Structural refinement, growth process, photoluminescence and photocatalytic properties of (Ba1-xPr2x/3)WO4 crystals synthesized by the coprecipitation method†
L. S.
Cavalcante
*ab,
F. M. C.
Batista
c,
M. A. P.
Almeida
d,
A. C.
Rabelo
d,
I. C.
Nogueira
d,
N. C.
Batista
b,
J. A.
Varela
a,
M. R. M. C.
Santos
c,
E.
Longo
bd and
M.
Siu Li
e
aUniversidade Estadual Paulista, P.O. Box 355, CEP. 14801-907, Araraquara, SP, Brazil. E-mail: laeciosc@gmail.com
bUESPI, CCN, Departamento de Química, Rua João Cabral, P.O.Box 2231, 64002-150, Teresina-PI, Brazil
cLIMAV-CCN-Química, Universidade Federal do Piauí, 64049-550, Teresina, PI, Brazil
dUniversidade Federal de São Carlos, P.O. Box 676, CEP., 13565-905, São Carlos, SP, Brazil
eIFSC-Universidade de São Paulo, P.O. Box 369, 13560 970, São Carlos, SP, Brazil
First published on 8th June 2012
Abstract
Barium praseodymium tungstate (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) were prepared by the coprecipitation method. These crystals were structurally characterized by X-ray diffraction (XRD), Rietveld refinements, Fourier-transform Raman (FT-Raman) and Fourier-transform infrared (FT-IR) spectroscopies. The shape and size of these crystals were observed by field emission scanning electron microcopy (FE-SEM). Their optical properties were investigated by ultraviolet visible (UV-vis) absorption and photoluminescence (PL) measurements. Moreover, we have studied the photocatalytic (PC) activity of crystals for degradation of rhodamine B (RhB) dye. XRD patterns, Rietveld refinements data, FT-Raman and FT-IR spectroscopies indicate that all crystals exhibit a tetragonal structure without deleterious phases. FT-Raman spectra exhibited 13 Raman-active modes in a range from 50 to 1000 cm−1, while FT-IR spectra have 8 infrared active modes in a range from 200 to 1050 cm−1. FE-SEM images showed different shapes (bonbon-, spindle-, rice- and flake-like) as well as a reduction in the crystal size with an increase in Pr3+ ions. A possible growth process was proposed for these crystals. UV-vis absorption measurements revealed a decrease in optical band gap values with an increase of Pr3+ into the matrix. An intense green PL emission was noted for (Ba1-xPr2x/3)WO4 crystals (x = 0), while crystals with (x = 0.01 and 0.02) produced a reduction in the wide band PL emission and the narrow band PL emission which is related to f–f transitions from Pr3+ ions. High photocatalytic efficiency was verified for the bonbon-like BaWO4 crystals as a catalyst in the degradation of the RhB dye after 25 min under UV-light. Finally, we discuss possible mechanisms for PL and PC properties of these crystals.
Introduction
Barium tungstate (BaWO4) crystals were traditionally prepared by different methods: an oxide mixture or a solid state reaction,1–3 flux evaporation,4 the Czochralski method,5–8 top-seeded slow-cooling9 and melting at a high temperature.10 However, these preparation methods require high temperatures, long processing times and sophisticated equipment with high maintenance costs as well as the formation of deleterious phases.In recent years, several synthesis methods have been developed and used in the preparation of micro-, nano- and BaWO4 mesocrystals.11–16 Different synthesis methods reported in the literature to obtain BaWO4 crystals are: coprecipitation,17,18 electrochemical,19,20 a biomimetic system of a supported liquid membrane,21 sonochemical,22 conventional hydrothermal,23–25 solvothermal,26–28 microwave-hydrothermal29,30 and cyclic-microwave.31 These methods facilitate the minimization of problems encountered in traditional methods. However, new synthesis routes and processing can lead to the formation of crystals with different sizes, shapes and structures.32,33 In particular, the conventional hydrothermal method has recently been widely used in the preparation of various tungstates with different sizes and shapes.34,35 However, these method still require high temperatures (160–220 °C) and long processing times (12–48 h).36
Regarding their crystalline structure, tungstates (AWO4, A = Ca, Sr, Ba and Pb) have a scheelite-type tetragonal structure which is characterized by a space group of I41/a) and a point group symmetry of (C64h)37–40 while tungstates (AWO4, A = Zn, Mn, Co, Fe and Ni) exhibit a wolframite-type monoclinic structure with a space group of (P2/c) and a point group symmetry of (C42h).41–44 Consequently, both “scheelite and wolframite” structures follow a general formula (AWO4) since they are composed of cations (A2+) in the lattice. Depending on the electron density of these cations, the structure can be tetragonal or monoclinic.
In relation to crystal-growth mechanisms in aqueous solution systems, it is well known that the “Ostwald ripening process”45 and the “oriented attachment”46 are the two main processes involved. Ostwald ripening is a thermodynamically-driven spontaneous process that occurs because larger particles are more energetically favored than smaller particles. In this growth process, all smaller crystals are dissolved and deposited on other intermediate crystals, while larger crystals continue to grow. After a length of time, all crystals in this system are already grown to minimize the total surface area. In the “oriented attachment” mechanism, the bigger crystals are grown from small primary nanocrystals through an oriented merger process where adjacent nanocrystals are self-assembled by sharing a common crystallographic orientation and the docking of these crystals at a planar interface.46,47
In particular, the tungstates (AWO4; A = Ca, Sr, Ba, Sr) with scheelite-type tetragonal structures have recently attracted the attention of several researchers, mainly because of their excellent photoluminescence (PL) properties at room temperature.48–50 Among these tungstates, BaWO4 crystals have the best PL, cathodoluminescence and photocatalytic (PC) properties reported in the literature.51–53
In this paper, for the first time, we report the synthesis of barium praseodymium tungstate (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) prepared by the coprecipitation (CP) method at room temperature. The structural refinement, morphology, optical properties and PC activity for the degradation of rhodamine B (RhB) are discussed in detail. Finally, we propose models to explain the growth mechanism, PL and PC properties of these crystals.
Experimental details
Synthesis of (Ba1-xPr2x/3)WO4 crystals by the CP method
(Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) were prepared by the CP method at room temperature without surfactants in aqueous solutions. The typical BaWO4 crystal synthesis procedure is described as follows: 2 × 10−3 mol tungstate(VI) sodium dihydrate (Na2WO4·2H2O) (99% purity, Vetec) and 2 × 10−3 mol barium(II) acetate [Ba(CH3COO)2] (99% purity, Aldrich) were dissolved separately into two plastic tubes (Falcon) with 50 mL capacity of deionized water. The first solution with (2Na+ and WO2−4) ions was transferred to a beaker of 250 mL capacity under constant stirring for 2 min. Then the second solution with Ba2+ and 2CH3COO− ions was added to this system where a suspension rapidly formed, and a white powder precipitated. Reactions between (Ba2+ ←:WO2−4) ions resulted in the formation of crystalline BaWO4 precipitates. (Ba1-xPr2x/3)WO4 crystals with (x = 0.01 and 0.02) were synthesized as follows: 2 × 10−3 mol Na2WO4·2H2O, 99%, and 98% of the 2 × 10−3 mol [Ba(CH3COO)2] were dissolved separately as described above. However, 1% and 2% of the 2 × 10−3 mol praseodymium(III) nitrate hexahydrate [Pr(NO3)3·6H2O] was previously prepared by the dissolution of praseodymium (III,IV) oxide [Pr6O11] (99.999% purity, Aldrich) with 4 drops of nitric acid (65% Suprapur®, Merck) into a beaker with 5 mL (H2O) at 85 °C and 100 mL capacity. After the complete dissolution of [Pr6O11 → Pr2O3 (III) + 4PrO2 (IV)], these oxides in an aqueous solution are decomposed in the Pr3+ ions in acid medium and form only Pr(NO3)3·6H2O with a more stable valence state. Then 50 mL of the Ba2+ ion solution was added to the system which resulted in the homogenization of the two Pr3+ and Ba2+ ions. In sequence, these systems were transferred into a beaker of 250 mL capacity with WO2−4 ions under constant stirring for 2 min. Reactions between (Pr3+/Ba2+ ←:WO2−4) ions resulted in the formation of crystalline (Ba,Pr)WO4 precipitates as shown in eqn (1)–(5) below: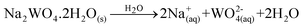 | (1) |
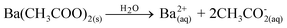 | (2) |
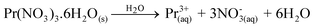 | (3) |
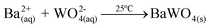 | (4) |
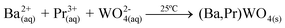 | (5) |
The resulting suspensions were washed with deionized water several times to remove any Na+ ions. Finally, these white powdered precipitates were collected and dried with acetone at 25 °C for some hours.
Characterization
After the CP at 25 °C, these (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) were structurally characterized by different techniques (ESI†).Photocatalytic activity measurement
After, the PC properties of these crystals (as an agent catalyst) for the degradation of [9-(2-carboxyphenyl)-6-diethylamino-3-xanthenylidene]-diethylammonium chloride, which is known as tetraethylated rhodamine or Rhodamine B (RhB) dye with a molecular formula [C28H31ClN2O3] (99.5% purity, Mallinckrodt) in an aqueous solution were tested under UV-light illumination. 50 mg catalyst crystals were placed in a 250 mL beaker; 50 mL of RhB solution (1 × 10−5 mol L−1) with pH = 4 was added. These suspensions were ultrasonicated for 10 min in a Branson (Model 1510) ultrasonic cleaner with a frequency of 42 kHz before illumination then were stored in the dark for 5 min to allow the saturated absorption of RhB onto the catalyst. The beakers were then placed in a photo-reactor at 20 °C and illuminated by six UV lamps (TUV Philips, 15 W, with a maximum intensity at 254 nm). The power light was measured by Coherent Power Max model No PM10 and the value for the optical energy density was 20 mW cm−2. At five-minute intervals, one 3 mL aliquot of these suspensions was removed and centrifuged at 9000 rpm for 5 min to remove crystals in suspension. Finally, variations of the absorption band maximum of supernatant solutions were monitored by UV-vis absorbance spectra measurements using a double-beam spectrophotometer with a double monochromator and a photomultiplier tube detector of JASCO (Model V-660, USA).Results and discussion
XRD and Rietveld refinement analyses
Fig. 1(a,b) show XRD patterns for (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) precipitates obtained by CP at 25 °C in a range from 10° to 70° and zoomed in normalized XRD patterns (from 27° to 35°), respectively.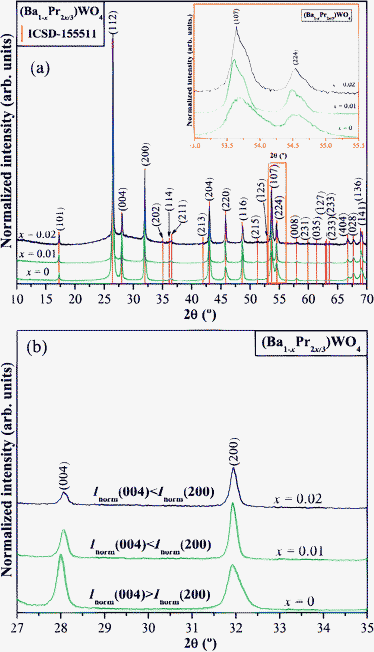 | ||
Fig. 1 (a) XRD patterns ranging from 10° to 70° of the (Ba1-xPr2x/3)WO4 crystals (x = 0, 0.01, and 0.02) precipitated at room temperature. Inset show the zoomed in XRD patterns from 53° to 55.5° (2θ range). The vertical lines indicate the position and relative intensity of the ICSD card N°. 155![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 511 for BaWO4 phase and (b) normalized XRD patterns (from 27° to 35°), respectively. 511 for BaWO4 phase and (b) normalized XRD patterns (from 27° to 35°), respectively. | ||
Fig. 1(a) demonstrates that precipitated BaWO4 crystals are crystalline and structurally ordered at long-range. When Pr3+ ions are incorporated into the BaWO4 lattice, a splitting of the XRD peaks occurs which is related to (107) and (224) planes (see Fig. 1(a) and the inset of Fig. 1(a)). This same figure also verifies that (Ba1-xPr2x/3)WO4 crystals with (x = 0.02) have a higher XRD pattern signal/noise ratio which infers possible roughness in these surface crystals. According to Robinson,54 it is possible to determine the surface roughness of crystals by X-ray crystallographic determinations. Also, we verified that all XRD patterns of these crystals correspond to a scheelite-type tetragonal structure which is in agreement with the respective Inorganic Crystal Structure Database (ICSD) N°. 155511.55 To verify and confirm that the structure of these (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) are really tetragonal, a structural refinement was conducted by the Rietveld method.56Fig. 1(b) shows that (Ba1-xPr2x/3)WO4 crystals with (x = 0) have a different normalized intensity between (004) and (200) planes in relation to (Ba1-xPr2x/3)WO4 crystals with (x = 0.01, and 0.02). Since these crystals are polycrystalline, it is possible that the synthesis method of CP and dopant can cause different effects in the crystallographic orientation. Therefore, the differences of normalized intensity between the (004) and (200) planes for these crystals were compared to verify that the (Ba1-xPr2x/3)WO4 crystals with x = 0 have a Inorm(004) > Inorm(200) plane, while (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 possess a Inorm(004) < Inorm(200) plane. This characteristic indicates that the pure BaWO4 crystal has a crystallographic orientation different from standard diffraction patterns (ICSD N°. 155511) while (Ba1-xPr2x/3)WO4 crystals with x = 0.01, and 0.02 have very similar XRD patterns as can be verified in the following section from Rietveld refinement data.
The lattice parameters, unit cell volume and atomic positions were obtained from a GSAS program.57,58 Structural refinement plots for (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01 and 0.02) crystals prepared by the CP method at 25 °C for 2 min are illustrated in Fig. 2(a–c).
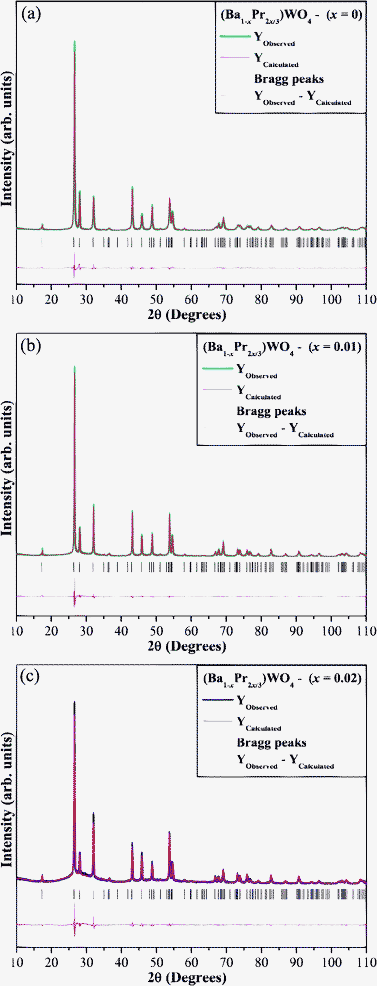 | ||
| Fig. 2 Rietveld refinement plots of the (Ba1-xPr2x/3)WO4 crystals precipitate at room temperature: (a) x = 0; (b) x = 0.01, and (c) x = 0.02. | ||
The results obtained from the Rietveld refinement method show good agreement between observed XRD patterns and theoretical results (see Fig. 2(a–c)). Moreover, the difference between XRD pattern profiles experimentally observed and theoretically calculated data display small differences near to zero in the intensity scale as illustrated by a line (Yobserved − Ycalculated). More details regarding structural refinement are shown in Table 1.
| ■Atoms | Wycroff | Site | S.O.F | x | y | z | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| a S.O.F = Site occupancy factor; (Ba1-xPr2x/3)WO4 crystals with (x = 0; ■), (x = 0.01; ●), and (x = 0.02; ▲) obtained by CP method; *generated from Rietveld refinements in ESI,† Table S1(a–c)). | |||||||||||
| Ba | 4b | −4 | 1 | 0 | 0.25 | 0.625 | |||||
| W | 4a | −4 | 1 | 0 | 0.25 | 0.125 | |||||
| O | 16f | 1 | 1 | 0.24566 | 0.12581 | 0.03991 | |||||
| a = b = 5.60433(1) Å; c = 12.7338(4) Å; V = 399.949(2) Å3; Rwp = 10.82%; Rp = 8.12%; Rb = 2.80%; χ2 = 5.299 and S = 2.032 | |||||||||||
| ●Atoms | Wycroff | Site | S.O.F | x | y | z | |||||
| Ba | 4b | −4 | 0.9813 | 0 | 0.25 | 0.625 | |||||
| Pr | 4b | −4 | 0.01247 | 0 | 0.25 | 0.625 | |||||
| W | 4a | −4 | 1 | 0 | 0.25 | 0.125 | |||||
| O | 16f | 1 | 1 | 0.25209 | 0.14395 | 0.04443 | |||||
| a = b = 5.61125(9) Å; c = 12.73071(3) Å; V = 400.841(1) Å3; Rwp = 10.39%; Rp = 7.50%; Rb = 3.46%; χ2 = 4.388 and S = 2.095 | |||||||||||
| ▲Atoms | Wycroff | Site | S.O.F | x | y | z | |||||
| Ba | 4b | −4 | 0.97616 | 0 | 0.25 | 0.625 | |||||
| Pr | 4b | −4 | 0.02256 | 0 | 0.25 | 0.625 | |||||
| W | 4ª | −4 | 1 | 0 | 0.25 | 0.125 | |||||
| O | 16f | 1 | 1 | 0.23923 | 0.12208 | 0.05307 | |||||
| a = b = 5.61140(7) Å; c = 12.7288(4) Å; V = 400.802(1) Å3; Rwp = 10.64%; Rp = 8.04%; Rb = 4.54%; χ2 = 4.018 and S = 2.004 | |||||||||||
In this table, fitting parameters (Rwp, Rp, Rb, χ2, and S) indicate good agreement between refined and observed XRD patterns for (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01, and 0.02 crystals. Structural refinement data confirm that these crystals are crystallized in a scheelite-type tetragonal structure with a space group of (I41/a). However, some variations in the atomic positions related to oxygen atoms were observed while barium, praseodymium and tungsten atoms have fixed atomic positions. These results indicate that the position of oxygen atoms is very disturbed in the lattice. Therefore, we believe these variations in atomic positions of oxygen atoms can lead to the formation of three types of distortions on [Ba–O], [Pr–O] and/or [W–O] bonds and consequently promotes different levels of distortions on the [BaO8], [PrO8] and/or [WO4] clusters in the lattice.
Representation of the (Ba1-xPr2x/3)WO4 unit cells
Fig. 3(a–c) illustrate schematic representations for tetragonal (Ba1-xPr2x/3)WO4 unit cells with (x = 0, 0.01 and 0.02) modeled from Rietveld refinement data.![Schematic representation of tetragonal (Ba1-xPr2x/3)WO4 unit cells with (a) x = 0; (b) x = 0.01, (c) x = 0.02 and its [BaO8]–[PrO8]–[WO4] clusters, respectively.](/image/article/2012/RA/c2ra20266b/c2ra20266b-f3.gif) | ||
| Fig. 3 Schematic representation of tetragonal (Ba1-xPr2x/3)WO4 unit cells with (a) x = 0; (b) x = 0.01, (c) x = 0.02 and its [BaO8]–[PrO8]–[WO4] clusters, respectively. | ||
These unit cells were modeled through the Diamond Crystal and Molecular Structure Visualization (Version 3.2 g for Windows) program59 using lattice parameters and atoms positions obtained from Rietveld refinement data presented in Table 1. BaWO4 crystals belong to scheelite-type tetragonal structures with a space group of (I41/a), a point-group symmetry of (C64h) and four molecular formula per unit cell (Z = 4).60Fig. 3(a) shows that bonds between O–W–O and O–Ba–O atoms were projected out of the unit cell. In these unit cells, tungsten (W) atoms are coordinated to four oxygen atoms which form [WO4] clusters with a tetrahedral configuration, a symmetry group (Td) and tetrahedron polyhedra (4 vertices, 4 faces and 6 edges).61 These [WO4] clusters are slightly distorted in the lattice and for each (Ba1-xPr2x/3)WO4 crystal. These differences in the (O–W–O) bond angles can lead to different levels of order-disorder and/or distortions in the (Ba1-xPr2x/3)WO4 crystal lattice with x = 0.01 and 0.02; (see Fig. 3(b,c)). We believe that this behavior can be due to the effect of Pr in the BaWO4 crystal lattice. In addition, for all unit cells, barium (Ba) atoms are bonded to eight oxygen atoms, which result in [BaO8] clusters with a deltahedral configuration, a symmetry group of (D2d) and snub-dispenoide polyhedra (8 vertices, 12 faces and 18 edges).62,63 Thus, [PrO8] clusters have the same electronic configuration as [BaO8] clusters in the A-site. Moreover, we note possible distortions on [BaO8] clusters through different bond angles between the (O–Ba–O). However, this situation is much more complicated and needs to be analyzed and explained in more detail.
FT-Raman/infrared spectroscopies analyses
According to group theory calculations, tungstates with a scheelite-type tetragonal exhibit 26 different (Raman and infrared) vibrational modes which are represented by eqn (6) below:64| Γ(Raman+Infrared) = 3Ag + 5Au + 5Bg + 3Bu + 5Eg + 5Eu | (6) |
| Γ(Raman) = 3Ag + 5Bg + 5Eg | (7) |
According to the literature,67,68 vibrational modes detected in tungstate Raman spectra can be classified into two groups: external and internal modes. Vibrational external modes are related to the lattice phonon or motion of [BaO8] clusters, and vibrational internal modes are caused by the vibration of [WO4] clusters (the mass center is in the stationary state). An isolated [WO4] cluster has a cubic symmetry point (Td),69 and its vibrations are composed of four modes (ν1(A1), ν2(E1), ν3(F2)) and ν4(F2)), one free rotation mode νf.r(F1) and one translational mode (F2). On the other hand, when [WO4] clusters are located in the scheelite structure, its point symmetry is reduced to S4.70
In infrared spectra, we can expect 13 infrared vibrational modes after excluding 13 Raman vibrational modes (see eqn (6)). Thus, the infrared modes can be described as shown in eqn (8) below:
| Γ(Infrared) = 5Au + 3Bu + 5Eu | (8) |
However, in vibrational infrared spectra, (1Au and 1Eu) are acoustic infrared modes, and (3Bu) are forbidden infrared modes; therefore, they are infrared-inactive modes. Thus, only 8 infrared-active vibrational modes remain which are 4Au modes that are perpendicular to the c-axis and 4Eu modes with the electric vector parallel to the c-axis (see eqn (9)):71,72
| Γ(Infrared) = 4Au + 4Eu | (9) |
Fig. 4 shows FT-Raman spectra for (Ba1-xPr2x/3)WO4 crystal precipitate obtained at 25 °C by the CP method.
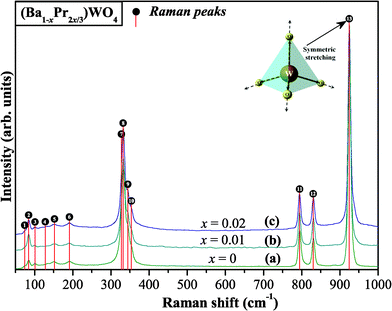 | ||
| Fig. 4 FT-Raman spectra in the range from 50 to 1000 cm−1 of the (Ba1-xPr2x/3)WO4 crystals precipitated at room temperature: (a) x = 0; (b) x = 0.01, and (c) x = 0.02. The vertical lines indicate the positions and relative intensities of Raman-active modes and inset shows the symmetric stretching of ←O←W→O→ bonds. | ||
Fig. 4(a–c) show that 13 Raman-active vibrational modes were detected experimentally. According to the literature,73 Raman spectra provide information on the degree of structural order-disorder at local short-range in ABO4 materials. Sharp and intense Raman-active modes indicate that (Ba1-xPr2x/3)WO4 crystals are structurally ordered at short-range with a strong interaction between clusters which arise from the symmetric stretching (←O←W→O→) (see inset in Fig. 4). Raman peak positions refer to active vibrational modes which are shown in Table 2 and are compared with active vibrational modes by other methods as reported in the literature.14,30,68,74,75
| M | T | t | B g | E g | E g | B g | A g | E g | A g | B g | B g | E g | E g | B g | A g | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (°C) | (min) |
|
|
|
|
|
|
|
|
|
|
|
|
|
||
| a M = method; T = temperature; t = time; Raman modes = (cm−1); MH = microwave-hydrothermal; CZ = Czochralski; CRM = chemical reaction peroxide; CP = coprecipitation of (Ba1-xPr2x/3)WO4 crystals (x = 0.0; * = 0.01 and ** = 0.02); and × = this work. | ||||||||||||||||
| MH | 140 | 6 | 62 | 74 | 101 | 132 | 149 | 190 | 330 | 332 | 344 | 354 | 793 | 830 | 924 | 14 |
| MH | 140 | 120 | 62 | 74 | 101 | 132 | 149 | 190 | 330 | 332 | 344 | 354 | 793 | 830 | 924 | 30 |
| MH | 1200 | 2400 | 63 | 75 | 101 | 132 | 150 | 191 | 332 | 332 | 345 | 353 | 795 | 831 | 925 | 68 |
| CZ | 1000 | 1440 | 63 | 74 | 101 | 133 | 150 | 191 | 331 | 332 | 344 | 352 | 795 | 831 | 926 | 74 |
| CRP | 25 | 180 | 60 | 72 | 98 | — | — | 188 | 330 | — | — | — | 790 | 829 | 920 | 75 |
| CP | 25 | 2 | 72 | 84 | 101 | 134 | 151 | 192 | 332 | 334 | 344 | 354 | 795 | 830 | 924 | × |
| CP* | 25 | 2 | 72 | 84 | 101 | 133 | 150 | 191 | 332 | 334 | 344 | 353 | 795 | 830 | 924 | × |
| CP** | 25 | 2 | 72 | 84 | 101 | 133 | 150 | 191 | 332 | 334 | 344 | 353 | 795 | 830 | 924 | × |






The results reported in Table 2 indicate that all Raman-active modes of (Ba1-xPr2x/3)WO4 crystals prepared by the CP method are related to a scheelite-type tetragonal structure which is in agreement with the research reported in the literature.14,30,68,74,75 In this table verifies that some relative positions of Raman modes have small shifts which can be caused by different factors such as: preparation methods, average crystal size, distortions on the (O–W–O)/(O–Ba–O) bonds, interaction forces between [WO4]–[BaO8]–[WO4] clusters and/or different degrees of structural order-disorder in the lattice at short-range. Relative positions for two Raman-active modes of (Ba1-xPr2x/3)WO4 crystals demonstrate a small shift in free rotation νf.r.(F1) Raman modes. These results can be linked to the preparation method at room temperature which is in agreement with Raman positions reported in ref. 75.
Fig. 5 illustrates FT-IR spectra for (Ba1-xPr2x/3)WO4 crystal precipitates obtained at 25 °C by the CP method.
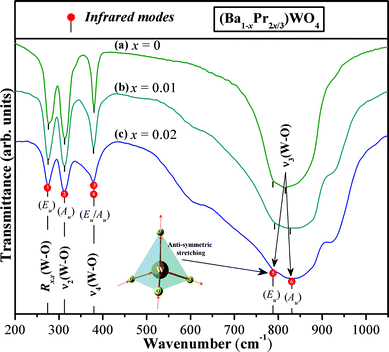 | ||
| Fig. 5 FT-IR spectra in the range 50 to 1000 cm−1 of the (Ba1-xPr2x/3)WO4 crystals precipitated at room temperature: (a) x = 0; (b) x = 0.01, and (c) x = 0.02. The vertical lines dotted indicate the positions and relative intensities of infrared-active modes and inset shows the anti-symmetric stretching →O→W→O→ bonds. | ||
As previously discussed in the text, tungstates with a scheelite-type tetragonal structure show 8 stretching and/or bending vibrational modes in FT-IR spectra.76,77 In our case, it was possible to identify no more than 6 modes [2(Au), 1(Eu)/1(Au) and 2(Eu)] which were found and identified in specific positions in the spectra (see Fig. 5(a–c)). First, strong absorption bands with two modes located at 788/792 cm−1 and 816/827 cm−1 are visible for our (Ba1-xPr2x/3)WO4 crystal (x = 0, 0.01, and 0.02) precipitates, respectively. Vertical lines are used to for easier identification (see Fig. 5(a–c)). These two bands are related to ν3[(1Eu) and (1Au)] internal modes which are assigned to (→O→W→O→) anti-symmetric stretching vibrations in [WO4] clusters (see inset in Fig. 5). The ν4[1(Eu)/1(Au)] modes are ascribed to the anti-symmetric bending of bonds in [WO4] clusters.77 These modes are located in the same position at 380–378 cm−1 for our (Ba1-xPr2x/3)WO4 crystal (x = 0, 0.01, and 0.02) precipitates, respectively. The ν2(Au) internal mode located at the same position (313 cm−1) is relative to the symmetric bending of [WO4] clusters. Finally, for other Rx,y(1Eu) external modes, R is related to the torsional motion of [WO4] clusters in the lattice. The relative positions for infrared-actives modes are shown in Table 3 and are compared with infrared-active modes for crystals prepared by other methods as reported in the literature.14,78,79
| M | T/°C | t/min | (Eu)
|
(Au)
|
(Eu)
|
(Au)
|
(Eu)
|
(Au)
|
Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a M = method; T = temperature; t = time; Raman modes = (cm−1); MH = microwave-hydrothermal, CZ = Czochralski; SSR = solid state reaction; CP = coprecipitation of (Ba1-xPr2x/3)WO4 crystals (x = 0.0; * = 0.01 and ** = 0.02); and × = this work. | |||||||||
| MH | 140 | 6 | — | — | — | — | 808 | 889 | 14 |
| CZ | 1250 | 1440 | 280 | 310 | 380 | 380 | 795 | 835 | 78 |
| SSR | 850 | 180 | — | — | — | — | 798 | 919 | 79 |
| CP | 25 | 2 | 276 | 313 | 380 | 380 | 788 | 816 | × |
| CP* | 25 | 2 | 275 | 313 | 378 | 378 | 792 | 827 | × |
| CP** | 25 | 2 | 274 | 313 | 378 | 378 | 792 | 827 | × |




These data (shown in Table 3) indicate a slight shift in the position of the stretching modes between (Ba1-xPr2x/3)WO4 crystal precipitates with x = 0 and 0.01 and 0.02. We believe that this behavior can possibly be related to different interaction strengths and stretching or angles between [O–W–O] bonds due to distortions on [WO4] clusters into the lattice. Other factors can be considered such as the replacement of [BaO8] by [PrO8] clusters which can influence bonds of neighboring [WO4] clusters. Moreover, we can verify a good agreement between the IR-active mode obtained in this work with IR-active mode reported in the literature.78,79
FE-SEM analyses
Fig. 6(a–f) show FE-SEM images of the (Ba1-xPr2x/3)WO4 crystal precipitates at room temperature.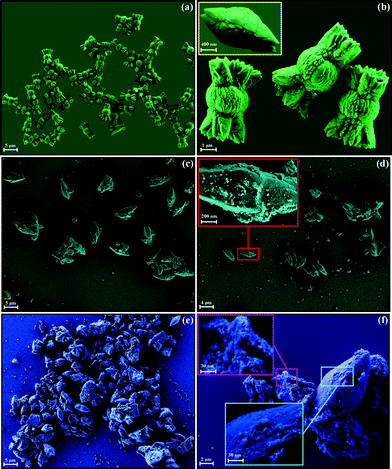 | ||
| Fig. 6 FE-SEM images of the (Ba1-xPr2x/3)WO4 crystals precipitate at room temperature: (a,b) x = 0; (c,d) x = 0.01, and (e,f) x = 0.02. | ||
The FE-SEM images indicate several bonbon-like BaWO4 microcrystals with an agglomerate nature as well as quasi-monodisperse in shape and polydisperse shapes in a size distribution (see Fig. 6(a) and ESI† in Fig. SD1(a)). These images also indicate that these microcrystals are formed and grow quickly in an aqueous solution after the precipitation reaction. Fig. 6(b) clearly shows these superstructures in high magnification FE-SEM images. The inset in Fig. 6(b) displays a microcrystal intermediate with its two extremities elongated before enabling the growth of the cuttings. After the final precipitation reaction stage occurs, the growth of these cuttings on the center part with a quasi-spherical shape for bonbon-like BaWO4 microcrystals. Bonbon-like BaWO4 microcrystals exhibit an average size distribution in the range from 5 to 6.75 μm. In this system, it was estimated that 30% of these superstructures have an average size of approximately 5.75 μm (see ESI,† Fig. SD-2(a)). When 1% of Ba2+ is replaced by Pr3+ in the BaWO4 lattice, FE-SEM images reveal a change in the shape to spindle-like microcrystals/flake-like nanocrystals and a reduction in the average microcrystal size (see Fig. 6(c)). These spindle-like microcrystals have an average size distribution in the range from 3 to 4.75 μm. In this system, it was estimated that 28% of these superstructures have an average size of approximately 3.75 μm (see Fig. 6(c) and ESI,† Fig. SD-1,2(b)). Moreover, FE-SEM images illustrated in Fig. 6(d) verify that a probable dissolution of microcrystals can occur due to small flake-like nanocrystals on the microcrystals surfaces (highlighted by rectangles; see inset in Fig. 6(d)). We believe that this behavior can be caused by the use of 4 drops of HNO3 at the start of the synthesis. Another possible explain is related to the effect of the introduction of earth-rare Pr3+ into an A-site where [BaO8] clusters are located, since [PrO8] clusters have a minor electronic density which promotes a reduction in the average crystal size. These results are in agreement with recent research reported in the literature for tungstates and molybdates doped with earth-rare.80,81 Finally, FE-SEM images show that (Ba1-xPr2x/3)WO4 crystal precipitates with x = 0.02 were dissolved due to a large quantity of small flake-like nanocrystals, imperfections on crystal surfaces and defects in shape (Fig. 6(e)). Moreover, the FE-SEM images shown in Fig. 6(f) indicate the presence of two shapes (rice-like microcrystals and flake-like nanocrystals). A high magnification of FE-SEM images (highlighted by rectangles, see inset of Fig. 6(f)) verifies that the increase of Pr3+ in the BaWO4 lattice promotes a clear reduction in the average crystal size. These rice-like microcrystals have an average size distribution in the range from 2 to 3.75 μm. In this system, it was verified that 25% of these superstructures have an average size of approximately 2.75 μm (see Fig. 6(c) and ESI,† Fig. SD-1,2(c)).
Growth process
Fig. 7(a–d) is a schematic representation of all stages involved in the synthesis and growth process of (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01 and 0.02) synthesized by the CP method at room temperature.![Schematic representation of the growth mechanism for the (Ba1-xPr2x/3)WO4 crystals precipitated at room temperature: (a) chemical synthesis (coprecipitation method) and all the stages involved in the growth process of the [(b) x = 0, (c) x = 0.01, (d) x = 0.02)] bonbon-, spindle-, rice-like and flake-like crystals precipitate.](/image/article/2012/RA/c2ra20266b/c2ra20266b-f7.gif) | ||
| Fig. 7 Schematic representation of the growth mechanism for the (Ba1-xPr2x/3)WO4 crystals precipitated at room temperature: (a) chemical synthesis (coprecipitation method) and all the stages involved in the growth process of the [(b) x = 0, (c) x = 0.01, (d) x = 0.02)] bonbon-, spindle-, rice-like and flake-like crystals precipitate. | ||
Fig. 7(a) shows the initial synthesis method employed in the preparation of (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02) by the addition of stoichiometric amounts of respective starting reagents [Ba(CH3CO2)2, Pr(NO3)3·6H2O and Na2WO4·2H2O]. These reagents were dissolved in order in two Falcon tubes containing 50 mL of deionized water under constant stirring. In this solution, the H2O molecule solvation energy promotes a fast salt dissociation where the Ba2+, Pr3+ and WO2−4 ions are quickly solvated by H2O molecules. Partial negative charges on the H2O molecules are electrostatically attracted by Ba2+/Pr3+ ions while the partial positive charges on the H2O molecules are electrostatically attracted by WO2−4 ions. However, due to the difference in the electronic density between Ba2+/Pr3+ and WO2−4 ions, a strong electrostatic force attraction occurs between both ion, which results in the formation of the first (Ba1-xPr2x/3)WO4 nuclei with (x = 0, 0.01, and 0.02) (see Fig. 7(b–d)). The increase in the precipitation rate leads to the aggregation process for small nuclei, and these small nuclei self-assemble due to specific conditions of the chemical reaction such as: the counter-ion effect, pH and solvent (see Fig. 7(b–d)). In the first case, these n⋯units of small BaWO4 crystals are formed and can be merged through an assembly process that leads to the growth of intermediate BaWO4 superstructures (see inset in Fig. 6(b) and Fig. 7(b)). In second and third case, the n⋯units of particles of these (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 which are formed as previously described have an assembly process as well as an Ostwald-ripening (OR) process. As can be observed in intermediate superstructures and nanostructures, a possible OR mechanism also governs the growth process of these crystals (see Fig. 7(c,d)). Since we verified a dissolution process of the microcrystals and a later recrystallization of nanocrystals and microcrystals, the OR process verifies that small crystals are dissolved and deposited on larger crystals. This thermodynamically-driven spontaneous process occurs because larger crystals are more energetically favored than are smaller crystals. In this mechanism, atoms on the crystal surface are energetically less stable than atoms in the interior.82,83 These results are in agreement with the explanations reported by Tian et al.84 about the growth mechanism of nanostructured CaWO4 microcrystals. Finally, after further growth of these intermediate (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01 and 0.02 for around 2 min, bonbon-, spindle-, and rice-like microcrystal precipitates are formed (see Fig. 7(b–d)).
UV-vis absorption spectroscopy analyses
The optical band gap energy (Egap) was calculated by the method proposed by Kubelka and Munk.85 This methodology is based on the transformation of diffuse reflectance measurements to estimate Egap values with good accuracy86 and is particularly effective in limiting cases of an infinitely thick sample layer. The Kubelka–Munk equation for any wavelength is described as: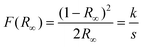 | (10) |
| αhv = C1(hv − Egap)n | (11) |
| [F(R∞)hv]2 = C2(hv − Egap) | (12) |
Therefore, by finding the F(R∞) value from eqn (10) and plotting a graph of [F(R∞)hv]2 against hν and C2, it was possible to determine the Egap of (Ba1-xPr2x/3)WO4 crystals.
Fig. 8(a–c) show UV-vis spectra of (Ba1-xPr2x/3)WO4 crystals with: (a) x = 0, (b) x = 0.01, and (c) x = 0.02 which were synthesized by the CP method.
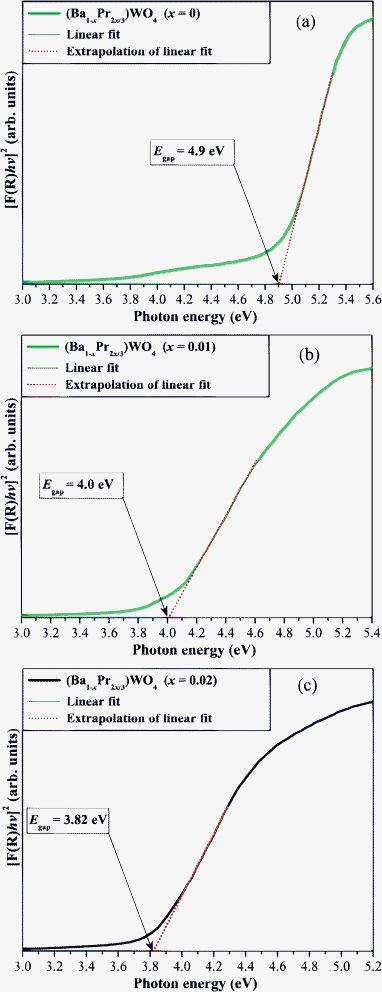 | ||
| Fig. 8 UV-vis absorbance spectra of (Ba1-xPr2x/3)WO4 crystals with (a) x = 0; (b) x = 0.01, and (c) x = 0.02. | ||
Fig. 8(a–c) illustrate different Egap values with a reduction in values because of the replacement of Ba2+ by Pr3+ in the BaWO4 lattice. The profile of UV-vis absorption curves indicates that Pr3+ ions induce the appearance of new intermediary energy levels within the optical band gaps, since the praseodymium shows (4f orbitals), while the barium exhibits (6s orbitals) in the valence band.90 This effect is promoted by 4f orbitals of [PrO8] clusters which leads to distortions on [BaO8]/[WO4] clusters that are interlinked in the tetragonal lattice ([BaO8]⋯[PrO8]⋯[WO4]). Fig. 8(a) shows that pure BaWO4 crystals have an Egap of 4.9 eV while (Ba1-xPr2x/3)WO4 crystals (x = 0.01) have a Egap = 4.0 eV (see Fig. 8(b)). A further increase in Pr3+ in the BaWO4 tetragonal lattice leads to a decrease in the Egap value to 3.82 eV (see Fig. 8(c)) which indicates the influence of [PrO8] clusters on the electronic structure of these crystals; data are listed in Table 4. Moreover, this table illustrates a comparison between the Egap obtained in our work with the Egap values reported in the literature.13,60,90–93
| M | Shape | T/°C | t/min | E gap /eV | Ref. |
|---|---|---|---|---|---|
| a M = method; T = temperature; t = time; Egap = energy band gap; MF = molten flux, CZ = Czochralski; SSR = solid state reaction; PP = polymeric precursors, PP = solid state metathetic; CP = coprecipitation of (Ba1-xPr2x/3)WO4 crystals (x = 0; ■), (x = 0.01; ●), and (x = 0.02; ▲); and × = this work. | |||||
| MF | Octahedrons-like microcrystals | 550 | 720 | 3.8 | 13 |
| CZ | Monocrystals | 1000 | 600 | 4.8 | 60 |
| CZ | Monocrystals | 1200 | 1440 | 4.9 | 90 |
| SSR | Bulk ceramics | 1400 | 2880 | 3.8 | 91 |
| PPM | Nanopowders | 600 | 120 | 4.1 | 92 |
| SSM | Rods-like microcrystals | — | — | 4.8 | 93 |
| CP■ | Bonbon-like microcrystals | 25 | 2 | 4.9 | × |
| CP● | Spindle-like microcrystals | 25 | 2 | 4.0 | × |
| CP▲ | Rice-like microcrystals | 25 | 2 | 3.82 | × |
Table 4 shows that Egap values for our (Ba1-xPr2x/3)WO4 crystals (x = 0, 0.01 and 0.02) differ slightly from the values reported in the literature for pure BaWO4 crystals.13,91,92 These small discrepancies can be attributed to the fact that Egap is very dependent upon the synthesis method, different defect levels defects in the band gap, shape, average crystal size, orientation and distortions in the lattice. According to Tyagi et al.,90 BaWO4 crystals have a direct band gap with less dispersion between the valence band (VB) and conduction band (CB). Therefore, our direct energy band gap values for bonbon-like BaWO4 microcrystals are very close to pure BaWO4 monocrystals values reported in the literature in ref. 60,90 (see Table 4). This behavior can be related to similar electronic densities in these BaWO4 crystals. (Ba1-xPr2x/3)WO4 crystals (x = 0.01 and 0.02) have intermediate electronic levels which are related to the addition of (4f orbitals) of praseodymium that promote a consequent reduction in Egap values. Moreover, another important point to considered is that the replacement of trivalent ions (Pr3+) of praseodymium into an A-site normally occupied by divalent ions (Ba2+) leads to a negative charge compensation in the BaWO4 lattice. This characteristic can be explained by the [2Pr•Ba, Va′′Ba and 3OxO] species.
PL emission, energy diagram levels (Pr3+) and wide band model of (Ba1-xPr2x/3)WO4 crystals absorption spectroscopy analyses
Fig. 9(a–c) illustrate PL emission spectra of (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01 and 0.02 synthesized by the CP method. The insets shows: (a) digital photos of PL emission for these crystals excited by the laser (λ = 350 nm) wavelength at room temperature; (b) the energy level diagram for the f–f transitions arising from Pr3+ ions; and (c) a wide band model proposed to explain the PL behavior of the crystals, respectively.![(a) PL emission of the (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and = 0.02, (b) energy level scheme for all the observed emission f–f transitions of Pr3+ ions and (c) model proposed in order to explain the origin of the intense visible PL emission at room temperature in the (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and 0.02; I—wavelength of laser employed in the excitation process of the crystals; II—presence of pair (distorted [WO]xd clusters and ordered [WO]xo clusters) into the lattice able to charge transference; III—wide band model with intermediary energy levels (oxygen (O)-2p, tungsten (W)-5d and praseodymium (Pr)-4f states) within the band gap; IV—transitions oxygen-2p → tungsten-5d and 4f–4f transitions before excitation, V—excitation process and formation of self-trapped excitons (STE's), internal f–f transitions of Pr3+ ions and recombination of e′–h˙ pair and VI—PL emission spectra of (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and = 0.02 precipitated at room temperature.](/image/article/2012/RA/c2ra20266b/c2ra20266b-f9.gif) | ||
| Fig. 9 (a) PL emission of the (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and = 0.02, (b) energy level scheme for all the observed emission f–f transitions of Pr3+ ions and (c) model proposed in order to explain the origin of the intense visible PL emission at room temperature in the (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and 0.02; I—wavelength of laser employed in the excitation process of the crystals; II—presence of pair (distorted [WO]xd clusters and ordered [WO]xo clusters) into the lattice able to charge transference; III—wide band model with intermediary energy levels (oxygen (O)-2p, tungsten (W)-5d and praseodymium (Pr)-4f states) within the band gap; IV—transitions oxygen-2p → tungsten-5d and 4f–4f transitions before excitation, V—excitation process and formation of self-trapped excitons (STE's), internal f–f transitions of Pr3+ ions and recombination of e′–h˙ pair and VI—PL emission spectra of (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and = 0.02 precipitated at room temperature. | ||
Table 4 and Fig. 9(a) verify that (Ba1-xPr2x/3)WO4 crystals with (x = 0) exhibit an intense PL emission at room temperature with a maximum at 520 nm in the green region. However, with the replacement of Ba2+ by Pr3+ ions (x = 0.01), a considerable decrease in PL emission is verified (Fig. 9(a)). This behavior can be justified by the formation of barium vacancies (V′′Ba) and complex oxygen vacancies which are able to show three different charge forms: neutral (VxO), singly ionized (V•O) and double ionized (V••O) and can donate up to two electrons. The VxO are able to donate up to two electrons, the V•O are donors or capture only one electron and V••O are not able to donate electrons, but can receive up to two electrons. These complex oxygen vacancies act as defects in the lattice of (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and can be stabilized by means of charge compensation into the lattice in agreement with the following eqn (13):
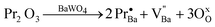 | (13) |
In addition to the wide band PL emission in these crystals, two small peaks of PL emission (at 449 and 486 nm) can be verified which are related to 4f–4f transitions of Pr3+ ions. The visible luminescence of Pr3+ ion transitions requires appropriate host materials when excited with UV wavelengths corresponding to 4fn-15d.94–96 The ground-state of these kinds of ions is characterized by Xe–4f36s2 valence electrons and PL properties of Pr3+ ions are ascribed to a 4f15d1 transition.97 According to Raju et al.,98 the first electronic transition is ascribed to (3P2 → 3H4), and the second transition corresponds to (3P1 → 3H4). The 3P2 → 3H4 transition is hypersensitive and is subject to experimental measurements with a dependence on the coordination environment, the vibronic effect and the host matrix.99–101 Therefore, this transition is suitable for a study of vibronic lines due to a coupling with high-frequency vibrations.102 The 3P1 → 3H4 transition is assigned to zero-phonon lines at the A-site of the BaWO4 lattice. These results are in agreement with other research reported in the literature for KSrMoO4:Pr3+ and BaMoO4:Pr3+.103,104Fig. 9(a) shows that the increase in the replacement of Ba2+ by Pr3+ ions (x = 0.02) promotes enhanced wide band intensity PL emission. Moreover, four PL emission peaks are visible at 444, 469, 486 and 612 nm which are related to 4f–4f transitions of Pr3+ ions. We can verify that increasing the Pr3+ ion content in the BaWO4 host matrix leads to an enhancement of 3P2 → 3H4 and 3P1 → 3H4 transitions at 444 and 486 nm, respectively. These two bands are sharper in relation to the same transitions observed in (Ba1-xPr2x/3)WO4 crystals with x = 0.01. Based on this information, depending on the concentration of Pr3+ ions in the BaWO4 lattice, a possible exchange of virtual phonons between coupled Pr3+ ion pairs occurs.105,106 The other two (3I6 → 3H4 and 3P0 → 3H6) transitions are small and are detected at 466 and 612 nm, respectively. These transitions also have been observed in PL emission spectra of Pr3+-doped glass host matrices with reasonable agreement.107,108 The energy level diagram corresponding to the f–f transitions ascribed to Pr3+ ions is shown in Fig. 9(b). However, in our (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02, we can observe only four such transitions because of the percentage of Pr3+ ions in the BaWO4 host lattice. This same behavior also is reported in the literature for the Pr3+-doped CaMoO4 and CaWO4.109,110 Another consideration is the low temperature of the CP method of crystal preparation, regarding vibrational progressions in the lattice that lead to displacement on the equilibrium position of f–f transitions (Pr3+ ions).101Fig. 9(c)-I shows the laser employed in the excitation process of the (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01, and 0.02, synthesized by the CP method. The use of different wavelengths promotes the excitation of electrons localized in different energy levels within the band gap. The Egap values for our crystals with x = 0, 0.01, and 0.02 are 4.9, 4.0 and 3.82 eV, respectively. Therefore, in this case, it was not possible to use a band-to-band emission process because the energy the laser  (3.54 eV) is smaller than the crystal Egap. Based on our previous research,73,89,92 where we calculated the electronic structure band of some scheelites, we have verified that after promoted displacements or distortions of the W/Mo atoms into the [WO4]/[MoO4] clusters, the appearance and redistribution of energy levels within the band gap are promoted. Therefore, we can postulate that our BaWO4 crystals prepared by CP method present some distortions on the disordered [WOxd] clusters that are able to promote charge transfer to ordered [WOxo] clusters. Moreover, [PrO8] clusters in the (Ba1-xPr2x/3)WO4 crystals with (x = 0.01 and 0.02) have electronic intra-4f-shell transitions of rare earths (see Fig. 9(c)-II). This polarization in the electronic structure by
(3.54 eV) is smaller than the crystal Egap. Based on our previous research,73,89,92 where we calculated the electronic structure band of some scheelites, we have verified that after promoted displacements or distortions of the W/Mo atoms into the [WO4]/[MoO4] clusters, the appearance and redistribution of energy levels within the band gap are promoted. Therefore, we can postulate that our BaWO4 crystals prepared by CP method present some distortions on the disordered [WOxd] clusters that are able to promote charge transfer to ordered [WOxo] clusters. Moreover, [PrO8] clusters in the (Ba1-xPr2x/3)WO4 crystals with (x = 0.01 and 0.02) have electronic intra-4f-shell transitions of rare earths (see Fig. 9(c)-II). This polarization in the electronic structure by 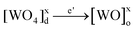 clusters leads to a non-homogeneous charge distribution and the formation of intermediate energy levels between the valence band (VB) and conduction band (CB). (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 have a 4f ↔ 4f transition which is intrinsic with Pr3+ ions (see Fig. 9(c)-III). Therefore, during the excitation process at room temperature, some electrons (e′) localized in these intermediary energy levels (oxygen-O 2p orbitals) and near the VB absorb photon energies (
clusters leads to a non-homogeneous charge distribution and the formation of intermediate energy levels between the valence band (VB) and conduction band (CB). (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 have a 4f ↔ 4f transition which is intrinsic with Pr3+ ions (see Fig. 9(c)-III). Therefore, during the excitation process at room temperature, some electrons (e′) localized in these intermediary energy levels (oxygen-O 2p orbitals) and near the VB absorb photon energies ( ) at this wavelength. Then these energetic levels are promoted to higher intermediate energy levels (tungsten-W 5d orbitals) near the CB, and the formation of holes (h˙) near the VB occurs. This mechanism results in the formation of self-trapped excitons (STE); i.e., the trapping of e′ by h˙. We believe that this phenomenon occurs in pure BaWO4 crystals. (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 have praseodymium-Pr 4f orbitals beyond these levels (see Fig. 9(c)-IV). As a consequence of this phenomenon, when electrons revert to lower energy levels (again via radiative return processes), the energies arising from this direct electronic transition are converted to photons (
) at this wavelength. Then these energetic levels are promoted to higher intermediate energy levels (tungsten-W 5d orbitals) near the CB, and the formation of holes (h˙) near the VB occurs. This mechanism results in the formation of self-trapped excitons (STE); i.e., the trapping of e′ by h˙. We believe that this phenomenon occurs in pure BaWO4 crystals. (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 have praseodymium-Pr 4f orbitals beyond these levels (see Fig. 9(c)-IV). As a consequence of this phenomenon, when electrons revert to lower energy levels (again via radiative return processes), the energies arising from this direct electronic transition are converted to photons ( ) with less energy than the initial (
) with less energy than the initial ( ) (see Fig. 9(c)-V). Thus, wide band PL emission is due to the absorption and emission processes of photons between O-2p and W-5d levels. For (Ba1-xPr2x/3)WO4 crystals with (x = 0.01 and 0.02), wide band and narrow band PL emission have been observed. The wide bands are due to 2p–5d transitions while the narrow band are related to intra 4f–4f transitions [(3P2 → 3H4), (3P1 → 3H4), (3I6 → 3H4), and (3P0 → 3H6)] represented by trivalent Pr3+ ions (see Fig. 9(c)-VI). Moreover, normalized PL emission analyses show that a narrowing of wide bands occurs due to the influence of 4f–4f transitions in the blue region.
) (see Fig. 9(c)-V). Thus, wide band PL emission is due to the absorption and emission processes of photons between O-2p and W-5d levels. For (Ba1-xPr2x/3)WO4 crystals with (x = 0.01 and 0.02), wide band and narrow band PL emission have been observed. The wide bands are due to 2p–5d transitions while the narrow band are related to intra 4f–4f transitions [(3P2 → 3H4), (3P1 → 3H4), (3I6 → 3H4), and (3P0 → 3H6)] represented by trivalent Pr3+ ions (see Fig. 9(c)-VI). Moreover, normalized PL emission analyses show that a narrowing of wide bands occurs due to the influence of 4f–4f transitions in the blue region.
Photocatalytic activity of (Ba1-xPr2x/3)WO4 crystals
Fig. 10(a–c) show the progress in the PC degradation of RhB by (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01, and 0.02 by monitoring the temporal changes of UV-vis absorbance spectra of the aqueous dye solution. The insets illustrate digital photos of the RhB aqueous dye solution after different times on the illumination UV light in the presence of the catalyst. The degradation rate (Cn/C0) of the RhB aqueous solution in the presence of different catalysts and without a catalyst is shown in Fig. 10(d).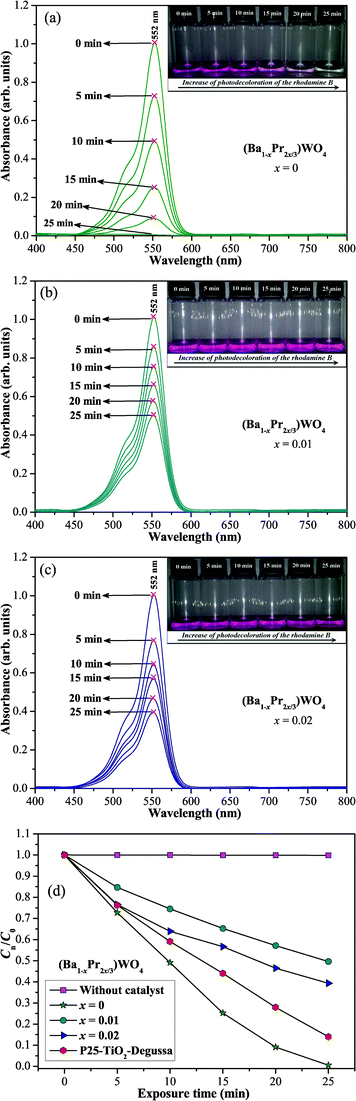 | ||
| Fig. 10 Evolution of UV-vis absorption spectra from after 25 min of illumination for photodegradation of RhB dye solution by the catalysts (Ba1-xPr2x/3)WO4 crystals with (a) x = 0; (b) x = 0.0; (c) x = 0.02 and (d) kinetic of weight-based photocatalytic degradation of RhB dye solution by the catalysts (Ba1-xPr2x/3)WO4 crystals with x = 0; 0.01, and = 0.02, TiO2–P25 (Degussa) and without catalysts. Inset shows a photograph of the photodegradation of RhB dye solution after different times of illumination on UV-lamps. | ||
Fig. 10(a) verifies a significant reduction in maximum absorption spectra of RhB aqueous solutions during the photodegradation process by the catalyst (Ba1-xPr2x/3)WO4 crystals with x = 0. Before irradiation, the N,N,N',N'-tetraethylated rhodamine molecule RhB dye has one band with a maximum absorption centered at λmax = 552 nm. The photodecoloration of RhB dye occurs due to an oxidative attack by one of the active oxygen species on the N-ethyl group.111 We have not noted displacements of the maximum absorption of RhB dye to other wavelength positions of its major absorption band which moved toward the N,N,N'-tri-ethylated rhodamine (λmax = 539 nm), N,N'-di-ethylated rhodamine, (λmax = 522 nm); N-ethylated rhodamine, (λmax = 510 nm) and rhodamine (λmax = 498 nm) species.112 We assumed that a high percentage of RhB dye was destroyed or photodegraded after 25 min under UV-illumination (see inset of Fig. 10(a)). Moreover, we have verified that our catalyst BaWO4 crystals are more efficient for the degradation of RhB dye than other scheelite-type crystals, such as: PbMoO4, SrMoO4, SrWO4, CdMoO4, PbWO4, and CdWO4 under UV-illumination.113–118 However, our catalyst (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 do not have a high photodegradation efficiency of RhB dye as a function of UV-irradiation time in comparison with the pure catalyst (see Fig. 10(b,c)). We believe that this behavior is due to the formation of V′′Ba and the low capability of [PrO8] clusters to trap photogenerated electrons. According to the literature,119–122 the main problem and/or factor responsible for the low photocatalytic efficiency of catalyst crystals is the high recombination rate between photogenerated electrons and holes on the crystal surface. Therefore, we can attribute that the h˙ generated by [PrO8] clusters is not as effective in the reduction of the time and the rate of recombination of electron–hole pairs. Fig. 10(d) verifies that after 25 min under UV-illumination, the RhB dye undergoes no degradation. As an efficiency experiment, we compared our catalyst (Ba1-xPr2x/3)WO4 crystals (x = 0, 0.01 and 0.02) with commercially available TiO2 photocatalyst (Degussa P25, Brazil) under photocatalytic reaction conditions. Based on the results obtained by kinetic weight-based (Cn/C0) photocatalytic degradation of RhB dye solutions for the catalysts, we found that pure BaWO4 crystals have a higher efficiency than TiO2–P25, and the degradation is complete after 25 min under UV-illumination.
Rate constants of catalyst (Ba1-xPr2x/3)WO4 crystals for the degradation of RhB dye
The photocatalytic efficiency of our catalyst (Ba1-xPr2x/3)WO4 crystals (x = 0, 0.01 and 0.02) were calculated and compared with the commercially available TiO2–P25 photocatalyst. The rate constant (k) obtained for degradation of RhB aqueous solutions is illustrated in Fig. 11(a–d).![First-order kinetics (a) without catalysts, (b) TiO2–P25, (c–e) (Ba1-xPr2x/3)WO4 crystals [x = 0 (c); 0.01 (d), and = 0.02 (e)]. The insets illustrate the normalized kinetics constant (k) values obtained by linear regression.](/image/article/2012/RA/c2ra20266b/c2ra20266b-f11.gif) | ||
| Fig. 11 First-order kinetics (a) without catalysts, (b) TiO2–P25, (c–e) (Ba1-xPr2x/3)WO4 crystals [x = 0 (c); 0.01 (d), and = 0.02 (e)]. The insets illustrate the normalized kinetics constant (k) values obtained by linear regression. | ||
In general, a catalyst crystal should be evaluated to verify that its photocatalytic activity is better than the TiO2–P25 (Degussa) photocatalyst.123,124 This photocatalytic test was conducted in this work to prove the efficiency of our crystals with the possibility for future industrial applications as a photocatalyst due to easy preparation by CP at room temperature. Therefore, in order to quantitatively understand the reaction kinetics for the degradation of the RhB dye by catalyst crystals, we applied the pseudo-first order model as expressed by eqn (14) to obtain the rate constant (k) of catalyst crystals.125
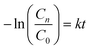 | (14) |
A comparative study of the kinetic parameters (k{absolute}/k{normalized}) of catalyst crystals for RhB aqueous solution degradation reactions and SBET are presented in Table 5.
| Samples | k{ }/min−1 | S BET /m2 g−1 | k[ ]/min−1 m−2 g−1 | K{ }/K{P25} | K[ ]/K[P25] |
|---|---|---|---|---|---|
| a wc = without catalyst, P25 = TiO2-Degussa; (Ba1-xPr2x/3)WO4 crystals with (x = 0, 0.01, and 0.02); { } = absolute and [ ] normalized by SBET. | |||||
| wc | 7.3781.10−5 | — | — | 9.8374. 10−4 | — |
| P25 | 0.075 | 50 | 0.0015 | 1 | 1 |
| x = 0 | 0.1889 | 1.52 | 0.1242 | 2.52 | 82.8 |
| x = 0.01 | 0.0106 | 9.95 | 0.001065 | 0.1413 | 0.71 |
| x = 0.02 | 0.0359 | 22.77 | 0.001576 | 0.4786 | 1.05 |
The results presented in this table indicate that knormalized values are smaller than k{absolute} values; i.e., each catalyst crystal has a specific surface area (SBET). Therefore, it is necessary normalize the k{absolute} values obtained. These k{normalized} values were obtained by dividing k{absolute} by the specific area surface (SBET) of each catalyst (see ESI,† Fig. SD-3(a–c)). After normalization, rate constants of the catalyst crystals obey the following ascending order: k[x=0] > k[x=0.02] > k[P25] > k[x=0.01] > k[wc]. Moreover, it can be observed that in this table, after comparing several relationships between k values of catalyst (Ba1-xPr2x/3)WO4 crystals (x = 0, 0.01, and 0.02) with the values of k values of the commercial catalyst (TiO2-P25, Degussa), it was found that normalized k[x=0] values are approximately 83 times higher than the normalized k[P25].
Rate constants of catalyst (Ba1-xPr2x/3)WO4 crystals for the degradation of RhB dye
Fig. 12(a–c) illustrate the schematic representation of all stages involved in experimental tests of photocatalysis, interactions of crystal surfaces, the RhB dye and a possible photocatalytic mechanism of the crystals with the species generated for the degradation of a RhB aqueous solution.![Proposal of photocatalytic reaction mechanism for the degradation of the RhB dye solution by the catalysts (Ba1-xPr2x/3)WO4 crystals with (x = 0; 0.01, and = 0.02): (a) Adsorption–desorption equilibrium of the RhB dye solution on the crystals surface, (b) Activation of the RhB* dye solution by defect on the crystal surface and distorted [WO4]xd and [WO4]xo clusters, and (c) distortions/defects in the electronic structure promoting the formation of intermediate energy levels within the Egap and electron (e′) transference between clusters. The [WO4]·o clusters adsorb on the (RhB*) and water (H2O) in valence band, while the [WO4]′o clusters adsorb reacts with oxygen molecule (O2) to form oxoanion (O′2) in conduction band.](/image/article/2012/RA/c2ra20266b/c2ra20266b-f12.gif) | ||
| Fig. 12 Proposal of photocatalytic reaction mechanism for the degradation of the RhB dye solution by the catalysts (Ba1-xPr2x/3)WO4 crystals with (x = 0; 0.01, and = 0.02): (a) Adsorption–desorption equilibrium of the RhB dye solution on the crystals surface, (b) Activation of the RhB* dye solution by defect on the crystal surface and distorted [WO4]xd and [WO4]xo clusters, and (c) distortions/defects in the electronic structure promoting the formation of intermediate energy levels within the Egap and electron (e′) transference between clusters. The [WO4]·o clusters adsorb on the (RhB*) and water (H2O) in valence band, while the [WO4]′o clusters adsorb reacts with oxygen molecule (O2) to form oxoanion (O′2) in conduction band. | ||
In our photocatalytic testing, the initial stage is extremely important for the optimization of this process by heterogeneous photocatalysis which is a very efficient technique for the degradation of organic pollutants such as “RhB dyes”.127Fig. 12(a), illustrates that before UV-light in suspension of crystals and RhB dyes, it is necessary to have an optimal dispersion of the system. Therefore, 50 mg of our catalyst crystals was added to 1 × 10−5 mol L−1 of the RhB aqueous solution and was well dispersed through an ultrasonic process for 10 min. We assume that this step is of fundamental importance for the reproducibility of these results and that the system reaches a perfect adsorption–desorption equilibrium. Fig. 12(b) illustrates the second state where this well dispersed system was stirred for 5 min inside a dark box followed by the collection of the first 3 mL aliquot. Then the six UV lights were triggered to start photocatalysis. During all these stages, the RhB aqueous solution is always kept at 20 °C with a thermostatic bath which is aided by a cooler exhaust. After the illumination of this system by UV light, the excitation of catalyst (Ba,Pr)WO4 crystals occurs, and the RhB dye molecules are adsorbed on the crystal surfaces128 As noted by previous analyses, (Ba,Pr)WO4 crystals which are obtained have different orientations, shapes and Egap values (see Fig. 1(b), Fig. 6(a–f) and Fig. 8(a–c)). These associated factors may also positively or negatively affect the photocatalytic properties of catalyst crystals. Moreover, we believe that each defect on the crystal surface can act as active site for the degradation of RhB dyes which is in agreement with recent research reported in the literature.129Fig. 12(c) illustrates a proposed a model as a possible explanation for photocatalytic mechanisms of our catalyst crystals for the photooxidation of a RhB aqueous solution. In this model, we assume that before the excitation processes (i.e., before the arrival of the UV-light in the system), our catalyst (Ba,Pr)WO4 crystals already have the ability to generate pairs of electrons (e′) and holes (h•). This characteristic is due to intrinsic defects in the lattice of materials with scheelite-type tetragonal structures. Therefore, these defects which are caused by distorted [WO4]d clusters can polarize in the lattice and lead to possible electronic transitions between disordered [[WO4]xd] and ordered [WO4]xo clusters. When the UV light is absorbed by the crystals, the following processes can occur as expressed in eqn (15) and (16) below:
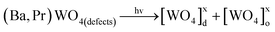 | (15) |
 | (16) |
 | (17) |
In the following step, due to an intermediary energy level between the VC and the BC of these crystals, electrons near them can be excited when illuminated by UV-light (λ = 254 nm ≈ 4.88 eV). This UV light energy can promote these excited electrons from the VB to the CB of the crystals. This process leads the formation of pairs (h•–e′) within the crystal band gap (see Fig. 12(c)). During photooxidation processes, the species generated [WO4]•d cluster can interact with the RhB* and H2O molecules130–133 as shown in eqn (18) and (19), which is a possible process that occurs in the VB:
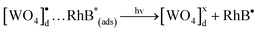 | (18) |
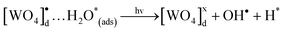 | (19) |
The [BaO8]•o clusters can react with the H* to generate (H+) while, the species generated [WO4]′o clusters react with oxygen molecules in an aqueous solution,134,135 as shown in eqn (20) which is a possible process that occurs in the CB:
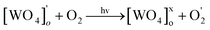 | (20) |
These cycles occur continuously while the system is exposed to the UV light. Finally, after several cycles of photooxidation, the degradation of RhB dye by the formed oxidant species occurs as indicated by eqn (21).
After 25 min of UV-illumination:
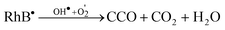 | (21) |
Based on our photocatalytic mechanisms, we assume that the defects on the crystal surface and electronic structure ([WO4]•d and [WO4]'o clusters) play an important role in producing OH• and O'2 radicals, which are the most oxidizing species in this process. Moreover, we believe that other generated species such as [BaO8]•d and [PrO8]•d clusters can participate in the mechanism of photocatalysis, but the [PrO8]•d clusters do not act effectively to produce oxidizing radicals (see Fig. 12(c)).
Conclusions
In summary, we have reported the easy production of (Ba1-xPr2x/3)WO4 crystals with x = 0, 0.01, and 0.02 by the CP method at room temperature. XRD patterns, Rietveld refinement data and FT-Raman spectra indicate that these crystals have a scheelite-type tetragonal structure without deleterious phases with the addition of Pr3+ ions. Normalized XRD patterns and structural refinement analyses indicate that BaWO4 crystals are polycrystalline with a crystallographic orientation in the (004) plane while (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 do not show any crystallographic orientation which is in agreement with the standard ICSD card. Structural refinement data were employed to model [BaO8], [PrO8] and [WO4] clusters by lattice parameters and atomic positions. FT-Raman spectra exhibited 13 modes which indicate that all crystals are structurally ordered at short range. FT-IR spectra showed that Pr3+ ions promote a widening in the vibrational modes due to [O–Pr–O] bonds. FE-SEM images revealed that Pr3+ ions affect the shape and cause a reduction in the crystal size. The growth process proposed for these crystals indicate that growth occurs by the self-assembly of small nanocrystals, and the further growth of intermediate superstructures and nanostructures leads to the formation of bonbon-, spindle-, rice- and flake-like crystals. UV-vis absorption spectra indicate that the replacement of Ba2+ by Pr3+ ions promotes a decrease in optical band gap values due to appearance of intermediary energy levels within the band gap. These levels are basically composed of oxygen 2p orbitals (above the VB) and tungsten 5d orbitals (below the CB). A wide band model was used to explain the wide and narrow PL behavior of (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 crystals synthesized by the CP method due to p–d transitions (defects at medium range in the lattice) and f–f transitions (Pr3+ ions), respectively. A high efficiency in the degradation of RhB dye was observed for our BaWO4 catalyst crystals compared to a commercially available TiO2-P25 photocatalyst while (Ba1-xPr2x/3)WO4 crystals with x = 0.01 and 0.02 showed a low efficiency for the degradation of RhB dyes under UV light as well as a reduction in PL intensity. Kinetic parameters which were obtained indicate that normalized k[x=0] values are 83 times higher than normalized k[P25] values. Finally, we propose photocatalytic mechanisms to explain the fast degradation of RhB dye by our crystals under UV light. Based in resulted obtained, we assumed that the following factors: considerable crystallographic orientation in the (004) plane, optical band gap values near the energy (4.88 eV) of UV-illumination, defects on the crystal surface and electronic structure ([WO4]•d, [BaO8]•d, [BaO8]'o and [WO4]'o clusters) plays an important role to produce OH• and O'2 radicals. Therefore, these factors associated are responsible by the fast degradation of RhB dyes by catalyst BaWO4 crystals after 25 min of illumination under UV-light.Acknowledgements
The authors acknowledge the financial support of the Brazilian research financing institutions: FAPESP (No.2009/53189-8), CNPq (No.159710/2011-1), CAPES, FAPEPI-GERATEC (No.01.08.0506.00) and LIMAV-UFPI.References
- R. C. Hughes, P. P. Coppola and H. T. Evans, J. Appl. Phys., 1952, 23, 635 CrossRef CAS.
- Q. Guo and O. J. Kleppa, Thermochim. Acta, 1997, 303, 183 CrossRef CAS.
- V. I. Smirnov and V. V. Aleksandrov, J. Eng. Phys. Thermophys., 1993, 65, 1117 CrossRef.
- A. R. Patel and S. K. Arora, J. Cryst. Growth, 1974, 23, 95 CrossRef CAS.
- S. Lin, J. L. Chen, N. F. Zhuang, B. Zhao and J. Z. Chen, J. Cryst. Growth, 2005, 277, 223 CrossRef.
- D. Ran, H. Xia, S. Sun, Z. Ling, W. Ge and H. Zhang, Mater. Sci. Eng., B, 2006, 130, 206 CrossRef CAS.
- A. K. Chauhan, J. Cryst. Growth, 2003, 254, 481 CrossRef.
- W. Ge, H. Zhang, J. Wang, J. Liu, X. Xu, X. Hu, J. Li and M. Jiang, J. Cryst. Growth, 2004, 270, 582 CrossRef CAS.
- J. Wang, H. Zhang, Z. Wang, W. Ge, J. Zhang and M. Jiang, J. Cryst. Growth, 2006, 292, 377 CrossRef CAS.
- Y. K. Voronko and A. A. Sobol, Inorg. Mater., 2005, 41, 420 CrossRef CAS.
- A. Phuruangrat, T. Thongtem and S. Thongtem, J. Phys. Chem. Solids, 2009, 70, 955 CrossRef CAS.
- N. Li, F. Gao, L. Hou and D. Gao, J. Phys. Chem. C, 2010, 114, 16114 CAS.
- P. Afanasiev, Mater. Lett., 2007, 61, 4622 CrossRef CAS.
- L. S. Cavalcante, J. C. Sczancoski, L. F. Lima Jr, J. W. M. Espinosa, J. A. Varela, P. S. Pizani and E. Longo, Cryst. Growth Des., 2009, 9, 1002 CAS.
- Y. Liu and Y. Chu, Mater. Chem. Phys., 2005, 92, 59 CrossRef CAS.
- S. Mann, Nat. Mater., 2009, 8, 781 CrossRef CAS.
- X. Wang, H. Xu, H. Wang and H. Yan, J. Cryst. Growth, 2005, 284, 254 CrossRef CAS.
- S. Hou, Y. Xing, H. Ding, X. Liu, B. Liu and X. Sun, Mater. Lett., 2010, 64, 1503 CrossRef CAS.
- Z. Song, J. Ma, X. Li, Y. Sun, J. Fang, Z. Liu and C. Gao, J. Am. Ceram. Soc., 2009, 92, 1354 CrossRef CAS.
- H. An, Z. Yang, D. Xiao, P. Yu, Z. Liu and R. Xie, Ferroelectrics, 2009, 385, 61 CrossRef.
- F. Q. Dong, Q. S. Wu and Y. P. Ding, J. Alloys Compd., 2010, 476, 571 CrossRef.
- T. Thongtem, A. Phuruangrat and S. Thongtem, Appl. Surf. Sci., 2008, 254, 7581 CrossRef CAS.
- J. Liao, B. Qiu, H. Wen, W. You and Y. Xiao, J. Lumin., 2010, 130, 762 CrossRef CAS.
- J. Liao, B. Qiu, H. R. Wen, Y. Li, R. Hong and H. You, J. Mater. Sci., 2011, 46, 1184 CrossRef CAS.
- F. Zhang, S. P. Yang, H. M. Chen, Z. H. Wang and X. B. Yu, J. Cryst. Growth, 2004, 267, 569 CrossRef CAS.
- C. Zhang, E. Shen, E. Wang, Z. Kang, L. Gao, C. Hu and L. Xu, Mater. Chem. Phys., 2006, 96, 240 CrossRef CAS.
- B. A. Hernandez-Sanchez, T. J. Boyle, H. D. Pratt, M. A. Rodriguez, L. N. Brewer and D. R. Dunphy, Chem. Mater., 2008, 20, 6643 CrossRef CAS.
- T. Thongtem, S. Kaowphong and S. Thongtem, Solid State Phenom., 2007, 124–126, 315 CrossRef CAS.
- K. P. F. Siqueira, R.L. Moreira, M. Valadares and A. Dias, J. Mater. Sci., 2010, 45, 6083 CrossRef CAS.
- L. S. Cavalcante, J. C. Sczancoski, J. W. M. Espinosa, J. A. Varela, P. S. Pizani and E. Longo, J. Alloys Compd., 2009, 474, 195 CrossRef CAS.
- A. Phuruangrat, T. Thongtem and S. Thongtem, J. Ceram. Soc. Jpn., 2008, 116, 605 CrossRef CAS.
- D. Ye, D. Li, W. Zhang, M. Sun, Y. Hu, Y. Zhang and X. Fu, J. Phys. Chem. C, 2008, 112, 17351 CAS.
- Y. Yin, F. Yang, Y. Yang, Z. Gan, Z. Qin, S. Gao, B. Zhou and X. Li, Superlattices Microstruct., 2001, 49, 599 CrossRef.
- P. Siriwong, T. Thongtem, A. Phuruangrat and S. Thongtem, Cryst. Eng. Comm, 2011, 13, 1564 RSC.
- G. Tian and S. Sun, Cryst. Res. Technol., 2001, 46, 389 CrossRef.
- J. Huang and L. Gao, J. Am. Ceram. Soc., 2006, 89, 3877 CrossRef CAS.
- S. B. Kim, B. G. Kum, H. M. Jang, A. Lakshmanan and B. K. Kang, J. Lumin., 2011, 131, 1625 CrossRef CAS.
- L. D. Feng, X. B. Chen and C. J. Mao, Mater. Lett., 2010, 64, 2420 CrossRef CAS.
- L. Ma, Y. Sun, P. Gao, Y. Yin, Z. Qin and B. Zhou, Mater. Lett., 2010, 64, 1235 CrossRef CAS.
- G. Wan and G. Wang, Mod. Phys. Lett. B, 2010, 24, 3081 CrossRef CAS.
- T. T. Basiev, A. Y. Karasik, A. A. Sobol, D. S. Chunaev and V. E. Shukshin, Quantum Electron., 2011, 41, 370 CrossRef CAS.
- T. D. Nguyen, D. Mrabet, T. T. D. Vu, C. T. Dinh and T. O. Do, Cryst. Eng. Comm, 2011, 13, 1450 RSC.
- S. Rajagopal, D. Nataraj, O. Y. Khyzhun, Y. Djaoued, J. Robichaud and D. Mangalaraj, J. Alloys Compd., 2010, 493, 340 CrossRef CAS.
- Z. Song, J. Ma, H. Sun, W. Wang, Y. Sun, L. Sun, Z. Liu and C. Gao, Ceram. Int., 2009, 35, 2675 CrossRef CAS.
- L. Ratker and P. W. Voorhees, Growth and coarsening: Ostwald ripening in material processing, Springer, 2002, 117–118 Search PubMed.
- R. L. Penn and J. F. Banfield, Am. Mineral, 1998, 83, 1077 CAS.
- J. C. Sczancoski, M. D. R. Bomio, L. S. Cavalcante, M. R. Joya, P. S. Pizani, J. A. Varela, E. Longo, M. S. Li and J. A. Andrés, J. Phys. Chem. C, 2009, 113, 5812 CAS.
- J. Liao, B. Qiu, H. Wen, J. Chen, W. You and L. Liu, J. Alloys Compd., 2009, 487, 758 CrossRef CAS.
- T. Thongtem, S. Kaowphong and S. Thongtem, Appl. Surf. Sci., 2008, 254, 7765 CrossRef CAS.
- M. Nikl, P. Bohacek, E. Mihokova, M. Kobayashi, M. Ishii, Y. Usuki, V. Babin, A. Stolovich, S. Zazubovich and M. Bacci, J. Lumin., 2000, 87–89, 1136 CrossRef CAS.
- G. Zhang, R. Jia and Q. Wu, Mater. Sci. Eng., B, 2006, 128, 254 CrossRef CAS.
- H. L. Li, Z. L. Wang and J. H. Hao, IOP Conf. Ser.: Mater. Sci. Eng., 2009, 1, 012010 CrossRef.
- Z. Shan, Y. Wang, H. Ding and F. Huang, J. Mol. Catal. A: Chem., 2009, 302, 54 CrossRef CAS.
- I. K. Robinson, Phys. Rev. B, 1986, 33, 3830 CrossRef CAS.
- D. Errandonea, J. Pellicer-Porres, F. J. Manjón, A. Segura, C. Ferrer-Roca, R. S. Kumar, O. Tschauner, J. López-Solano, P. Rodríguez-Hernández, S. Radescu, A. Mujica, A. Muñoz and G. Aquilanti, Phys. Rev. B, 2006, 73, 224103 CrossRef.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, General Structure Analysis System (GSAS), Los Alamos National Laboratory Report LAUR, 1994, 86, 748 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- http://www.crystalimpact.com/diamond/download.htm .
- D. Ran, H. Xia, S. Sun, P. Zhao, F. Liu, Z. Ling, W. Ge, H. Zhang and J. Wang, Cryst. Res. Technol., 2006, 41, 1189 CrossRef CAS.
- http://polyhedra.org/poly/show/0/tetrahedron .
- http://en.wikipedia.org/wiki/Deltahedron .
- http://polyhedra.org/poly/show/128/snub_disphenoid .
- T. T. Basiev, A. A. Sobol, Y. K. Voronko and P. G. Zverev, Opt. Mater., 2000, 15, 205 CrossRef CAS.
- T. T. Basiev, A. A. Sobol, P. G. Zverev, L. I. Ivleva, V. V. Osiko and R. C. Powell, Opt. Mater, 1999, 11, 309 Search PubMed.
- A. Jayaraman, B. Batlogg and L. G. Vanuitert, Phys. Rev. B, 1983, 28, 4774 CrossRef CAS.
- S. Desgreniers, S. Jandl and C. Carlone, J. Phys. Chem. Solids, 1984, 45, 1105 CrossRef CAS.
- S. P. S. Porto and J. F. Scott, Phys. Rev., 1967, 157, 716 CrossRef CAS.
- J. C. Sczancoski, L. S. Cavalcante, M. R. Joya, J. W. M. Espinosa, P. S. Pizani, J. A. Varela and E. Longo, J. Colloid Interface Sci., 2009, 330, 227 CrossRef CAS.
- G. Jia, C. Wang and S. Xu, J. Phys. Chem. C, 2010, 114, 17905 CAS.
- A. Golubović, R. Gajić, Z. Dohčević-Mitrović and S. Nikolić, J. Alloys Compd., 2006, 415, 16 CrossRef.
- A.S. Barker Jr., Phys. Rev., 1964, 135, A742 CrossRef.
- J. C. Sczancoski, L. S. Cavalcante, N. L. Marana, R. O. da Silva, R. L. Tranquilin, M. R. Joya, P. S. Pizani, J. A. Varela, J. R. Sambrano, M. S. Li, E. Longo and J. Andrés, Curr. Appl. Phys., 2010, 10, 614 CrossRef.
- (a) F. J. Manjón, D. Errandonea, N. Garro, J. Pellicer-Porres, P. Rodríguez-Hernández, S. Radescu, J. López-Solano, A. Mujica and A. Muñnoz, Phys. Rev. B, 2006, 74, 144111.75 Search PubMed; (b) R. C. Hughes, P. P. Coppola and H. T. Evans, J. Appl. Phys., 1952, 23, 635 CrossRef CAS.
- D. Rangappa, T. Fujiwara and M. Yoshimura, Solid State Sci., 2006, 8, 1074 CrossRef CAS.
- Z. C. Ling, H. R. Xia, D. G. Ran, F. Q. Liu, S. Q. Sun, J. D. Fan, H. J. Zhang, J. Y. Wang and L. L. Yu, Chem. Phys. Lett., 2006, 426, 85 CrossRef CAS.
- R. K. Khanna and E. R. Lippincott, Spectrochim. Acta, 1968, 21A, 905 CrossRef.
- G. M. Clark and W. P. Doyle, Spectrochim. Acta, 1966, 22, 1441 CrossRef CAS.
- L. A. Al-Hajji, M. A. Hasan and M. I. Zaki, J. Therm. Anal. Calorim., 2010, 100, 43 CrossRef CAS.
- D. Gao, Y. Li, X. Lai, Y. Wei, J. Bi, Y. Li and M. Liu, Mater. Chem. Phys., 2011, 126, 391 CrossRef CAS.
- Y. Zhou, J. Liu, X. Yang, X. Yu and L. Wang, J. Electrochem. Soc., 2011, 158, K74 CrossRef CAS.
- J. Zheng, F. Huang, S. Yin, Y. Wang, Z. Lin, X. Wu and Y. Zhao, J. Am. Chem. Soc., 2010, 132, 9528 CrossRef CAS.
- C. S. Xavier, J. C. Sczancoski, L. S. Cavalcante, C. O. Paiva-Santos, J. A. Varela, E. Longo and M. S. Li, Solid State Sci., 2009, 11, 2173 CrossRef CAS.
- Y. Tian, B. Chen, H. Yu, R. Hu, X. Li, J. Sun, L. Cheng, H. Zhong, J. Zhang, Y. Zheng, T. Yu and L. Huang, J. Colloid Interface Sci., 2011, 360, 586 CrossRef CAS.
- P. Kubelka and F. Munk-Aussig, Zeit. Für. Tech. Physik, 1931, 12, 593 Search PubMed.
- A. E. Morales, E. S. Mora and U. Pal, Rev. Mex. Fis. S, 2007, 53, 18 CAS.
- R. A. Smith, Semiconductors, 2nd ed. (Cambridge University Press, London, 1978, 434 Search PubMed.
- R. Lacomba-Perales, J. Ruiz-Fuertes, D. Errandonea, D. Martínez-García and A. Segura, Europhys. Lett., 2008, 83, 37002 CrossRef.
- V. S. Marques, L. S. Cavalcante, J. C. Sczancoski, A. F. P. Alcântara, M. O. Orlandi, E. Moraes, E. Longo, J. A. Varela, M. S. Li and M. R. M. C. Santos, Cryst. Growth Des., 2010, 10, 4752 CAS.
- M. Tyagi, S. G. Singh, A. K. Chauhan and S. C. Gadkari, Phys. Rev. B, 2010, 405, 4530 CAS.
- H. W. Eng, P. W. Barnes, B. M. Auer and P. M. Woodward, J. Solid State Chem., 2003, 175, 94 CrossRef CAS.
- M. Anicete-Santos, F. C. Picon, C. N. Alves, P. S. Pizani, J. A. Varela and E. Longo, J. Phys. Chem. C, 2011, 115, 12180 CAS.
- P. Parhi, T. N. Karthik and V. Manivannan, J. Alloys Compd., 2008, 465, 380 CrossRef CAS.
- J. Cao, Y. Wang, X. Ma, J. Li, Z. Zhu, Z. You, F. Yang, C. Sun, T. Cao, Y. Ji and C. Tu, J. Alloys Compd., 2001, 509, 185 CrossRef.
- P. S. Peijzel, A. Meijerink, R. T. Wegh, M. F. Reid and G. W. Burdick, J. Solid State Chem., 2005, 178, 448 CrossRef CAS.
- C. D. Cordero-Montalvo and N. Bloembergen, Phys. Rev. B, 1984, 30, 438 CrossRef CAS.
- A. Flórez, O. L. Malta, Y. Messaddeq and M. A. Aegerter, J. Non-Cryst. Solids, 1997, 213, 315 CrossRef.
- G. S. R. Raju, J. Y. Park, H. C. Jung, R. Balakrishnaiah, B. K. Moon and J. H. Jeong, Curr. Appl. Phys., 2011, 11, S292 CrossRef.
- R. Reisfeld, Radiative and nonradiative transition of rare earths in glasses, Struct. Bond, Springer-Verlag, New York, vol. 22, 1975,pp. 123–175 Search PubMed.
- R. D. Peacock, The intensities of lanthanide f-f transitions: Struct. Bond. , Springer-Verlag, New York, vol. 22, 1975, pp. 83–122 Search PubMed.
- H. Yersin, Transition metal and rare earth compounds III: excited states, transitions, interactions, Springer-Verlag, New York, 2004, 1edn, pp. 198–205 Search PubMed.
- C. M. Donegá and A. Meijerink, J. Lumin., 1993, 55, 315 CrossRef.
- Q. Li, J. Huang and D. Chen, Luminescence, 2011, 26, 349 CrossRef CAS.
- X. Yang, J. Liu, H. Yang, X. Yu, Y. Guo, Y. Zhou and J. Liu, J. Mater. Chem., 2009, 19, 3771 RSC.
- M. Galczynnski and W. Strek, J. Phys. Chem. Solids, 1991, 52, 681 CrossRef.
- M. Galczynnski, M. Blazej and W. Strek, Mater. Chem. Phys., 1992, 31, 175 CrossRef.
- G. Lakshminarayana and J. Qiu, J. Alloys Compd., 2009, 478, 630 CrossRef CAS.
- M. Olivier, P. Pirasteh, J. L. Doualan, P. Camy, H. Lhermite, J. L. Adam and V. Nazabal, Opt. Mater., 2011, 33, 980 CrossRef CAS.
- F. Zhu, Z. Xiao, F. Zhang, L. Yan and A. Huang, J. Lumin., 2011, 132, 22 CrossRef.
- F. Zhu, Z. Xiao, L. Yan, F. Zhang and A. Huang, Appl. Phys. A: Mater. Sci. Process., 2010, 101, 689 CrossRef CAS.
- Y. Zhao, C. Li, X. Liu and F. Gu, J. Alloys Compd., 2007, 440, 281 CrossRef CAS.
- T. Wu, G. Liu, J. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845 CrossRef CAS.
- J. Bi, L. Wu, Y. Zhang, Z. Li, J. Li and X. Fu, Appl. Catal., B, 2009, 91, 135 CrossRef CAS.
- G. J. Xing, R. Liu, C. Zhao, Y.-L. Li, Y. Wang and G. M. Wu, Ceram. Int., 2011, 37, 2951 CrossRef CAS.
- J. Yu, L. Qi, B. Cheng and X. Zhao, J. Hazard. Mater., 2008, 160, 621 CrossRef CAS.
- L. Zhen, W. S. Wang, C. Y. Xu, W. Z. Shao, M. M. Ye and Z. L. Chen, Scr. Mater., 2008, 58, 461 CrossRef CAS.
- H. Fu, C. Pan, L. Zhang and Y. Zhu, Mater. Res. Bull., 2007, 42, 696 CrossRef CAS.
- T. Yan, L. Li, W. Tong, J. Zheng, Y. Wang and G. Li, J. Solid State Chem., 2011, 184, 357 CrossRef CAS.
- A. Zhang and J. Zhang, J. Mater. Sci., 2010, 45, 4040 CrossRef CAS.
- A. Zhang and J. Zhang, Chin. J. Chem. Phys., 2010, 23, 73 CrossRef CAS.
- C. J. Philippopoulos, M. D. Nikolaki, 2010; Photocatalytic processes on the oxidation of organic compounds in water, New trends in technologies, Blandna Ramov (Ed.), ISBN: 978-953-7619-62-6, InTech, p. 97 Search PubMed.
- A. Zhang and J. Zhang, J. Hazard. Mater., 2010, 173, 265 CrossRef CAS.
- E. Y. Kim, D. S. Kim and B. T. Ahn, Bull. Korean Chem. Soc., 2009, 30, 193 CrossRef CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855 CrossRef CAS.
- A. R. Malagutti, H. A. J. L. Mourão, J. R. Garbin and C. Ribeiro, Appl. Catal., B, 2009, 90, 205 CrossRef CAS.
- J. Bi, L. Wu, Z. Li, Z. Ding, X. Wang and X. Fu, J. Alloys Compd., 2009, 480, 684 CrossRef CAS.
- N. Barka, S. Qourzal, A. Assabbane, A. Nounah and Y. Ait-Ichou, J. Photochem. Photobiol., A, 2008, 195, 346 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735 CrossRef CAS.
- T. Saison, N. Chemin, C. Chanéac, O. Durupthy, V. Ruaux, L. Mariey, F. Maugé, P. Beaunier and J. P. Jolivet, J. Phys. Chem. C, 2011, 115, 5657 CAS.
- J. M. Wu and T. W. Zhang, J. Photochem. Photobiol., A, 2004, 162, 171 CrossRef CAS.
- H. A. J. L. Mourão, V. R. de Mendonça, A. R. Malagutti and C. Ribeiro, Quim. Nova, 2009, 32, 2181 CrossRef.
- K. Rajeshwar, M. E. Osugi, W. Chanmanee, C. R. Chenthamarakshan, M. V. B. Zanoni, P. Kajitvichyanukul and R. Krishnan-Ayer, J. Photochem. Photobiol., C, 2008, 9, 171 CrossRef CAS.
- T. Montini, V. Gombac, A. Hameed, L. Felisari, G. Adami and P. Fornasiero, Chem. Phys. Lett., 2010, 498, 113 CrossRef CAS.
- S. Banerjee, J. Gopal, P. Muraleedharan, A. K. Tyagi and B. Raj, Curr. Sci, 2006, 90, 1378 CAS.
- K. Yu, S. Yang, H. He, C. Sun, C. Gu and Y. Ju, J. Phys. Chem. A, 2006, 113, 10024 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization details and figures for supplementary information. CCDC reference numbers 871280–871282, 873869, 873870, 873871. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20266b |
| This journal is © The Royal Society of Chemistry 2012 |