD (24700 U/g) is a crude enzyme preparation containing dextrin as diluents and PCL IM was immobilised in diatomaceous earth (943 U/g), both were generously given by Amano Enzyme Europe Ltd. All other reagents were obtained from different commercial sources and used without further purification. Anhydrous solvents were dried over an adequate desiccant under nitrogen. Column chromatography was performed using silica gel 60 (230–240 mesh). Melting points were obtained in open capillary tubes and are uncorrected. IR spectra were recorded on NaCl plates or as KBr pellets on a Perkin-Elmer Spectrum 100 FT-IR. 19F, 1H, 13C NMR, DEPT and bidimensional spectra were recorded on a Bruker NAV-300 or DPX-300 spectrometer at 282 (19F), 300 (1H) and 75 (13C) MHz. The chemical shifts are given in parts per million (ppm) and the coupling constants (J) in Hertz (Hz). Mass spectra (MS) were obtained by APCI+, ESI+ or ESI− on a HP1100 chromatograph mass detector. Measurement of the optical rotation was done on a Perkin-Elmer 241 polarimeter. High performance liquid chromatography (HPLC) analyses were carried out on a Hewlett Packard 1100 chromatograph with a UV detector at 210 nm using Daicel Chiralcel OB–H, OJ–H and Chiralpak AS (25 cm × 4.6 mm) columns and hexane–2-propanol mixtures. Microwave reactions were carried out with a CEM Discover system S-Class microwave; the conditions for all the microwave reactions were: temperature 150 °C, power 200 W and pressure 250 psi with medium to high stirring. Further detailed data are provided in the ESI.†
(±)-3-(1H-Imidazol-1-yl)cyclohexanone (3)
Imidazole (2, 1.362 g, 20.0 mmol) was dissolved in acetonitrile (10.0 mL) in a microwave vessel and cyclohex-2-en-1-one (1, 3.87 mL, 40.0 mmol) was added. After 2 h and 30 min of the microwave reaction (150 °C, 200 W and 250 psi), the solution mixture was transferred to a round bottom flask, the solvent was evaporated under reduced pressure and the yellow oil was left under high vacuum. The resulting solid was crushed to a powder and washed with Et2O (3 × 10 mL) to obtain a beige fine powder (3.145 g, 96%).(±)-cis-3-(1H-Imidazol-1-yl)cyclohexanol (cis-4)
A solution of the ketone 3 (6.50 g, 39.6 mmol) dissolved in anhydrous MeOH (12 mL) was cooled to −78 °C and NaBH4 (1.05 g, 27.7 mmol) was added. The reaction mixture was stirred at this temperature for 15 h, then left to warm to room temperature and quenched with HCl 1 N (5 mL). The solvent was evaporated, the product was extracted with CH2Cl2, dried over Na2SO4, filtered and concentrated under reduced pressure to afford the pure product as a white powder (6.419 g, 98%) with a cis/trans ratio of 95:5 (90% dr) as measured by 1H NMR. After recrystallisation from CH2Cl2, the desired racemic alcohol diastereomer cis-4 was obtained as white solid crystals (>99% dr, 4.686 g, 74%).
(±)-cis-3-(1H-Imidazol-1-yl)cyclohexyl acetate (cis-5)
To a solution of 4-dimethylaminopyridine (DMAP) (14.7 mg, 0.12 mmol) and Ac2O (228 μL, 2.41 mmol) in anhydrous CH2Cl2 (15 mL), the alcohol 4 (200 mg, 1.20 mmol) and Et3N (507 μL, 3.61 mmol) were added. The reaction mixture was stirred for 30 min, the solvent was then evaporated and the crude product was purified by column chromatography (5% MeOH–CH2Cl2), affording the pure product (237 mg, 95%) as a clear yellow oil.PCL IM catalysed kinetic resolution of (±)-cis-4 through an acetylation reaction
To a solution of (±)-cis-4 (1.0 g, 6.0 mmol) and PCL IM (1.0 g) in anhydrous THF (60 mL), vinyl acetate (1.664 mL, 18.0 mmol) was added and the reaction mixture was left in an orbital shaker at 250 rpm and 30 °C. The reaction was followed by taking aliquots at regular intervals and analysed by HPLC. After 23 h, 50% conversion was obtained and the reaction was stopped by filtering the enzyme out and washing it with CH2Cl2 (3 × 5 mL). The solvent was then evaporated under reduced pressure and the crude product was purified by column chromatography (5–10% MeOH–CH2Cl2), obtaining the alcohol (1S,3R)-4 (492 mg, 98%, 99% ee) as a white powder and the acetate (1R,3S)-5 (614 mg, 98%, 97% ee) as a clear yellow liquid.(1R,3S)-cis-3-(1H-Imidazol-1-yl)cyclohexanol (cis-4)
Acetate (1R,3S)-5 (2.50 g, 12.0 mmol) was dissolved in MeOH (12 mL) and K2CO3 (4.976 g, 36.0 mmol) was added. The mixture was stirred overnight at room temperature, then the solvent was evaporated and the crude product was purified by flash column chromatography (10% MeOH–CH2Cl2) to obtain the alcohol (1R,3S)-4 as a pure white powder (1.95 g, 98%, 97% ee).(±)- or (1R,3R)-trans-3-(1H-Imidazol-1-yl)cyclohexanol (trans-4)
To a solution of the alcohol (±)-cis-4 (500 mg, 3.01 mmol) in anhydrous THF (15 mL), p-nitrobenzoic acid (1.005 g, 6.02 mmol) and triphenylphosphine (1.578 g, 6.02 mmol) were added. Diethyl azodicarboxylate (DEAD) (1.084 mL, 6.02 mmol) was added dropwise leaving a clear yellow solution. The reaction mixture was stirred at room temperature for 2 h. Then, the solvent was evaporated and a solution of NaOMe (15 mL) was added. The reaction was stirred at room temperature for 2 h. Afterwards, the solvent was evaporated and the crude product was purified by column chromatography (5% MeOH–CH2Cl2), affording the pure product (255 mg, 51%) as a white powder.General procedure for the synthesis of bromide salts (IM-OH-R-Br)
To a solution of the alcohol 4 (200 mg, 1.20 mmol) in acetonitrile (1 mL), the corresponding halide (1.44 mmol) was added inside a sealed tube. The reaction mixture was stirred at 70 °C for 2 h 30 min (for BnBr) or at 100 °C for 24 h (for n-BuBr and n-OctBr) in a sand bath and then cooled to room temperature. Et2O (5 mL) was added and the resulting white solid precipitate was further washed with Et2O (3 × 10 mL) to remove the excess of unreacted halide, thus obtaining the corresponding bromide salts (98% in all cases) as white powders.General procedure for the synthesis of IM-OH-Bn-Cl
To a solution of the alcohol 4 (200 mg, 1.20 mmol) in acetonitrile (1 mL), benzyl chloride (1.384 mL, 12.0 mmol) was added in a sealed tube. The reaction mixture was stirred at 70 °C in a sand bath for 7 h and then cooled to room temperature. Et2O (5 mL) was added and the resulting white solid precipitate was further washed with Et2O (3 × 10 mL) to obtain IM-OH-Bn-Cl (352.2 mg, 98%) as a white powder.General procedure for the synthesis of IM-OH-R-Cl (R = Bu, Oct)
To a solution of the alcohol 4 (450 mg, 2.71 mmol) in acetonitrile (2 mL), the corresponding halide (27.1 mmol) was added. The reaction was done using microwave conditions over 1 h. Et2O (5 mL) was added and the resulting clear colourless oil was further washed with Et2O (3 × 10 mL) to remove the excess of unreacted chloride, obtaining IM-OH-R-Cl (82–90%) as a clear colourless oil.General procedure for the anion exchange reaction in the synthesis of IM-OH-R-X (R = Bn, Bu, Oct; and X = BF4, NTf2)
A solution of LiNTf2 or NaBF4 (0.445 mmol) in H2O (300 μL) was added to a solution of the bromide salt IM-OH-R-Br (0.297 mmol) in MeOH (7.0 mL) and the reaction mixture was stirred for 24 h at room temperature. The solvent was then evaporated under reduced pressure and the product was extracted with CH2Cl2 (3 × 10 mL), dried over Na2SO4 and concentrated to afford the corresponding IM-OH-R-X salt (74–98%).General procedure for the anion exchange reaction in the synthesis of IM-OH-R-Pro (R = Bn, Bu, Oct)
The corresponding chloride salt IM-OH-R-Cl (0.512 mmol) was dissolved in distilled H2O (5.0 mL) and two scoops of Amberlite IRA-440-OH resin (previously washed with distilled water and methanol) were added. The mixture was stirred for 5 min, then the resin was filtered out and L-proline (70.8 mg, 0.615 mmol) was added to the solution. The reaction mixture was stirred at room temperature for 24 h. The solvent was then evaporated under reduced pressure and the oil was left under high vacuum. The oil was then dissolved in minimal amounts of 10% MeOH–MeCN and the resulting precipitate observed was filtered out with a short pipette cotton plug, thus removing any excess of proline. The solvent was evaporated and the product dried under high vacuum for 48 h, obtaining a clear light yellow oil (82–98%).General procedure for the Michael addition of diethyl malonate to trans-chalcone catalysed by imidazolium salts
The imidazolium salt IM-OH-R-X (0.036 mmol, 0.5 equiv.), trans-chalcone (7, 15 mg, 0.072 mmol, 1 equiv.), dry K2CO3 (30 mg, 0.216 mmol, 3 equiv.) and diethyl malonate (6, 13 μL, 0.086 mmol, 1.2 equiv.) were dissolved using a mixture of CH2Cl2–toluene (300/60 μL) in an Eppendorf tube. The mixture was stirred on an orbital shaker at 900 rpm and at 25 °C for 24 h. Once the reaction was stopped, the conversion value was calculated by 1H-NMR analysis of the crude reaction product. The enantiomeric excess of the final product was calculated by HPLC with the chiral column Chiralpak IA at 40 °C n-hexane:EtOH (95:5) as eluent with a flow of 0.8 mL min−1: tR = 14.5 min for the (S)-8 enantiomer and 17.1 min for the (R)-8 enantiomer.
Acknowledgements
This project was supported by the BIOTRAINS Marie Curie Initial Training Network, financed by the European Union through the 7th Framework People Programme (grant agreement number 238531). Financial support from the Spanish MICINN (Projects CTQ-2007-61126, MAT-2006-01997 and MAT-2010-15094) is also gratefully acknowledged. V. G. -F. and I. L. acknowledge MICINN for their research contracts under the Ramón y Cajal Program.References
- (a) S. Chowdhury, R. S. Mohan and J. L. Scott, Tetrahedron, 2007, 63, 2363–2389 CrossRef; (b) V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS; (c) Š. Toma, M. Mèeiearová and R. Šebesta, Eur. J. Org. Chem., 2009, 321–327 CrossRef CAS.
- (a) T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS; (b) Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- (a) X. Han and D. W. Armstrong, Acc. Chem. Res., 2007, 40, 1079–1086 CrossRef CAS; (b) M. F. Costa Gomes and P. Husson, in Ionic Liquids: from Knowledge to Application, ed. N. V. Plechkova, R. D. Rogers and K. R. Seddon, Oxford University Press, Washington, 2009, pp. 223–238 Search PubMed.
- (a) S. Chun, S. V. Dzyuba and R. A. Bartsch, Anal. Chem., 2001, 73, 3737–3741 CrossRef CAS; (b) A. Berthod, M. J. Ruiz-Angel and S. Carda-Broch, J. Chromatogr., A, 2008, 1184, 6–18 CrossRef CAS; (c) M.-C. Tseng, M.-J. Tseng and Y.-H. Chu, Chem. Commun., 2009, 7503–7505 RSC; (d) G. W. Meindersma and A. B. de Haan, in Ionic Liquids: from Knowledge to Application, ed. N. V. Plechkova, R. D. Rogers and K. R. Seddon, Oxford University Press, Washington, 2009, pp. 255–272 Search PubMed.
- (a) K. P. Doyle, C. M. Lang, K. Kim and P. A. Kohl, J. Electrochem. Soc., 2006, 153, A1353–A1357 Search PubMed; (b) D. R. Macfarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS.
- (a) D. W. Armstrong, L. Zhang, L. He and M. L. Gross, Anal. Chem., 2001, 73, 3679–3686 CrossRef CAS; (b) S. Liangliang, T. Dingyin, H. Bin, M. Junfeng, Z. Guijie, L. Zhen, S. Yichu, Z. Lihua and Z. Yukui, Anal. Bioanal. Chem., 2011, 399, 3387–3397 Search PubMed; (c) P. J. Dyson, M. A. Henderson and J. S. McIndoe, in Ionic Liquids: from Knowledge to Application, ed. N. V. Plechkova, R. D. Rogers and K. R. Seddon, Oxford University Press, Washington, 2009, pp. 135–146 Search PubMed.
- P. Sun and D. W. Armstrong, Anal. Chim. Acta, 2010, 661, 1–16 CrossRef CAS.
- (a) For recent revisions in the field of chiral ionic liquids, see: A. Winkel, P. V. G. Reddy and R. Wilhelm, Synthesis, 2008, 999–1016 Search PubMed; (b) X. Chen, X. Li, A. Hu and F. Wang, Tetrahedron: Asymmetry, 2008, 19, 1–14 CrossRef CAS; (c) J.-C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A.-C. Gaumont, Chem. Rev., 2008, 108, 5035–5060 CrossRef CAS; (d) K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235–3250 CrossRef CAS; (e) P. Domínguez de Maria, Angew. Chem., Int. Ed., 2008, 47, 6960–6968 CrossRef; (f) R. Šebesta, I. Kmentova and S. Toma, Green Chem., 2008, 10, 484–496 RSC; (g) M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688 RSC; (h) S. Luo, L. Zhang and J.-P. Cheng, Chem.–Asian J., 2009, 4, 1184–1195 CrossRef CAS; (i) M. H. G. Prechtl, J. D. Scholten, B. A. D. Neto and J. Dupont, Curr. Org. Chem., 2009, 13, 1259–1277 CrossRef CAS; (j) B. Ni and A. D. Headley, Chem.–Eur. J., 2010, 16, 4426–4436 CrossRef CAS; (k) A.-C. Gaumont, Y. Génisson, F. Guillen, V. Zgonnik and J.-C. Plaquevent, in Catalytic Methods in Asymmetric Synthesis, ed. M. Gruttadauria and F. Giacalone, John Wiley & Sons, Hoboken, 2011, pp. 323–344 Search PubMed.
- P. M. Dean, J. M. Pringle and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2010, 12, 9144–9153 RSC.
- M. Petkovic, K. R. Seddon, L. P. N. Rebelo and C. Silva Pereira, Chem. Soc. Rev., 2011, 40, 1383–1403 RSC.
- (a) N. Srivastava and B. K. Banik, J. Org. Chem., 2003, 68, 2109–2114 CrossRef CAS; (b) K. R. Reddy and N. S. Kumar, Synlett, 2006, 2246–2250 Search PubMed; (c) M. Kawatsura, S. Aburatani and J. Uenishi, Tetrahedron, 2007, 63, 4172–4177 Search PubMed; (d) C. Mukherjee and A. K. Misra, Lett. Org. Chem., 2007, 4, 54–59 Search PubMed; (e) M. I. Uddin, K. Nakano, Y. Ichikawa and H. Kotsuki, Synlett, 2008, 1402–1406 CAS.
- C. O. Kappe, D. Dallinger and S. S. Murphee, Practical Microwave Synthesis for Organic Chemists, Wiley-VCH, Weinheim, 2008 Search PubMed.
- (a) Recent bibliography: G. W. Huisman, J. Liang and A. Krebber, Curr. Opin. Chem. Biol., 2010, 14, 122–129 Search PubMed; (b) E. García-Urdiales, I. Alfonso and V. Gotor, Chem. Rev., 2011, 111, PR110–PR180 Search PubMed; (c) F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2313 RSC; (d) M. M. Musa and R. S. Phillips, Catal. Sci. Technol., 2011, 1, 1311–1323 RSC.
- (a) E. Marcantoni, S. Alessandrini and M. Malavolta, J. Org. Chem., 1999, 64, 1986–1992 CrossRef CAS; (b) L. M. Levy and V. Gotor, J. Org. Chem., 2004, 69, 2601–2602 CrossRef CAS.
- (a) V. Gotor, I. Alfonso and E. García-Urdiales, Asymmetric Synthesis with Enzymes, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) B. M. Nestl, B. A. Nebel and B. Hauer, Curr. Opin. Chem. Biol., 2011, 15, 187–193 CrossRef CAS.
- (a) For recent revisions: T. Hudlicky and J. W. Reed, Chem. Soc. Rev., 2009, 38, 3117–3132 Search PubMed; (b) R. Yuryev and A. Liese, ChemCatChem, 2010, 2, 103–107 Search PubMed; (c) S. Wenda, S. Illner, A. Mell and U. Kragl, Green Chem., 2011, 13, 3007–3047 RSC.
- A. Ghanem, Tetrahedron, 2007, 63, 1721–1754 CrossRef CAS.
- (a) O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS; (b) T. Y. S. But and P. H. Toy, Chem.–Asian J., 2007, 2, 1340–1355 CrossRef CAS; (c) K. C. K. Swamy, N. N. B. Kumar, E. Balaraman and K. V. P. P. Kumar, Chem. Rev., 2009, 109, 2551–2651 CrossRef.
- (a) E. Busto, V. Gotor-Fernández, N. Ríos-Lombardía, E. García-Verdugo, I. Alfonso, S. García-Granda, A. Menéndez-Velázquez, M. I. Burguete, S. V. Luis and V. Gotor, Tetrahedron Lett., 2007, 48, 5251–5254 CrossRef CAS; (b) N. Ríos-Lombardía, E. Busto, V. Gotor-Fernández, V. Gotor, R. Porcar, E. García-Verdugo, S. V. Luis, I. Alfonso, S. García-Granda and A. Menéndez-Velázquez, Chem. Eur. J., 2010, 16, 836–847 Search PubMed.
- K. Fukumoto, M. Yoshizawa and H. Ohno, J. Am. Chem. Soc., 2005, 127, 2398–2399 CrossRef CAS.
- Crystal data: C16H21BrN2O, M = 337.26, orthorhombic, a = 8.48954(9) Å, b = 13.2411(2) Å, c = 14.0540(2) Å, V = 1579.84(3) Å3, T = 298(2)K, space group P212121, Z = 4, 8728 reflections measured, 2948 unique (Rint = 0.033), which were used in all calculations. The final wR(F2) was 0.0622 (all data). Crystallographic data (excluding structure factors) for the structure (1S,3R)-cis-IM-OH-Bn-Br have been deposited with the Cambridge Crystallographic Data Centre publication number CCDC858900. Copies of the data can be obtained free of charge from the Cambridge Crystallographic Data Centre from http://www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi.
- (a) M. López-García, I. Alfonso and V. Gotor, Chem.–Eur. J., 2004, 10, 3006–3014 CrossRef CAS; (b) L. M. Levy, G. de Gonzalo and V. Gotor, Tetrahedron: Asymmetry, 2004, 15, 2051–2056 CrossRef CAS.
- K. Maruoka, Org. Process Res. Dev., 2008, 12, 679–697 Search PubMed.
- N. Ríos-Lombardía, R. Porcar, E. Busto, I. Alfonso, J. Montejo-Bernardo, S. García-Granda, V. Gotor, S. V. Luis, E. García-Verdugo and V. Gotor-Fernández, ChemCatChem, 2011, 3, 1921–1928 Search PubMed.
- (a) See for example: A. S. Wells and V. T. Coombe, Org. Process Res. Dev., 2006, 10, 794–798 Search PubMed; (b) J. Ranke, S. Stolte, R. Stormann, J. Arning and B. Jastorff, Chem. Rev., 2007, 107, 2183–2206 CrossRef CAS; (c) S. V. Malhorta and V. Kumar, Bioorg. Med. Chem. Lett., 2010, 20, 581–585 CrossRef CAS; (d) L. Myles, R. Gore, M. Špulák, N. Gathergood and S. J. Connon, Green Chem., 2010, 12, 1157–1162 RSC; (e) G. V. S. M. Carrera, R. F. M. Frade, J. Aires-de-Sousa and C. A. M. Afonso, Tetrahedron, 2010, 66, 8785–8794 CrossRef CAS.
- (a) M. Rebros, H. Q. N. Gunaratne, J. Ferguson, K. R. Seddon and G. Stephens, Green Chem., 2009, 11, 402–408 RSC; (b) N. Wood, J. L. Ferguson, H. Q. N. Gunaratne, K. R. Seddon, R. Goodacre and G. Stephens, Green Chem., 2011, 13, 1843–1851 RSC.
- R. F. M. Frade, A. A. Rosatella, C. S. Marques, L. C. Branco, P. S. Kulkarni, N. M. M. Mateus, C. A. M. Afonso and C. M. M. Duarte, Green Chem., 2009, 11, 1660–1665 RSC.
- P. Balczewski, B. Bachowska, T. Bialas, R. Biczak, W. M. Wieczorek and A. Balinska, J. Agric. Food Chem., 2007, 55, 1881–1892 CrossRef CAS.
- N. Wood and G. Stephens, Phys. Chem. Chem. Phys., 2010, 12, 1670–1674 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Full characterisation of novel compounds with spectroscopic data and HPLC analysis are given. Toxicity studies are also reported including tables with data for the better understanding of the manuscript. CCDC reference number 858900. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20876h |
| This journal is © The Royal Society of Chemistry 2012 |