DOI:
10.1039/C2RA20630G
(Paper)
RSC Adv., 2012,
2, 6464-6477
Highly efficient base catalysis and sulfide oxidation reactions over new functionalized mesoporous polymers
Received
5th April 2012
, Accepted 7th May 2012
First published on 9th May 2012
Abstract
Base catalyzed aldol condensation reactions and selective partial oxidation of sulfides are very important classes of reactions in synthetic organic chemistry. We have prepared a new alkali exchanged form of mesoporous polytriallylamine, OH-MPTA-1 and a Cr-grafted material Cr-MPTA-1 via the simple and facile in situ radical polymerization of a triallylamine template by organic–organic self-assembly of anionic surfactant systems. The materials have been characterized by elemental analysis, powder X-ray diffraction (XRD), transmission electron microscope (TEM), Fourier transform infrared spectroscopy (FT-IR), UV-vis diffuse reflectance spectroscopy (DRS), thermogravimetric analysis (TGA) and scanning electron microscopy (SEM). Powder XRD and TEM image analyses suggests the presence of mesophase and disordered wormhole-like mesopores in these samples. Our experimental results reveal that the alkali exchanged material OH-MPTA-1 acts as a very efficient base catalyst for the direct aldol reaction of different aromatic aldehydes with acetone. On the other hand, the Cr-grafted material Cr-MPTA-1, obtained through the reaction of potassium chromate with MPTA-1, acts as a very efficient catalyst for the liquid phase partial oxidation of sulfides to sulfoxides. Both OH-MPTA-1 and Cr-MPTA-1 can be recycled more than five times without an appreciable loss in activity for their respective catalytic reactions.
Introduction
Organic–inorganic hybrid mesoporous materials possessing a high-content of organic groups are a unique class of materials and they are widely studied in the areas of catalysis, separation science, adsorption, environmental remediation, sensing and electronics.1–3 Mesoporous materials with a tuneable pore structure and tailored framework composition can satisfy this area owing to their distinctive properties like high surface area, uniform pore size distribution (PSD), plenty of well distributed active sites and ease of diffusion for large reacting molecules through their pore channels.4 These materials are largely explored as adsorbents,5 exchangers,6 acid–base catalysts,7 light-harvesting materials,8 energy storage and conversion materials,9 and biological applicatons.10 Soft templating approachs employing a supramolecular assembly formed by the liquid crystalline array of surfactant molecules under mild sol–gel synthesis conditions have attracted increasing attention in designing these materials over a decade.11–13 A large variety of mesoporous frameworks have been fabricated, where soft templates like surfactants or hard templates like mesoporous silica or carbon played crucial role in directing organized porous structures through self-aggregation process.14
The aldol reaction is one of the most powerful methods of forming carbon–carbon bonds and it has tremendous synthetic applications.15 Biological systems have perfected this stereospecific transformation by using enzymes: type I aldolases function via the formation of an enamine intermediate with a lysine residue, whereas type II aldolases activate substrates by forming a zinc enolate. Despite their lack of large-scale compatibility and their narrow substrate specificity, aldolases represent a great source of inspiration for the development of catalysts. In general, the aldol reaction proceeds in homogeneous acid or basic solutions through carbocation or carbanion mechanisms.16 In recent years, several aldol reactions using acid or base catalysts have been reported,17 including iron-catalyzed cross-aldol reactions,18 proline-loaded catalysts for stereoselectivity,19,20 and aldol-type reactions of lithium enolate21 and silyl enolate.22 Currently, due to low toxicity, operational simplicity and efficiency, compared to traditional metal-based catalysts, there is much interest in organocatalysts.23 Organocatalysis is the acceleration of chemical reactions with a sub-stoichiometric amount of an organic compound, which does not contain a metal atom.24 Organocatalysts that can promote aldol reactions have recently been reported by several research groups.25 Some new direct intermolecular reactions such as Mannich,26 Michael27 and other analogous reactions have also been reported using organocatalysts. However, their drawbacks also have been realized. The major limitations using organocatalyst catalyzed reactions are high catalyst loadings (up to 20 mol%) and the difficulty of recovering the catalyst.28 An alternative strategy is to design recyclable and subsequently reusable versions of organocatalysts.29
On the other hand, in recent years interest in sulfoxide chemistry has been increasing at a very fast rate. Sulfoxides are the valuable intermediates in organic synthesis30 and in the formation of biologically active molecules.31 They play an important role as cardiotonic agents,32 anti-hypertensives,33 as well as vasodilators.34 The selective oxidation of sulfides is a simple technique that can be used to obtain sulfoxides and sulfones. Many reagents are available for the oxidation of sulfides such as halogen compounds, nitrates, transition metal oxides, oxygen and hydrogen peroxide.35 Among them, hydrogen peroxide has attracted much attention over the years because it forms harmless by-products and product work-up is simple. Previous research has shown that soluble transition metal based complexes are effective catalysts for the oxidation of sulfides when using hydrogen peroxide as an oxidant.36 Although homogenous catalysts have favourable catalytic abilities,37,38 heterogeneous catalysts are of considerable industrial interest as they can be easily separated from the reaction mixture and recycled. Some heterogeneous catalysts have been applied to the oxidation of sulfides.39 However, these catalytic systems suffer from at least one of the following shortcomings: long reaction time, low conversion of sulfide, bad selectivity between sulfoxide and sulfone, high catalyst quantities and excessive H2O2 consumption. Thus, an efficient heterogeneous catalytic system for the liquid phase selective partial oxidation of sulfides is desirable.
It is pertinent to mention here that we have recently developed a series of polymer anchored transition metal compounds,40 a few of which proved to be effective catalysts with good activity at ambient reaction conditions. Also, the complexes efficiently mediated oxidation and coupling reactions of organic substrates in aqueous media.41 In this context functionalized mesoporous materials containing donor functional groups at the surface of the pore can play an important role in designing an efficient catalytic material. Although there are several reports on the use of functionalized mesoporous materials as solid acid catalysts, there use in base catalyzed reactions is quite rare.42 Very recently, mesoporous polytriallylamine (MPTA-1) has been synthesized by using sodiumlaurylsulfate as template.43 Inspired by the above observations, in the present work we have designed a new alkali exchanged purely organic mesoporous polymer, OH-MPTA-1, by using the supramolecular assembly of surfactant in situ during the radical polymerization of triallylamine under a sol–gel-driven soft templating approach followed by its alkali exchange. This material has been utilized as a heterogeneous base catalyst for direct aldol condensation reactions of various aldehydes with acetone. We have also prepared the Cr-grafted derivative of this mesoporous polymer, Cr-MPTA-1 catalyst, by immobilized the metal on MPTA-1 surface and its catalytic activity in the selective oxidation of organic sulfides using H2O2 as an oxygen source has been explored.
Experimental section
Materials
Triallylamine (TAA) was obtained from Sigma Aldrich. N,N,N′,N′-tetramethylenediamine (TEMED) and ammonium persulfate (APS) were obtained from Loba Chemie, India. Sodium lauryl sulfate (SDS) was purchased from Loba Chemie and used as a structure-directing agent. Potassium chromate (K2CrO4) and conc. HCL were obtained from Merck, India.
Physical measurements
Powder X-ray diffraction (XRD) patterns of different samples were analyzed with a Bruker D8 Advance X-ray diffractometer using Ni-filtered Cu–Kα (λ = 0.15406 nm) radiation. Transmission electron microscopy (TEM) images of the mesoporous polymer were obtained using a JEOL JEM 2010 transmission electron microscope operating at 200 kV. FTIR spectra of the samples were recorded on a Perkin-Elmer FTIR 783 spectrophotometer using KBr Pellets. Nitrogen adsorption–desorption experiments were carried out using a Bel Japan Inc. Belsorp-HP surface area analyzer at 77 K. Prior to gas adsorption, the sample was degassed for 4 h at 393 K under high vacuum conditions. UV-vis spectra were taken using a Shimadzu UV-2401PC double beam spectrophotometer with an integrating sphere attachment for solid samples. Surface morphology of the samples was measured using a scanning electron microscope (SEM) (ZEISS EVO40, England) equipped with EDX facility. Analytical high performance liquid chromatography (HPLC) was carried out on an Agilent 1200 instrument. The reaction products were quantified (GC data) by Varian 3400 gas chromatograph equipped with a 30 m CP-SIL8CB capillary column and a flame ionization detector and identified by Trace DSQ II GC-MS equipped with a 60 m TR-50MS capillary column.
Preparation of the catalyst
Synthesis of mesoporous polytriallylamine MPTA-1.
MPTA-1 was synthesized through the aqueous-phase polymerization of triallylamine under hydrothermal conditions using APS as an initiator (Scheme 1).42 In a typical synthesis, 2.35 g (0.00814 mol) of SDS was dissolved in 40 ml of water with constant stirring followed by the addition of 2.23 g (0.01628 mol) of TAA. Then, 0.94 g (0.00814 mol) of TEMED was added in this mixture and conc. HCL (12N) was also added dropwise to obtain a clear solution. This helps the protonation of the N atoms of the triallylamine molecules, which facilitates the ionic interaction with the C13H25SO3 of the SDS molecules. The pH of the gel was maintained at ca 7.0. Finally, 7.42 g (0.03256 mol) of APS dissolved in 10 ml of water was added quickly into the solution with vigorous stirring and a white precipitate appeared immediately. The resultant slurry was stirred for another 1 h and then autoclaved at 348 K for 3 days without stirring. Finally, the pH of the synthesis gel was ca 4.0–5.0. The resultant precipitate was filtered and washed with deionized water, yielding the mesoporous polymer MPTA-1.
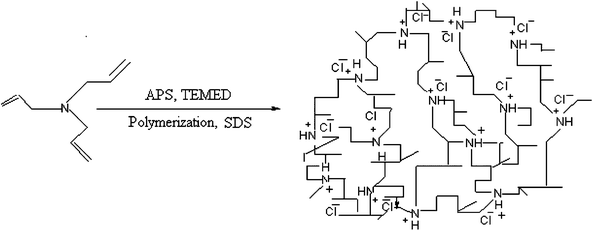 |
| | Scheme 1 Schematic diagram showing the formation of the mesoporous polymer MPTA-1. | |
Preparation of the OH-MPTA-1 material.
The mesoporous polymer MPTA-1 was functionalized with an –OH− group (Scheme 2). In an experiment, MPTA-1 was treated several times with NaHCO3 until MPTA-1 was neutralized. The neutralized MPTA-1 was stirred vigorously with 0.5 mmol of NaOH solution at room temperature for 12 h. As a result the Cl− ion of MPTA-1 was replaced by an OH− ion.
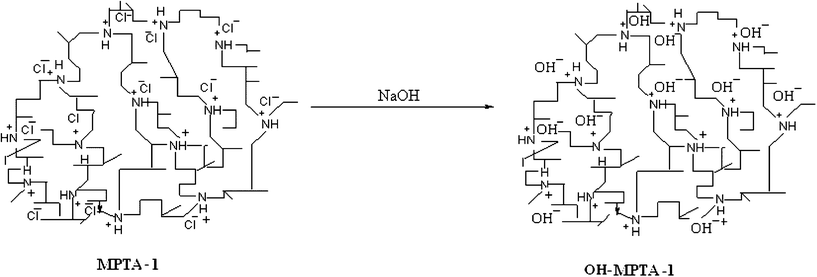 |
| | Scheme 2 Diagram showing formation of the OH-functionalized mesoporous polymer OH-MPTA-1. | |
Preparation of the Cr-MPTA-1 material.
The suspension of mesoporous polytriallylamine (500 mg), 10 ml double distilled water and potassium chromate (100 mg) was placed in a round bottom flask (100 ml) and kept it under a vigorous stirring condition for 24 h at room temperature. Finally, the resultant precipitate was filtered and washed with double distilled water and dried under vacuum.
General procedure for the direct aldol reaction.
The organo-catalyst OH-MPTA-1 was stirred with 10 mmol of acetone for 30 min at room temperature. To this mixture corresponding aldehyde (5 mmol) was added and the resulting mixture was stirred for an appropriate time (see Table 1). After completion of the reaction, the reaction mixture was filtered and the filtrate was extracted with ethyl acetate (3 × 10 mL). The organic layer was washed with water and dried over Na2SO4. After evaporation of the solvent under reduced pressure, the crude product was purified by column chromatography over silica gel using ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane (2:8) as the eluent to afford the pure the aldol product. The solid filtrate was washed with acetone (5 × 2 mL), and dried at 70 °C, being ready for a new catalytic reaction.
:hexane (2:8) as the eluent to afford the pure the aldol product. The solid filtrate was washed with acetone (5 × 2 mL), and dried at 70 °C, being ready for a new catalytic reaction.
Table 1 Direct aldol reaction of acetone and p-nitrobenzaldehyde catalyzed by catalyst OH-MPTA-1a
| Entry |
Wt % of catalyst |
Solvent |
Temperature |
Conversionb |
Aldol adductc |
|
Reaction conditions: p-nitrobenzaldehyde (5 mmol); acetone (10 mmol); time: 14 h.
Conversion and selectivity were determined by HPLC.
Isolated yields were obtained after silica gel column chromatography.
Reaction time 24 h.
|
| 1 |
2 |
Acetone |
25 °C |
65d |
74 |
| 2 |
4 |
Acetone |
25 °C |
65 |
76 |
|
3
|
6
|
Acetone |
25 °C |
96
|
88
|
| 4 |
6 |
ACN |
25 °C |
62 |
79 |
| 5 |
6 |
MeOH |
25 °C |
68 |
74 |
| 6 |
6 |
DMSO |
25 °C |
49 |
76 |
| 7 |
6 |
THF |
25 °C |
41 |
80 |
| 8 |
6 |
DMF |
25 °C |
67 |
73 |
| 9 |
6 |
Toluene |
25 °C |
7 |
59 |
| 10 |
6 |
Acetone |
10 °C |
35 |
90 |
| 11 |
6 |
Acetone |
0 °C |
No reaction |
— |
| 12 |
6 |
Acetone |
50 °C |
96 |
69 |
General experimental procedure for the oxidation reaction of sulfides using H2O2 as an oxidant.
A typical experimental procedure for the oxidation reaction of sulfides using Cr-MPTA-1 catalyst is described as follows: a mixture of sulfide (5.0 mmol), catalyst (0.050 g), and 30% hydrogen peroxide (10 mmol) in acetonitrile (10 mL) was stirred at ambient temperature. At the end of specified time, the reaction mixture was filtered and the filtrate was analyzed using a Varian 3400 gas chromatograph equipped with a 30 m CP-SIL8CB capillary column and a flame ionization detector. Peak position of various reaction products were compared and matched with the retention times of authentic samples. Identification of the products was also confirmed using GC-MS.
Formation mechanism of the mesoporous polymer MPTA-1.
The organic–organic supramolecular assembly of the anionic surfactant SDS or a mixture of anionic/non-ionic surfactants in the aqueous solution could direct the formation of these organic mesoporous materials. Here the organic polymerized moieties (which are positively charged through the protonation of the nitrogen atoms) preferentially interacted with the negatively charged C13H25SO3− head groups of the surfactants and condensed into a solid and continuous framework. The resulting arrays of organic mesophases upon removal of the surfactants through acid–solvent extractions retained their mesostructure and generated mesoporosity. Scheme 1 shows a schematic diagram of the polymerization of TAA and the formation of the polytriallylamine pore-wall. APS is a good radical initiator and reactive oxygen source under aqueous-phase polymerization of olefins44 under mildly acidic conditions. Since one TAA molecule contains three olefin double bonds in each of its branches, this polymerization process gives random cross-linking, leading to a high stability of the organic framework. In Scheme 3 a proposed reaction mechanism for the radical polymerization in the presence of APS is shown. Under mildly acidic conditions, APS behaves in a similar way, polymerizing olefins44b to yield the cross-linked MPTA-1 framework.
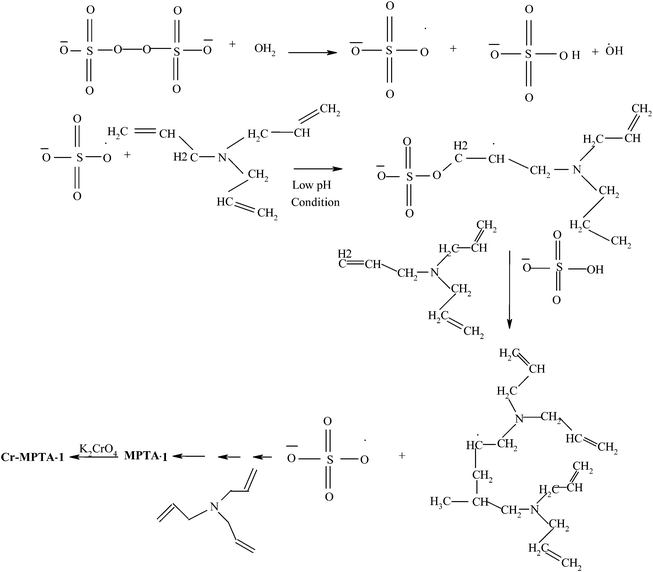 |
| | Scheme 3 Proposed reaction scheme for the synthesis of Cr-MPTA-1. | |
Results and discussion
Characterization of the OH-MPTA-1 and Cr-MPTA-1 materials
Spectroscopic analysis.
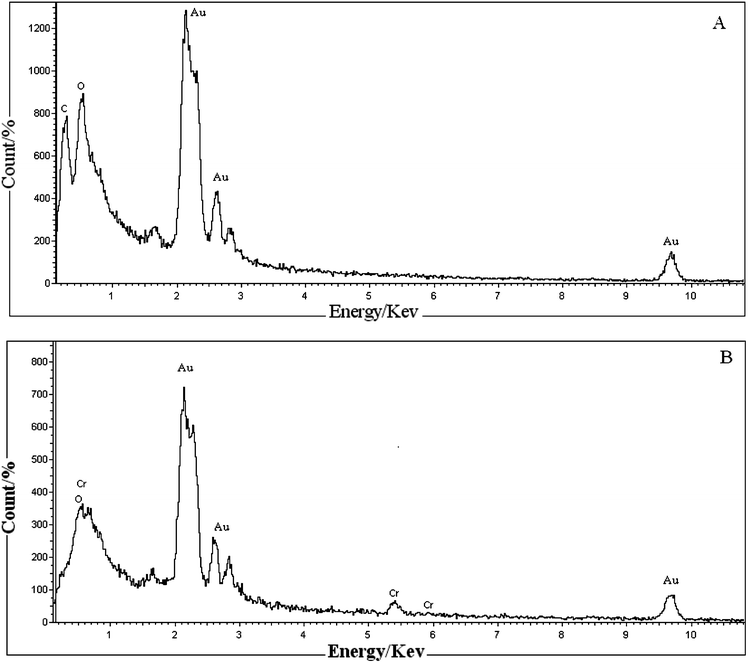
FT-IR spectra of the OH-MPTA-1 and Cr-MPTA-1 samples are shown in Fig. 1a and 1b, respectively. In the OH-MPTA-1 sample, two distinct C–H vibrations of the sp3 were observed in the 2800–2950 cm−1 region, corresponding to non-identical C–H bonds in the polytriallylamine matrices. Further, it exhibits a very broad and strong band at 3436 cm−1 (ν-N–H) and two additional sharp peaks at 1217 (ν-C–N) and 1467 cm−1 (ν-CH2;δ-N–H). Other stretching frequencies and skeletal vibrations agree quite well with the FT-IR spectra of an N-protonated triallylamine.45 Further, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C triallylamine vibration at 1840 cm−1 disappeared, and the intensity of another at 1630 cm−1 has diminished considerably (Fig. 1), which attributed near-completion of the polymerization reaction. Thus, the presence of these framework vibrations prove that protonated and saturated tertiary amine moieties are present in the OH-MPTA-1 samples. The band at 1650 cm−1, belonging to the amine group, is present in the OH-MPTA-1 sample. The intensity of this band is slightly decreased after complexation with chromium. The FTIR spectra of the Cr-MPTA-1 shows a band at 920 cm−1. This band is absent from the mesoporous polymer OH-MPTA-1 due to a Cr–O or CrO vibration from the Cr(VI) species in Cr-MPTA-1 materials.46 The bands at 623 and 569 cm−1 are found to be due to extra framework chromium oxide as reported earlier.47 UV-vis spectra of mesoporous Cr-MPTA-1 samples show two characteristic peak as shown in Fig. 2. The bands at 224 and 340 nm are assigned to the O to Cr(VI) charge transfer transitions of chromate species.48 The absence of Cr2O3 bands at 560 nm indicated that the trivalent chromium is absent in the sample.
C triallylamine vibration at 1840 cm−1 disappeared, and the intensity of another at 1630 cm−1 has diminished considerably (Fig. 1), which attributed near-completion of the polymerization reaction. Thus, the presence of these framework vibrations prove that protonated and saturated tertiary amine moieties are present in the OH-MPTA-1 samples. The band at 1650 cm−1, belonging to the amine group, is present in the OH-MPTA-1 sample. The intensity of this band is slightly decreased after complexation with chromium. The FTIR spectra of the Cr-MPTA-1 shows a band at 920 cm−1. This band is absent from the mesoporous polymer OH-MPTA-1 due to a Cr–O or CrO vibration from the Cr(VI) species in Cr-MPTA-1 materials.46 The bands at 623 and 569 cm−1 are found to be due to extra framework chromium oxide as reported earlier.47 UV-vis spectra of mesoporous Cr-MPTA-1 samples show two characteristic peak as shown in Fig. 2. The bands at 224 and 340 nm are assigned to the O to Cr(VI) charge transfer transitions of chromate species.48 The absence of Cr2O3 bands at 560 nm indicated that the trivalent chromium is absent in the sample.
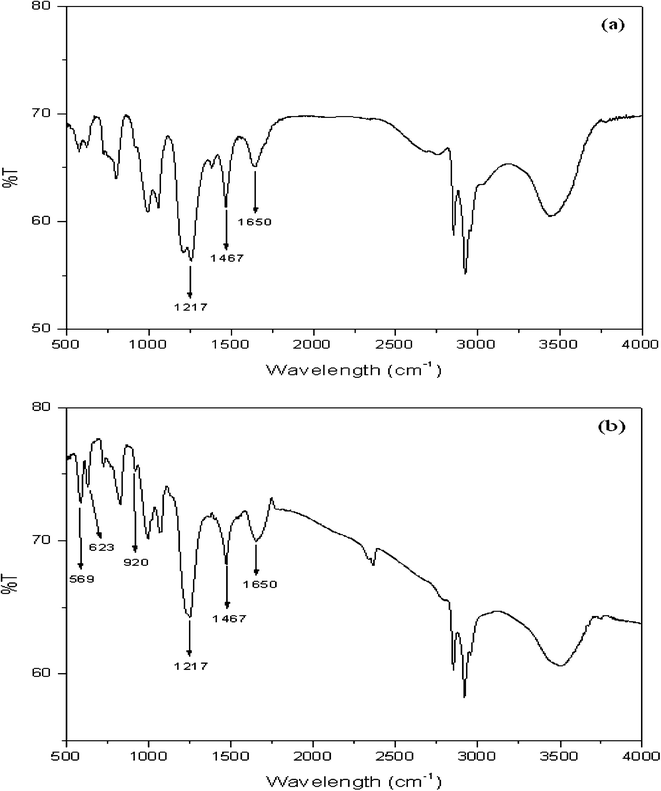 |
| | Fig. 1 FT-IR spectra of mesoporous OH-MPTA-1 (a) and Cr-MPTA-1 (b). | |
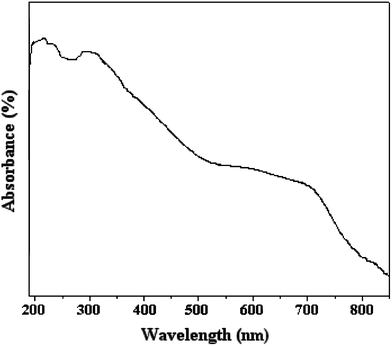 |
| | Fig. 2 DRS-UV visible absorption spectra of mesoporous Cr-MPTA-1. | |
N2 adsorption–desorption studies.
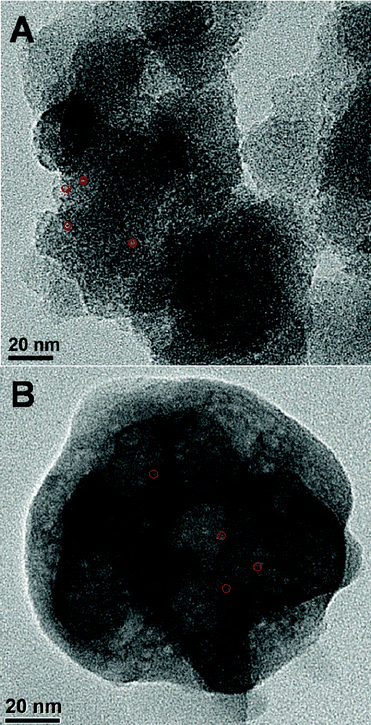
N2 adsorption–desorption studies of the mesoporous Cr-MPTA-1 and MPTA-1(OH) samples were carried out at 77 K. The BET surface areas for the Cr-MPTA-1 and MPTA-1(OH) samples were 11 m2 g−1 and 18 m2 g−1, respectively with corresponding pore volumes of 0.0209 cc g−1 and 0.0434 cc g−1, respectively. A considerable decrease in the BET surface area and pore volume from the parent MPTA-143 material suggests that OH−/CrO42− could have been anchored at the surface of the pores. The pore size distribution of both the samples calculated by using non-local density functional theory (NLDFT) method suggests pore dimensions in the mesopore length scale: ca 3.1 nm and 3.3 nm for OH-MPTA-1 and Cr-MPTA-1 samples, respectively.
X-ray diffraction.
In Fig. 3 the small angle powder XRD patterns of MPTA-1, OH-MPTA-1, Cr-MPTA-1 samples are shown. For all these samples a single high intensity diffraction peak in the small angle 2θ region of 2.0 to 3.0 was observed. This is attributed to the presence of a disordered mesostructure in these materials. When this parent MPTA-1 mesoporous polymer (Fig. 3a) was functionalized with OH− and CrO42− ions, a considerable decrease in the intensity of peak is observed, however a disordered mesophase has been retained. This decrease in the peak intensity on grafting of OH− and CrO42− at the surface could be attributed to more disorder of the respective mesophases.
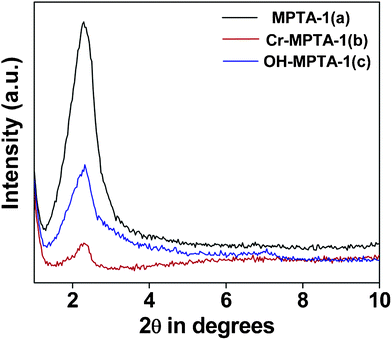 |
| | Fig. 3 Small-angle powder XRD patterns of MPTA-1 (a), Cr-MPTA-1 (b) and OH-MPTA-1 (c). | |
Catalytic activity
Since mesoporous supported metal systems exhibit high catalytic activity in a wide range of the industrially important processes and have been extensively studied, we decided to investigate the catalytic activity of the mesoporous polymer OH-MPTA-1 and Cr-MPTA-1 in the direct aldol reaction and sulphide oxidation reactions, respectively.
Direct aldol reaction of aldehydes.
The aldol reaction is one of the most important carbon–carbon bond forming processes utilized in organic synthesis. β-hydroxy carbonyl compounds are subunits of many natural products. The efficiency of OH-MPTA-1 as a base catalyst was first tested in aldol reaction. The OH-functionalized mesoporous material (OH-MPTA-1) has been used as a catalyst in the liquid phase aldol reactions over various electron-withdrawing and electron-donating aromatic aldehydes with acetone.
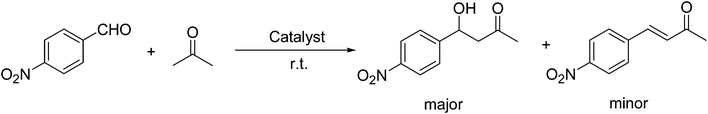
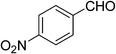
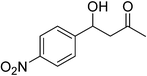
First, OH-MPTA-1 was chosen for optimization studies. As an initial test run, the aldol reaction between acetone and p-nitrobenzaldehyde was carried out (Scheme 4) under different experimental conditions such as solvent, temperature and the amount of catalyst and the results are summarized in Table 1. The temperature had a pronounced effect on the yield. In most cases, the yields ascend with decreasing temperature. However, as seen in Table 1, entries 10 and 11, conversion of 4-nitrobenzaldehyde, decreased at lower temperatures. The above reaction did not proceed at 0 °C even after several hours. At high temperature (50 °C), the yield of aldol adduct decreased. Therefore, the room temperature, 25 °C, was appropriate for the direct aldol reaction of p-nitrobenzaldehyde and acetone and the best yield was up to 96% (Table 1, entry 3).
 |
| | Scheme 4 Reaction pathway for the direct aldol reaction over OH-functionalized mesoporous polymer OH-MPTA-1. | |
Next, the effect of the solvent was examined. Performing the reaction in several polar solvents such as acetonitrile, MeOH, DMSO, THF, and DMF always resulted in diminished yields, compared to the result obtained with acetone as the solvent (Table 1, entries 3–8). When the solvent was changed to the non-polar toluene, the product was obtained with lower yield (Table 1, entry 9).
The above reaction was also carried out under the influence of 2, 4 and 6 wt% catalyst concentrations. When 4 wt% of catalyst was used, the reaction was accelerated and afforded the product in 65% yield (Table 1, entry 2). When the loading of the catalyst was decreased from 4 to 2 wt%, there was no change in the yield but the reaction took 24 h to afford similar results (Table 1, entry 1). When the reaction was conducted with 6 wt% of catalyst, the aldol product was obtained in highest yield (Table 1, entry 3).
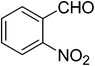
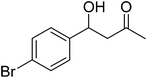
The optimal conditions were employed for the catalytic aldol reaction of acetone with a broad scope of aldehydes and the results of this study are summarized in Table 2. The reactions of various aromatic aldehydes, which bear an electron-withdrawing group on the benzene ring, proceeded smoothly in excellent yields (up to 96%). The electron-withdrawing group present in the para position of the aromatic ring increases the yield by activating the aldehyde group due to the −I effect. The catalytic system displays some sensitivity toward steric effects. When a –NO2 group is present in the ortho position of the aromatic ring the yield of the product was minimized. This could be attributed to the steric hindrance at the reaction site. On the other hand the yield for the –OH group containing substrate decreases a little bit due to the +R effect of the –OH group present in the ring. The halo and cyano substituted benzaldehydes were also effective for this conversion (Table 2, entries 4–6). In the case of naphthaldehyde, the yield was moderate (Table 2, entry 8). This catalytic system was proven to be ineffective for the aromatic aldehydes with an electron-donating group or without any substitution (Table 2, entries 7, 9 and 10). In the case of p-methoxybenzaldehyde and p-methylbenzaldehyde, the yields were very low (Table 2, entries 9 and 10). This may be due to the electron donating ability of the methoxyl and methyl group, which may increase the electron density at the carbonyl group. In several instances, dehydrated products were also obtained along with the aldol products. The ratio of dehydrated products increased with increase in temperature.
Table 2 Direct aldol reaction of different aldehydes and acetone over OH-MPTA-1a
| Entry |
Aldehydes |
Products |
Time (h) |
Conversion (%)b |
Selectivity of aldol adductc |
|
Reaction conditions: aldehyde (5 mmol); acetone (10 mmol); 6 wt% of catalyst, reaction T = 25 °C.
Conversion and selectivity were determined by HPLC.
Isolated yields were obtained after silica gel column chromatography.
|
| 1 |
|
|
14 |
96 |
88 |
| 2 |
|
|
14 |
74 |
75 |
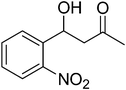
| 3 |
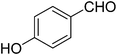
|
|
14 |
82 |
82 |
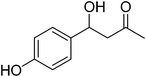
| 4 |

|
|
14 |
87 |
83 |
| 5 |
|
|
24 |
75 |
81 |
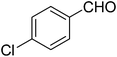
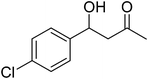
| 6 |
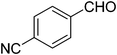
|
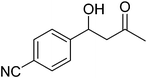
|
18 |
75 |
85 |
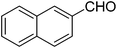
| 7 |

|
|
24 |
49 |
74 |
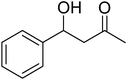
| 8 |
|
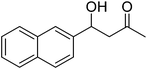
|
24 |
52 |
81 |
| 9 |
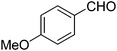
|
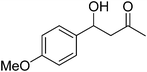
|
24 |
12 |
77 |
| 10 |
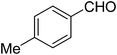
|
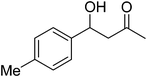
|
24 |
10 |
70 |
Oxidation of sulfides with H2O2 catalyzed by Cr-MPTA-1.
Oxidation of sulfides is the most straight forward method for the synthesis of sulfoxides and sulfones, both of which are important as commodity chemicals and, in some cases, as pharmaceuticals. To test the activity of the Cr-MPTA-1 as an oxidation catalyst, first the oxidation of sulfides was conducted with hydrogen peroxide.
For optimization of the reaction conditions, we chose oxidation of diphenylsulfide as a probe reaction (Scheme 5). Firstly, the oxidations of diphenylsulfide with 30% hydrogen peroxide using Cr-MPTA-1 as a catalyst were examined in several solvents (Table 3). The reactions were carried out using methanol, DMF, acetonitrile, CHCl3 and toluene as solvents. The data in Table 3 clearly shows that acetonitrile, fully miscible with the organic substrate and H2O2, are highly effective as a solvent affording product selectivity in the presence of the catalyst (Table 3, entry 3). The results also indicated a favorable effect of the protic solvent. It is seen that the less polar solvents constituting a heterogeneous solvent system with aqueous hydrogen peroxide were not effective (Table 3, entries 4, 5). Reactions were also conducted in solvents such as ethyl cyanide and iso-propanol, which yielded low conversions of diphenylsulfide within the range of 28–41% after 12 h of reaction time (Table 3, entries 6, 7).
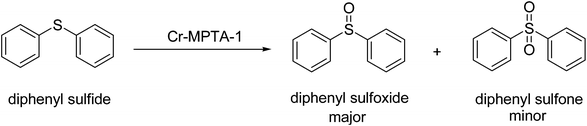 |
| | Scheme 5 Reaction pathway for the sulphide oxidation reaction over Cr-MPTA-1. | |
Table 3 Effect of different solvents on the oxidation of diphenyl sulfide with the mesoporous Cr-MPTA-1 catalysta
| Run |
Solvent |
Time (h) |
Temp. (°C) |
Conversion (%)b |
Selectivity of sulfoxide (%)bc |
|
Conditions: diphenyl sulfide (5 mmol); 30% aq H2O2 (10 mmol); solvent (10 mL); 0.050 g catalyst.
Conversion and selectivity were determined by GC.
Products were characterized by GC-MS.
Catalyst amount 0.025 g.
Catalyst amount 0.050 g.
5 mmol 30% H2O2 was used.
15 mmol 30% H2O2 was used.
|
| 1 |
Methanol |
5 |
40 |
71 |
83 |
| 2 |
DMF |
3 |
40 |
75 |
75 |
|
3
|
Acetonitrile |
3
|
40
|
97
|
90
|
| 4 |
CHCl3 |
12 |
40 |
11 |
48 |
| 5 |
Toluene |
12 |
40 |
7 |
50 |
| 6 |
CH3CH2CN |
8 |
40 |
41 |
81 |
| 7 |
iPrOH |
8 |
40 |
28 |
67 |
| 8 |
Acetonitrile |
3 |
25 |
69 |
92 |
| 9 |
Acetonitrile |
3 |
60 |
97 |
73 |
| 10d |
Acetonitrile |
3 |
40 |
76 |
89 |
| 11e |
Acetonitrile |
3 |
40 |
97 |
90 |
| 12f |
Acetonitrile |
6 |
40 |
63 |
86 |
| 13g |
Acetonitrile |
3 |
40 |
97 |
75 |
Temperature was found to play a crucial role in determining the activity and selectivity of the catalyst in the sulfide oxidation reactions. The reaction temperature was also optimized by repeating the reaction in various temperatures. At room temperature (25 °C), the product yields were moderate. With increasing the reaction temperature to 40 °C, the complete conversion of diphenylsulfide and the highest selectivity of corresponding sulfoxide were obtained. It was also observed that by increasing the temperature to 60 °C, the reaction led to more sulfone (27%) and the selectivity of the sulfoxide decreased.
The effect of the amount of H2O2 on the oxidation of diphenylsulfide was studied. At a lower concentration of hydrogen peroxide (5 mmol), the reaction took a longer time and low conversion of sulfide was obtained. Conversion increased when the amount of H2O2 increased from 5 mmol (run 12) to 10 mmol (run 3), while with a further increase in concentration to 15 mmol, no significant increase in conversion was observed.
In an effort to determine the optimum amount of the catalyst, we studied the catalyst amount ranging from 0.025 g to 0.075 g. An increase in initial catalyst concentration from 0.025 g to 0.05 g increased the yield of product, while further increases of the catalyst amount had no profound effect on the yield of the desired product (Table 3, entries 10 and 11). In a control experiment conducted in the absence of the catalyst, very little conversion of diphenylsulfide to sulfoxide or sulfone was observed (<10%) under otherwise similar reaction conditions. It is thus evident that the Cr-MPTA-1 catalyst plays a crucial role in facilitating the partial oxidation reactions.
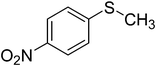
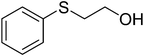
To examine the reactivity of the catalyst further, oxidation of other sulfides has also been investigated. Aliphatic and aromatic sulfides underwent clean and selective oxidation to the corresponding sulfoxide under the optimum condition, in impressive yields. The results are summarized in Table 4. As it can be seen most of the substrates are oxidized to give high yields and excellent sulfoxide selectivity. The reactivity and the conversion were dependent on the nature of the substituents present in the aryl moiety of ArSMe. The introduction of electron donating substituents in the phenyl ring of ArSMe accelerates the rate whereas the electron withdrawing substituents retard the rate (Table 4, entries 7 and 8). In addition, the yield of diphenyl sulfoxide was lower than that of methyl phenyl sulfoxide because of steric effects. The catalyst exhibited excellent chemoselectivity towards the sulfur group of substituted sulfides containing other oxidation prone functional groups such as C–C and OH (Table 4, entries 9 and 10).
Table 4 Oxidation of sulfides using 30% H2O2 catalyzed by mesoporous Cr-MPTA-1 catalysta
| Entry |
Sulfide |
Conversion (%)b/ (time, h) |
Selec. sulfoxide (%)bc |
|
Conditions: sulfide (5 mmol); 30% aq H2O2 (10 mmol); CH3CN (10 mL); 0.050 g catalyst.
Conversion and selectivity were determined by GC.
Products were characterized by GC-MS.
|
| 1 |
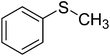
|
100 (3) |
95 |
| 2 |
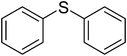
|
97 (3) |
90 |
| 3 |
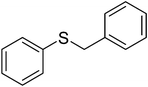
|
90 (5) |
92 |
| 4 |
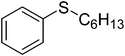
|
84 (6) |
94 |
| 5 |
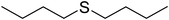
|
85 (4) |
85 |
| 6 |
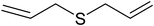
|
80 (4) |
82 |
| 7 |
|
100 (3) |
97 |
| 8 |

|
93 (3) |
95 |
| 9 |
|
91 (5) |
88 |
| 10 |
|
88 (5) |
89 |
| 11 |
|
87 (5) |
96 |
Heterogeneity test.
To check the leaching of metal into the solution during the reaction, a hot-filtration test was performed with Cr-MPTA-1 for the oxidation of diphenyl sulfide. After continuing the reaction for 1 h, the catalyst was removed by filtration and the determined conversion was 45%. The resulting filtrate was subjected to heating for further 3 h. The GC result reveals that no reaction occurred when the reaction was conducted with the filtrate obtained after removal of the solid catalyst. This is supported the observation that the active catalyst species did not leach from the solid catalyst. The absence of chromium in the filtrate was also confirmed by UV-vis spectroscopy and AAS analysis. The UV-vis spectra of the filtrate, did not show any absorption peaks characteristic of chromium metal, indicating that the leaching of metal did not take place during the course of the reaction and no chromium was detected by atomic absorption spectroscopic measurement of the filtrate after removal of catalyst. The IR spectrum of the used catalyst was quite similar to that of the fresh sample. These studies clearly demonstrated that chromium was intact to a considerable extent with the heterogeneous support, and no significant amount of leaching occurred during reaction.
Recycling of the catalyst.
The advantage of the heterogeneous catalysis is the possibility of recovering and reusing the catalyst during the reaction. The catalyst remains insoluble in the present reaction conditions and hence can be easily separated by simple filtration followed by washing. We studied the reusability of the present heterogeneous OH-MPTA-1 and Cr-MPTA-1 catalyst in the aldol reaction of p-nitrobenzaldehyde and oxidation of diphenyl sulfide respectively (Fig. 7). After completion of the reaction, the catalyst was recovered by simple filtration and washed with ethyl acetate followed by acetone then dried under reduced pressure at 40 °C. The recovered catalyst was employed in the next run with further addition of substrates in appropriate amount under optimum reaction conditions. As seen from Fig. 7, the recycled catalyst did not show any appreciable change in the activity indicating that the catalyst is stable and can be regenerated for repeated use.
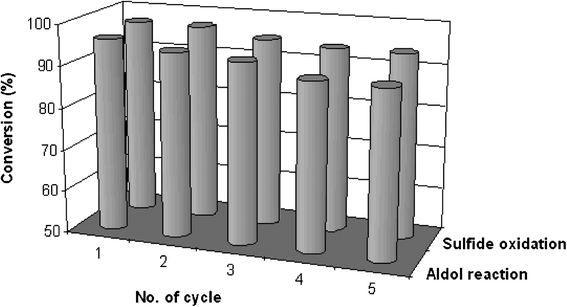 |
| | Fig. 7 Recycling efficiency for the aldol reaction of 4-nitrobenzaldehyde and oxidation reaction of diphenyl sulphide over OH-MPTA-1 and Cr-MPTA-1, respectively. | |
Conclusions
In conclusion, we have described herein the synthesis of an alkali exchanged mesoporous polymer, OH-MPTA-1, through in situ aqueous-phase radical polymerization of triallylamine using the supramolecular assembly of the anionic surfactant SDS as the structure-directing agent. This mesoporous organocatalyst OH-MPTA-1 shows excellent catalytic activity towards direct aldol reactions of different substituted aldehydes with acetone. Furthermore, the organocatalyst OH-MPTA-1 can be recovered by simple filtration and reused as the catalyst in the subsequent runs. We have also successfully synthesized mesoporous Cr-MPTA-1 by grafting of chromate on MPTA-1 surface. The Cr-MPTA-1 material shows high activity in selective partial oxidation of sulfide to sulfoxides. A variety of sulfoxides were obtained in high to excellent yields together with high chemoselectivity. Easy regeneration and reusability of the catalyst for several catalytic cycles without much change in its activity could make it an excellent heterogeneous catalyst for partial oxidation reactions. Excellent base catalysis and selective partial oxidation reactions over these mesoporous polymers could open new catalytic applications of mesoporous organic polymers in future.
Acknowledgements
We acknowledge the Department of Science and Technology (DST), the Council of Scientific and Industrial Research (CSIR) and the University Grant Commission (UGC), New Delhi, India for providing research grants. PM and JM thank CSIR, New Delhi for their respective senior research fellowships.
References
-
(a) A. Corma, J. Catal., 2003, 216, 298 CrossRef CAS;
(b) I. K. Mbaraka and B. H. Shanks, J. Catal., 2003, 219, 32 CrossRef;
(c) I. K. Mbaraka, D. R. Radu, V. S. Lin and B. H. Shanks, J. Catal., 2003, 219, 365 CrossRef;
(d) A. Bhaumik, M. P. Kapoor and S. Inagaki, Chem. Commun., 2003, 470 RSC;
(e) D. Liu, A. Bhan, M. Tsapatsis and S. A. Hashimi, ACS Catal., 2011, 1, 7 CrossRef CAS.
-
(a) W. D. Wang, J. E. Lofgreen and G. A. Ozin, Small, 2010, 6, 2634 CrossRef CAS;
(b) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC.
-
(a) Y. L. Zhang, S. Wei, Y. Y. He, F. Nawaz, S. Liu, H. Y. Zhang and F. S. Xiao, J. Mater. Chem., 2010, 20, 4609–4614 RSC;
(b) K. Ariga, S. Ishihara, H. Abe, M. Li and J. P. Hill, J. Mater. Chem., 2012, 22, 2369–2377 RSC.
-
(a) R. Liu, X. Wang, X. Zhao and P. Feng, Carbon, 2008, 46, 1664 CrossRef CAS;
(b) A. C. Dimian, Z. W. Srokol, M. C. Mittelmeijer-Hazeleger and G. Rothenberg, Top. Catal., 2010, 53, 1197 CrossRef CAS.
-
(a) K. F. Lam, K. L. Yeung and G. McKay, Microporous Mesoporous Mater., 2007, 100, 191 CrossRef CAS;
(b) A. K. Sinha, K. Suzuki, M. Takahara, H. Azuma, T. Nonaka and K. Fukumoto, Angew. Chem., Int. Ed., 2007, 46, 2891 CrossRef CAS;
(c) J. Li, L. Wang, T. Qi, Y. Zhou, C. Liu, J. Chu and Y. Zhang, Microporous Mesoporous Mater., 2008, 110, 442 CrossRef CAS.
-
(a) B. T. Holland, P. K. Isbester, C. F. Blanford, E. J. Munson and A. Stein, J. Am. Chem. Soc., 1997, 119, 6796 CrossRef CAS;
(b) A. Bhaumik and S. Inagaki, J. Am. Chem. Soc., 2001, 123, 691 CrossRef CAS;
(c) H. Yoshitake, T. Yokoi and T. Tatsumi, Chem. Mater., 2002, 14, 4603 CrossRef CAS.
-
(a) K. Nakajima, I. Tomita, M. Hara, S. Hayashi, K. Domen and J. N. Kondo, Adv. Mater., 2005, 17, 1839 CrossRef CAS;
(b) P. Botella, A. Corma, S. Iborra, R. Monton, I. Rodríguez and V. Costa, J. Catal., 2007, 250, 161 CrossRef CAS.
- M. Waki, N. Mizoshita, Y. Maegawa, T. Hasegawa, T. Tani, T. Shimada and S. Inagaki, Chem.–Eur. J., 2012, 18, 1992–1998 CrossRef CAS.
- S. Agarwala, M. Kevin, A. S. W. Wong, C. K. N. Peh, V. Thavasi and G. W. Ho, ACS Appl. Mater. Interfaces, 2010, 2, 1844–1850 CAS.
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273–3286 CrossRef CAS.
-
(a) A. K. Sinha and K. Suzuki, Angew. Chem., Int. Ed., 2005, 44, 271 CrossRef CAS;
(b) J. C. Yu, G. Li, X. Wang, X. Hu, C. W. Leunga and Z. Zhang, Chem. Commun., 2006, 2717 RSC;
(c) X. Cui, H. Li, L. Guo, D. He, H. Chen and J. Shi, Dalton Trans., 2008, 6435 RSC;
(d) Z. Zheng, K. Gao, Y. Luo, D. Li, O. Meng, Y. Wang and D. Zhang, J. Am. Chem. Soc., 2008, 130, 9785 CrossRef CAS.
-
(a) H. Fan, Q. Xu, Y. Guo and Y. Cao, Ind. Eng. Chem. Res., 2006, 45, 5009 CrossRef CAS;
(b) M. Nandi, R. Gangopadhyay and A. Bhaumik, Microporous Mesoporous Mater., 2008, 109, 239 CrossRef CAS;
(c) P. Paik, A. Gedanken and Y. Mastai, J. Mater. Chem., 2010, 20, 4085–4093 RSC.
-
(a) Y. Wang, C.-M. Yang, W. Schimdt, B. Spliethoff, E. Bill and F. Schuth, Adv. Mater., 2005, 17, 53 CrossRef CAS;
(b) W. Yue and W. Zhou, J. Mater. Chem., 2007, 17, 4947 RSC;
(c) H. Chen, X. Dong, J. Shi, J. Zhao, Z. Hua, J. Gao, M. Ruan and D. Yan, J. Mater. Chem., 2007, 17, 855 RSC.
-
(a) L. Y. Song, D. Feng, C. G. Campbell, D. Gu, A. M. Forster, K. G. Yager, N. Fredin, H. J. Lee, R. L. Jones, D. Y. Zhao and B. D. Vogt, J. Mater. Chem., 2010, 20, 1691 RSC;
(b) Y. P. Zhai, Y. Q. Dou, D. Y. Zhao, P. F. Fulvio, R. T. Mayes and S. Dai, Adv. Mater., 2011, 23, 4828 CrossRef CAS;
(c) A. P. Katsoulidis and M. G. Kanatzidis, Chem. Mater., 2012, 24, 471 CrossRef CAS.
- B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC.
- R. Mestres, Green Chem., 2004, 6, 583 RSC.
- M. L. Kantam, B. M. Choudary, C. V. Reddy, K. K. Rao and F. Figueras, Chem. Commun., 1998, 1033 RSC.
- H.-J. Li, W.-J. Li and Z.-P. Li, Chem. Commun., 2009, 3264 RSC.
-
(a) F. Calderon, R. Fernandez, F. Sanchez and A. Fernandez-Mayoralas, Adv. Synth. Catal., 2005, 347, 1395 CrossRef CAS;
(b) E. G. Doyaguez, F. Calderon, F. Sanchez and A. Fernandez-Mayoralas, J. Org. Chem., 2007, 72, 9353 CrossRef.
- M. R. M. Andreae and A. P. Davis, Tetrahedron: Asymmetry, 2005, 16, 2487 CrossRef CAS.
- Y. Wei and R. Bakthavatchalam, Tetrahedron Lett., 1991, 32, 1535 CrossRef CAS.
- B. C. Ranu and R. Chakraborty, Tetrahedron, 1993, 49, 5333 CrossRef CAS.
-
(a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS;
(b) K.N. Houk and B. List, Acc. Chem. Res., 2004, 37, 487 CrossRef CAS;
(c) B. List, Chem. Commun., 2006, 819 RSC;
(d) G. Guillena and D. J. Ramo'n, Tetrahedron: Asymmetry, 2006, 17, 1465 CrossRef CAS.
- M. Nandi, J. Mondal, K. Sarkar, Y. Yamauchi and A. Bhaumik, Chem. Commun., 2011, 47, 6677 RSC.
-
(a) S. S. Chimni, D. Mahajan and V. V. S. Babu, Tetrahedron Lett., 2005, 46, 5617 CrossRef CAS;
(b) D. Font, C. Jimeno and M. A. Pericàs, Org. Lett., 2006, 8, 4653 CrossRef CAS;
(c) Z. Jiang, Z. Liang, X. Wu and Y. Lu, Chem. Commun., 2006, 2801 RSC;
(d) W.-P. Huang, J.-R. Chen, X.-Y. Li, Y.-J. Cao and W.-J. Xiao, Can. J. Chem., 2007, 85, 208 CrossRef CAS;
(e) X. Wu, Z. Jiang, H.-M. Shen and Y. Lu, Adv. Synth. Catal., 2007, 349, 812 CrossRef CAS;
(f) F. Giacalone, M. Gruttadauria, A. M. Marculescu and R. Noto, Tetrahedron Lett., 2007, 48, 255 CrossRef CAS;
(g) H. Zhou, Y. Xie, L. Ren and K. Wang, Adv. Synth. Catal., 2009, 351, 1284 CrossRef;
(h) J. Li, N. Fu, X. Li, S. Luo and J.-P. Cheng, J. Org. Chem., 2010, 75, 4501 CrossRef CAS;
(i) A. Ricci, L. Bernardi, C. Gioia, S. Vierucci, M. Robitzer and F. Quignard, Chem. Commun., 2010, 46, 6288 RSC.
-
(a) B. List, J. Am. Chem. Soc., 2000, 122, 9336 CrossRef CAS;
(b) B. List, P. Pojarliev, W. T. Biller and H. J. Martin, J. Am. Chem. Soc., 2002, 124, 827 CrossRef CAS.
- B. List, P. Pojarliev and H. J. Martin, J. Org. Chem., 2001, 2423 CAS.
- S. H. Youk, S. H. Oh, H. S. Rho, J. E. Lee, J. W. Lee and C. E. Song, Chem. Commun., 2009, 2220 RSC.
-
(a) W. Wang, Y. Mei, H. Li and J. Wang, Org. Lett., 2005, 7, 601 CrossRef CAS;
(b) L. Zu, H. Li, J. Wang, X. Yu and W. Wang, Tetrahedron Lett., 2006, 47, 5131 CrossRef CAS;
(c) L. Zu, J. Wang, H. Li and W. Wang, Org. Lett., 2006, 8, 3077 CrossRef CAS;
(d) T. Miura, K. Imai, M. Ina, N. Tada, N. Imai and A. Itoh, Org. Lett., 2010, 12, 1620 CrossRef CAS.
- H. L. Holland, Chem. Rev., 1998, 88, 473 CrossRef.
- M. C. Carreno, Chem. Rev., 1995, 95, 1717 CrossRef CAS.
- W. J. Parsons, V. Ramkumar and G. L. Stiles, Mol Pharmacol, 1988, 34, 37 CAS.
- B. Kotelanski, R. J. Grozmann and J. N. Cohn, Clin. Pharmacol. Ther, 1973, 14, 427 CAS.
- S. Padmanabhan, R. C. Lavin and G. J. Durant, Tetrahedron: Asymmetry, 2000, 11, 195 CrossRef.
-
(a) M. G. Buonomenna, A. Figoli, I. Spezzano, M. Davoli and E. Drioli, Appl. Catal., B, 2008, 80, 185 CrossRef CAS;
(b) K. Jeyakumar, R. D. Chakravarthy and D. K. Chand, Catal. Commun., 2009, 10, 1948 CrossRef CAS;
(c) D. Betz, W. A. Herrmann and F. E. Kühn, J. Organomet. Chem., 2009, 694, 3320 CrossRef CAS;
(d) C. J. Qiu, Y. C. Zhang, Y. Gao and J. Q. Zhao, J. Organomet. Chem., 2009, 694, 3418 CrossRef CAS;
(e) T. Hida and H. Nogusa, Tetrahedron, 2009, 65, 270 CrossRef CAS;
(f) A. Pordea, D. Mathis and T. R. Ward, J. Organomet. Chem., 2009, 694, 930 CrossRef CAS;
(g) M. Bakavoli, A. M. Kakhky, A. Shiri, M. Ghabdian, A. Davoodnia and H. M. Eshghi, Chin. Chem. Lett., 2010, 21, 651 CrossRef CAS;
(h) A. Rostami and J. Akradi, Tetrahedron Lett., 2010, 51, 3501 CrossRef CAS;
(i) M. K. Panda, M. M. Shaikh and P. Ghosh, Dalton Trans., 2010, 39, 2428 RSC.
-
(a) K. Jeyakumar and D. K. Chand, Tetrahedron Lett., 2006, 47, 4573 CrossRef CAS;
(b) F. Vin' Tkhai, A. V. Tarakanova, O. V. Kostyuchenko, B. N. Tarasevich, N. S. Kulikov and A. V. Anisimov, Theor. Found. Chem. Eng., 2008, 42, 636 CrossRef;
(c) M. R. Maurya, A. Arya, A. Kumar and J. C. Pessoa, Dalton Trans., 2009, 2185 RSC;
(d) I. Lippold, K. Vlay, H. Görls and W. Plass, J. Inorg. Biochem., 2009, 103, 480 CrossRef CAS;
(e) A. R. Supale and G. S. Gokavi, React. Kinet. Catal. Lett., 2009, 96, 83 CrossRef CAS.
- K. Sato, M. Hyodo, M. Aoki, X. Q. Zheng and R. Noyori, Tetrahedron, 2001, 57, 2469 CrossRef CAS.
- M. Chiarini, N. D. Gillitt and C. A. Bunton, Langmuir, 2002, 18, 3836 CrossRef CAS.
-
(a) Y. Sasaki, K. Ushimaru, K. Iteya, Y. S. Nakayama and J. Ichihara, Tetrahedron Lett., 2004, 45, 9513 CrossRef CAS;
(b) D. H. Koo, M. Kim and S. Chang, Org. Lett., 2005, 7, 5015 CrossRef CAS;
(c) A. Jimtaisong and R. L. Luck, Inorg. Chem., 2006, 45, 10391 CrossRef CAS;
(d) M. Al-Hashimi, E. Fisset, A. C. Sullivan and J. R. H. Wilson, Tetrahedron Lett., 2006, 47, 8017 CrossRef CAS;
(e) K.L. Prasanth and H. Maheswaran, J. Mol. Catal. A: Chem., 2007, 268, 45 CrossRef CAS.
-
(a) S. M. Islam, P. Mondal, K. Tuhina, A. S. Roy, S. Mondal and D. Hossain, J. Organomet. Chem., 2010, 695, 2284 CrossRef;
(b) S. M. Islam, A. S. Roy, P. Mondal, M. Mubarak, S. Mondal and D. Hossain, J. Mol. Catal. A: Chem., 2011, 336, 106 CrossRef CAS;
(c) S. M. Islam, P. Mondal, S. Mukherjee and A. S. Roy, Polym. Adv. Technol., 2011, 22, 933 CrossRef CAS;
(d) S. M. Islam, A. S. Roy, P. Mondal, K. Tuhina, M. Mobarak and J. Mondal, Tetrahedron Lett., 2012, 53, 127 CrossRef CAS;
(e) S. M. Islam, S. Mondal, P. Mondal, A. S. Roy, K. Tuhina, N. Salam and M. Mobarak, J. Organomet. Chem., 2012, 696, 4264 CrossRef CAS.
-
(a) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS;
(b) S. M. Islam, P. Mondal, A. S. Roy and K. Tuhina, Synthesis, 2010, 2399 CrossRef;
(c) S. M. Islam, A. S. Roy, P. Mondal and N. Salam, J. Mol. Catal. A: Chem., 2012, 358, 38 CrossRef CAS.
-
(a) S. Huh, H. T. Chen, J. W. Wiench, M. Pruski and V. S. Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 1826–1830 CrossRef CAS;
(b) A. Corma and H. Garcia, Adv. Synth. Catal., 2006, 348, 1391–1412 CrossRef CAS;
(c) S. L. Jain, A. Modak and A. Bhaumik, Green Chem., 2011, 13, 586–590 RSC.
- D. Chandra and A. Bhaumik, J. Mater. Chem., 2009, 19, 1901 RSC.
-
(a) D. Chandra, B. K. Jena, C. R. Raj and A. Bhaumik, Chem. Mater., 2007, 19, 6290 CrossRef CAS;
(b)
G. Moad and D. H. Solomon, The chemistry of free radical polymerization, Elsevier, Amsterdam, 2nd edn, 2006 Search PubMed.
- R. Langner and G. Zundel, J. Phys. Chem. A, 1998, 102, 6635 CrossRef CAS.
- M. A. Vuurman, I. E. Wachs, D. J. Stufkens and A. J. Oskam, J. Mol. Catal., 1993, 80, 209 CrossRef CAS.
-
J. A. Gadsden, Infrared spectra of minerals and related inorganic compounds, Butterworth and Co. Publishers Ltd., London, 1975, p. 44 Search PubMed.
-
(a) S. De Rossi, M. P. Casaletto, G. Ferraris, A. Cimino and G. Minelli, Appl. Catal., A, 1998, 167, 257 CrossRef CAS;
(b) L. Zhang, Y. Zhao, H. Dai, H. He and C. T. Au, Catal. Today, 2008, 131, 42 CrossRef CAS;
(c) Y. Shao, L. Wang, J. Zhang and M. Anpo, Microporous Mesoporous Mater., 2008, 109, 271 CrossRef CAS;
(d) A. P. Singh, N. Torita, S. Shylesh, N. Iwasa and M. Arai, Catal. Lett., 2009, 132, 492 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.