Complexing AIEE-active tetraphenylthiophene fluorophore to poly(N-isopropyl acrylamide): fluorescence responses toward acid, base and metal ions†
Yi-Wen
Lai
,
Shiao-Wei
Kuo
and
Jin-Long
Hong
*
Department of Materials and Optoelectrical Science, National Sun Yat-Sen University, Kaohsiung, 80424, Taiwan. E-mail: Fax: +886 07 5254099 Tel: +886 07 5252000ext4065; Fax: +886 07 5254099; Tel: +886 07 5252000ext4065
First published on 7th August 2012
Abstract
In this article, a multiple-responsive polymer micelles system was constructed by using an ionic bond as the link to connect the hydrophobic tetraphenylthiophene (TP) fluorophores having aggregation-induced emission enhancement (AIEE), and the hydrophilic poly(N-isopropyl acrylamide) (PNIPAM). The susceptibility of the ionic ammonium-sulfonate (Am-Sul) bond towards metal ions, acid and base triggered the AIEE-operative fluorescence (FL) response. To exercise the idea, PNIPAM with a sulfonate terminal was primarily prepared to react with an ammonium-functionalized TP derivative to generate a polymer complex of TP-PNIPAM. When in water, the polymer complex TP-PNIPAM formed micelles with the aggregated TP core interconnecting the hydrophilic PNIPAM shell by the ionic Am-Sul bonds. With the operative AIEE effect, the aggregated TP core in the micelles fluoresced strongly but upon the additions of metal ions, acid and base, the ionic bonds dissociated to result in the collapse of the micelles and the corresponding emission quenching. A novel fluorogenic sensor capable of responding to multi-stimuli was therefore constructed.
Introduction
Micelles fabricated from polymeric systems1 responsive to external signals have attracted considerable research interest due to their potential applications in drug delivery2 and catalysis.3 The simplicity of micelle formation by self-assembly of amphiphilic block copolymer molecules is extremely attractive and much effort4 has been directed toward engineering “smart” micelle systems from polymers featuring stimuli-responsive hydrophilic/hydrophobic properties. The hydrophilic part of the amphiphilic block copolymers is preferentially immersed in the water, while the hydrophobic part tends to resides in the air or in the non-solvent. Therefore, the amphiphilic block copolymers are aggregated to form different molecular assemblies by the repelling and coordinating action between the hydrophilic and hydrophobic parts to the surrounding environment. Currently, redox,5 temperature,6 pH,7 photochemical,8 and enzyme9 sensitive groups have been incorporated with polymer backbones to generate different responsive micelle systems.Among the varieties of stimuli-responsive systems, polymers exhibiting phase transformation above LCST have attracted much interest in view of the potential applications in cell immobilization, purification and drug delivery systems. PNIPAM with an LCST of ∼32 °C is the most extensively studied system due to its dramatic and reversible transition behavior across the LCST.10f A possible model relating to the coil-to-globule collapse of PNIPAM in water10g–10i has been proposed and the temperature-driven single-chain conformational transformation and concurrent macroscopic phase separation reflect rather subtle changes in polymer/water hydrogen-bonded interactions. A previous study10i has indicated that minor changes in the chemical composition of PNIPAM caused significant alternations on the phase diagram of PNIPAM in water.
Conventional organic fluorophores enjoy high fluorescence (FL) in dilute solutions but suffer from the detrimental aggregation-caused emission quenching in the condensed aggregated state. In contrast to conventional organic fluorophores, Tang's groups discovered that the silole molecule has the reverse emission property that it emitted strongly in the condensed aggregated state despite the observed weak emission in dilute solution.11a This interesting phenomenon was designated as “aggregation-induced emission” (AIE) and was rationalized as a result of restricted intramolecular rotation (RIR)11b–11e of the non-conplanar silole molecule in the aggregated state. Since the discovery of the silole system, lots of organic and polymeric materials12a–12l with AIE or AIE enhancement (AIEE) properties have been prepared and well characterized. A PNIPAM chain12l with an AIEE-active tetraphenylthiophene (TP) center was prepared in our laboratory and its FL response toward LCST was characterized. In water medium, micelles from the TP-derived PNIPAM emitted strongly due to the aggregated TP core but at temperature above LCST the dissociation of the aggregated core resulted in the emission quenching.
In this study, we reported the successful engineering of novel responsive polymer micelles by using ionic ammonium sulfonate (Am-Sul) bonds, which are susceptible to metal ions, acid and base, as the linkage between the hydrophobic AIEE-operative fluorophore and the hydrophilic PNIPAM chain. Primarily, PNIPAM with an anionic sulfonate terminal was synthesized to complex with AIEE-active TP fluorophore containing two cationic ammonium groups through the facile ionic interaction (Scheme 1). In aqueous solution, the resultant polymer complexes will self-assemble into micelles with an architecture where the hydrophobic AIEE-active core was connected with the hydrophilic PNIPAM shell through ionic Am-Sul linkages. Within the central aggregated core, the fluorophores emit with the expected AIEE effect but upon the operations of the metal ions, acid and base additives, the ionic bonds dissociate to launch the disintegration of the micelle structure, which in turn leads to the release of the AIEE-active fluorophores into the aqueous phase and the reduced FL emission. A novel AIEE-active fluorescent micelle system capable of responding to the stimuli of acid, base and metal ions was thereby constructed.
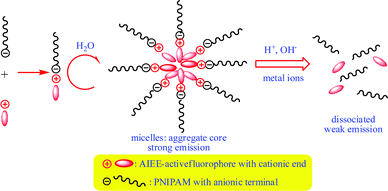 | ||
| Scheme 1 Complexation of AIEE-active cationic fluorophore with PNIPAM with anionic terminal to form self-assembled micelles in water and the subsequent dissociation of micelles under the operation of acid, base and metal ions. | ||
Experimental
Materials
Reagent grade benzyl chloride, sulfur powder, ethanol, ethyl acetate, glacial acetic acid, nitric acid, stannous chloride dihydrate, hydrochloric acid, 2-bromoethanesulfonate, sodium azide, propargylamine, α-bromoisobutyryl bromide, triethyl amine and 2-propanol were purchased from Aldrich Chemical Co. and used directly without further purification. N-Isopropylacrylamide (NIPAM) was purchased from Acros and recrystallized from hexane. CuBr (98%, Aldrich) was stirred overnight in acetic acid, filtered and washed with ethanol and diethyl ether before dried in vacuo. N,N-Dimethylformamide (DMF) was refluxed over CaH2 under nitrogen for 5 h before distillation for use. Tetrahydrofuran (THF) and dichloromethane were refluxed over sodium/benzophenone under nitrogen for more than 2∼3 days before distillation for use. Organic molecules of propargyl 2-bromo-2-methylpropionamide12l (BMP), PNIPAM,12l TP13,14 and tris(2-dimethylaminoethyl)amine (Me6-TREN)15 were prepared according to the reported procedures. Other organic molecules and polymers shown in Scheme 2 were prepared by the detailed procedures given below.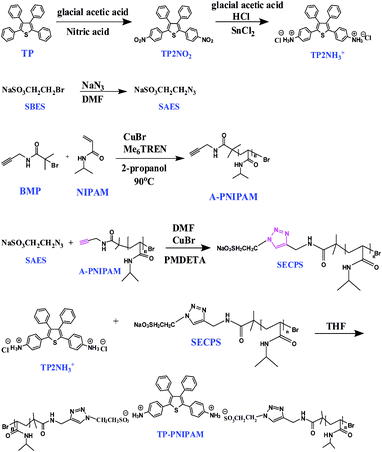 | ||
| Scheme 2 Syntheses of small-mass organic molecules of TP2NH3+, SAES and polymers of A-PNIPAM, SECPS and their subsequent complexation reaction to form TP-PNIPAM. | ||
Synthesis of 2,5-bis(4-nitrophenyl)-3,4-diphenylthiophene (TP2NO2)
TP (10 g, 25.8 mmol) and glacial acetic acid (150 mL) were placed in a 250 mL two-necked flask equipped with a magnetic stirrer and a condenser. The mixture was vigorously stirred at 110 °C to obtain a suspension. A mixture of glacial acetic acid (25 mL) and conc. nitric acid (10 mL) was then added quickly. After stirring at 110 °C for an additional 3 h, the yellow suspension was filtered and washed with water until pH = 7. The resultant product was eluted with hexane to yield the final product (10.24 g; 83% yield). Mp: 220 °C; IR (KBr pellet, cm−1) 3061, 1591, 1515, 1346, 1115, 853, 700; 1H NMR (500 MHz, CDCl3) δ 7.95 (d, 4H, Ha), 7.16 (d, 4H, Hb), 7.05–7.13 (m, 8H, Hc, Hd), 6.85 (d, 2H, He) (Fig. S1A, ESI†); MS m/z: calcd for C28H18N2O4S, 478.1; found, 478.9 (M+); Anal. Calcd for C28H18N2O4S : C, 70.28; H, 3.79; N, 5.85; O, 13.37; S, 6.7. Found: C, 70.56; H, 3.11; N, 5.76; O, 13.67; S, 6.9.Synthesis of TP2NH3+
TP2NO2 (2.51 g; 5.23 mmol) and glacial acetic acid (25 mL) were placed in a 100 mL two-necked flask equipped with a magnetic stirrer and a condenser. Solution of stannous chloride dehydrate (12.5 g; 55.4 mmol) in conc. hydrochloric acid (4 mL) was added at once. The mixture was stirred and refluxed at 100 °C for 5 h before cooling to −10 °C to give the solid dihydrochloride salt. The resultant salt was filtered and re-dissolved in 15 mL of hot water, to which 5 mL of conc. hydrochloric acid was added. The ammonium salt was then pressed and dried to give the final product (1.64 g; 64% yield). Mp: 271 °C; IR (KBr pellet, cm−1) 3447, 3420, 3345, 3212, 3057, 3026, 1618, 1546, 1482, 1442, 1284, 1179, 834, 770, 700; 1H NMR (500 MHz, (CD3)2SO) δ 7.12 (t, 6H, Hd, He), 6.93 (d, 4H, Hc), 6.81 (d, 4H, Hb), 6.38 (d, 4H, Ha), 5.18 (s, 6H, Hf) (Fig. S1B, ESI†); MS m/z: calcd for C28H24Cl2N2S, 490.1; found, 490.96 (M+); Anal. Calcd for C28H18N2O4S : C, 68.43; H, 4.92; N, 5.7; S, 6.52. Found: C, 68.7; H, 4.5; N, 5.88; S, 6.69.Synthesis of sodium 2-azidoethanesulfonate (SAES)
Solution of 2-bromoethanesulfonate (1 g; 4.76 mmol) in dried DMF (25 mL) was placed in a 50 mL two-necked flask equipped with a magnetic stirrer and a condenser. Sodium azide (0.32 g; 4.92 mmol) was then added before heating to 80 °C for 24 h. After cooling to room tempeature, the solvent was removed by a rotary evaporator to obtain the final product (0.82 g; 82% yield). Mp: 78 °C; IR (KBr pellet, cm−1) 2981, 2953, 2929, 2900, 2124, 1613, 1294, 1190, 1054, 808, 600; 1H NMR (500 MHz, D2O) δ 3.72 (t, 2H, Hb), 3.19 (t, 2H, Ha) (Fig. S2A, ESI†); MS m/z: calcd for C2H4N3NaO3S, 172.99; found, 172.73 (M+); Anal. Calcd for C2H4N3NaO3S : C, 13.88; H, 2.33; N, 24.27; O, 27.72; S, 18.52. Found: C, 14.15; H, 1.9; N, 24.47; O, 27.54; S, 18.65.Synthesis of SECPS
Solution of NaSO3CH2CH2N3 (0.2 g; 1.16 mmol) and PNIPAM (3.78 g; 1.05 mmol) in dried DMF (30 mL) was placed in a 50 mL two-neck flask equipped with a magnetic stirrer and a condenser. After the addition of CuBr (0.44 g; 3.07 mmol), all the mixtures were vigorously stirred and degassed by argon gas for 1 h and the PMDETA (0.2 mL) ligand was added and degassed for another 1 h. The resultant solution was then heated to 80 °C under N2 for 24 h. After cooling to room temperature, the resultant mixtures were vacuum dried at high temperature to remove DMF. The residue was re-dissolved in chloroform and a small amount of the precipitates was filtered off. The resultant filtrate was passed through an alumina column to remove the copper catalyst. Finally, the solution was dried by a rotary evaporator to obtain the product (2.81 g, 71% yield). Tg: 159 °C; IR (KBr pellet, cm−1) 3447, 3298, 2974, 2933, 2874, 1651, 1546, 1458, 1388, 1367, 1264, 1171, 1127, 1115; 1H NMR (500 MHz, CDCl3) δ 6.11–6.87 (broad, 2H, NHCO), 4.01 (broad, 1H, CH(CH3)2), 3.69 (t, 2H, Hl), 3.55 (broad, 2H, Hg), 1.4–2.2 (broad, 3H, backbone Hs), 1.18 (broad, 6H, CH(CH3)2) (Fig. S2B, ESI†).Synthesis of TP-PNIPAM
Solution of TP2NH3+ (0.5 g; 1.02 mmol) in THF (5 mL) was added to a solution of SECPS (7.2 g; 2.04 mmol) in THF (5 mL). The resultant precipitates were filtered off before drying with a rotary evaporator to obtain the final product (6.5 g, 85% yield). Tg: 144 °C; IR (KBr pellet, cm−1) 3440, 3298, 3071, 2971, 2933, 2874, 1651, 1541, 1460, 1388, 1358, 1258, 1171, 1127, 1115; 1H NMR (500 MHz, CDCl3) δ 6.95–7.24 (m, 18H, aromatic Hs), 6.11–6.87 (broad, 2H, NHCO), 5.29 (s, 6H, Hk), 4.01 (broad, 1H, CH(CH3)2), 3.75 (t, 2H, Hl), 3.49 (t, 2H, Hg), 1.4–2.2 (broad, 3H, backbone Hs), 1.18 (broad, 6H, CH(CH3)2) (Fig. S5, ESI†).Measurements
UV-vis absorption spectra were recorded with an Ocean Optics DT 1000 CE 376 spectrophotometer. FL spectra were obtained from a LabGuide X350 fluorescence spectrophotometer using a 450W Xe lamp as a continuous light source. 1H NMR spectra were recorded with a VarianVXR-500 MHz FT-NMR instrument. Tetramethylsilane was used as internal standard. FT-IR spectra were obtained on a Bruker Tensor 27. Particle sizes of TP2NH3+ in THF/water mixtures at different conditions were measured by dynamic light scattering (DLS) using a Brookhaven 90 plus spectrometer equipped with a temperature controller. An argon ion laser operating at 658 nm was used as a light source. Elemental analyses were performed on an Elementary Vario EL-III C, H, N, O and S analyzer. A mass spectrum was obtained by using a Bruker Daltonics Autoflex III MALDI-TOF mass spectrometer. The melting point (Tm) of the organic compound and the glass transition temperature (Tg) of the polymer were determined from a TA Q-20 differential scanning calorimeter (DSC) calorimeter with a scan rate of 20 °C min−1 under nitrogen. TEM was obtained from a JEM-2100 electron microscope with LaB6 as the light source.Results and discussion
Synthesis
As illustrated in Scheme 2, the desired polymeric complex TP-PNIPAM was prepared from the complexation of the ammonium (–NH3+) function in the cationic TP2NH3+ to the anionic sulfonate (–SO3−) terminal in the polymeric SECPS. To obtain the cationic TP2NH3+ with the AIEE feature, a two-step synthesis pathway including nitration of the TP molecule to generate TP2NO2 and the subsequent reduction/acidification to yield the cationic –NH3+ function in the desired TP2NH3+, was conducted. On the other hand, the required sulfonate terminal in SECPS was introduced by the facile Click reaction between the azide-functionalized SAES and the alkyne-terminated polymer A-PNIPAM. In this case, a nucleophilic attack of sodium azide (NaN3) to SBES resulted in compound SAES while the atom transfer radical polymerization (ATRP) of NIPAM monomer by an alkyne-functionalized initiator BMP directly afforded the polymeric A-PNIPAM. The chemical structures of all organic intermediates and products were carefully characterized and confirmed by 1H-NMR (Fig. S1, S2, ESI†), mass spectroscopy and elemental analysis.Click reaction between SAES and A-PNIPAM yielded the key polymer precursor of SECPS. Primarily, a successful Click reaction can be assured from the corresponding FTIR spectra (Fig. S3, ESI†), in which the azide –N3+ peak of SAES at 2100 cm−1 was absent in the spectrum of the SECPS product. Molecular weights of A-PNIPAM and SECPS polymers can be evaluated from the MALDI-TOF mass and the 1H NMR (Fig. S4 and S5, ESI†) spectra and the results were summarized and compared in Table 1. For polymer SECPS, the corresponding degree of polymerization (n) evaluated from the mass spectrum is 28, which is slightly lower than that (= 31) calculated from the 1H NMR spectrum. The resultant SECPS was further complexed with TP2NH3+ to afford the complex TP-PNIPAM and in the complex, the average complexation number (CN) of SECPS chain to each TP2NH3+ fluorophore can be also formulated from the mass and the 1H NMR spectra and the resultant CNs (2.00 and 2.01) are well correlated with the theoretical value of 2.04 evaluated from the experimental feed ratio. All three values discussed here are in close vicinity, which indicates that the complexation between the ammonium cation and the sulfonate anion proceeded efficiently and thoroughly. Successful incorporation of TP2NH3+ into the polymer complex can also be verified from the metal-chelation experiment given later.
| from MALDI-TOFa | from 1H NMR | ||||||
|---|---|---|---|---|---|---|---|
| M n a | M w a | PDIa | Nb | CNc | Nb | CNc | |
| a M n = number average molecular weight, Mw = weight-average molecular weight, PDI = polydispersity. b N = number of repeat unit incorporated per each PNIPAM chain. c CN (complexation number) = the average number of PNIPAM chain incorporated to each TP2NH3+ fluorophore, evaluated from MALDI-TOF and 1H NMR spectrum. | |||||||
| A-PNIPAM | 3400 | 3600 | 1.06 | ||||
| SECPS | 3000 | 3500 | 1.17 | 28 | 31 | ||
| TP-PNIPAM | 7200 | 7500 | 1.04 | 2.00 | 2.01 | ||
AIEE characterizations on the model compound TP2NH3+ and the polymer complex TP-PNIPAM
Primarily, the AIEE feature of the model TP2NH3+ was characterized by its emission behavior in solvent/poor solvent mixtures. The hydrophilic ammonium functions of TP2NH3+ do not render its solubility in water; on the contrast, the high fraction of hydrophobic aromatic rings in TP2NH3+ directly leads to its insolubility in water. The AIEE effect of TP2NH3+ was therefore conducted in a solvent pair of THF/water (solvent/poor solvent). As illustrated in Fig. 1, pure solution (10−5 M) of TP2NH3+ in THF emitted with the overlapped bands attributed to the monomer (at 444 nm) and aggregated (at 484 nm) emissions, respectively. With increasing the water content from 30 to 80 vol.% (TP2NH3+ precipitated from the solution when the water content reached 90 vol.%), we observed a gradual increase of the emission intensity. It was noted here that the emission enhancement is mostly due to the intensified aggregate emission, which coincided with the general character of AIEE effect. Aggregate formation upon water inclusion can be confirmed by the turbid appearance of the resultant mixture solutions. DLS analysis (Fig. S7A, ESI†) indicated that the aggregate particles formed during water inclusion shrunk in size with increasing water content in the solvent mixtures. The volume shrinkage will impose steric hindrance for free molecular rotations of the TP fluorophores inside the aggregated particles, which blocked the non-radiative decay pathway and resulted in the emission enhancement.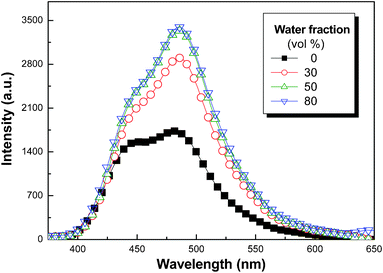 | ||
| Fig. 1 FL emission spectra of TP2NH3+ (10−5 M) in THF/water mixtures of different composition (λex = 325 nm). | ||
In aqueous solution, the TP-PNIPAM chains self-assembled into micelles with an architecture where the aggregated TP core and the hydrophilic PNIPAM shell are connected by the ionic Am-Sul bonds. With the aggregated TP core, the micelles formed in aqueous solution are supposed to exhibit the AIEE feature; however, our initial attempt to characterize the AIEE effect by using THF/water solvent pair was unsuccessful due to the good solubility of PNIPAM in water. Reasonable results were therefore obtained from another solvent pair of ethanol/acetone and in this case, acetone serves as poor solvent for PNIPAM and TP2NH3+. Fig. 2 showed the fundamental feature of AIEE-operative emission that the FL emission intensity increased with increasing acetone content in the solution mixtures. The resolved monomer and aggregate emissions are also correlated with those (cf. Fig. 1) observed in the model TP2NH3+ solutions. The DLS analyses (Fig. S7B, ESI†) on the corresponding solution mixtures also suggested that the detected particle sizes decreased with increasing the acetone content in the solution mixtures. Basically, the size shrinkage of the resultant particles imposed restrictions on the molecular rotations of the inside fluorophores, which in turn led to emission enhancement due to the blockage of non-radiative decay pathways.
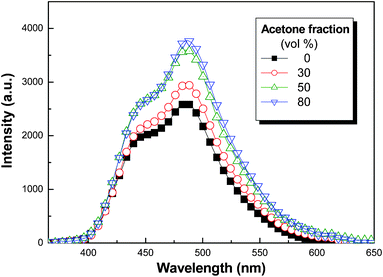 | ||
| Fig. 2 FL emission spectra of TP-PNIPAM (10−5 M) in ethanol/acetone mixtures of different compositions (λex = 350 nm). | ||
TP-PNIPAM as fluorogenic metal-ion sensor
The visual response for the aq. TP-PNIPAM solution upon introduction of NaCl can be used as indirect proof of micelle formation. In water, the TP-PNIPAM chains self-assembled into micelles with their sizes large enough to scatter the incident light, and therefore the corresponding aq. solution (Fig. 3A) is turbid. The micelle structure can also be verified by the TEM image (Fig. S8, ESI†) of the solid sample cast from the aq. TP-PNIPAM solution. Upon addition of aq. NaCl solution, the turbid solution shown in Fig. 3A became clear but with lots of white precipitates scattered over the surfaces of the container. These solid precipitates luminesced upon irradiation by ultraviolet light and were separately identified to be TP2NH3+ by 1H NMR. It is envisaged that upon the reaction of the sodium ion the ionic Am-Sul bonds were dissociated to result in the release of TP2NH3+ species into the water matrix. The fluorogenic sensitivity of TP-PNIPAM toward the sodium ion can be demonstrated by the complete FL quenching after introduction of NaCl (Fig. 3B). The FL response indicates that TP-PNIPAM is a FL sensor toward the sodium ion. With the strong electrostatic interaction forces, the ionic NaCl is capable of penetrating into the core region of the micelles to dissociate the ionic Am-Sul bonds and rupture the micelles, to end in the release and precipitation of the TP2NH3+ species in water. With no aggregated TP core to exert the AIEE-operative emission, the collapsed micelles are no longer luminescent due to the scarcity of AIEE effect.![(A) Photographs of the aq. TP-PNIPAM solutions before (up) and after (bottom) the addition of NaCl and (B) the corresponding emission response of the aq. TP-PNIPAM solution toward NaCl addition. ([TP-PNIPAM] = 1.335 × 10−4 M; [NaCl] = 1 M; λex = 350 nm).](/image/article/2012/RA/c2ra20857a/c2ra20857a-f3.gif) | ||
| Fig. 3 (A) Photographs of the aq. TP-PNIPAM solutions before (up) and after (bottom) the addition of NaCl and (B) the corresponding emission response of the aq. TP-PNIPAM solution toward NaCl addition. ([TP-PNIPAM] = 1.335 × 10−4 M; [NaCl] = 1 M; λex = 350 nm). | ||
Fig. 4 shows the FL response of the aq. TP-PNIPAM solution to various metal ions. Similar to NaCl case, the added metal ions dissociated certain ionic bonds inside the micelles and led to the precipitation of TP2NH3+ species. In all cases, both the monomer and the aggregate emissions reduced their intensities upon inclusion of metal ions. When exposed to the monovalent K+, Rb+ and Cs+ ions, the FL emission became weaker but to a lesser extent than those exposed to the bivalent Ca2+, Ni2+, Cu2+ and Zn2+ ions. The bivalent ions exerted more complexation power and are therefore more efficient in dissociating the ionic Am-Sul bonds than the monovalent metal ions.
![FL emission spectra of the aq. TP-PNIPAM solutions in the presence of different metal salts. ([TP-PNIPAM] = 6.676 × 10−5 M; [metal salts] = 1.315 × 10−1 M (λex = 350 nm).](/image/article/2012/RA/c2ra20857a/c2ra20857a-f4.gif) | ||
| Fig. 4 FL emission spectra of the aq. TP-PNIPAM solutions in the presence of different metal salts. ([TP-PNIPAM] = 6.676 × 10−5 M; [metal salts] = 1.315 × 10−1 M (λex = 350 nm). | ||
pH-Driven emission variations
The ionic interaction between ammonium and sulfonate bond was affected by the pH value in the surrounding environment. Under the operation of strong HCl acid, the ionic bonds will be ruptured to result in the collapse of micelles, which can be reflected by the slow decay of the AIEE-operative emission during the disassembling process.To verify our hypothesis, the FL response of the TP-PNIPAM solution toward HCl addition was investigated. Different amounts of HCl were added to the aq. TP-PNIPAM solution to attain solutions of varied pH values (while keeping the concentration of TP-PNIPAM constant). As shown in Fig. 5A, both the monomer and the aggregate emissions gradually lost their intensities with increasing the acidity to pH = 3. The emission reductions at this stage were also accompanied by precipitation of the TP2NH3+ species. Further addition of HCl to pH = 2, however, caused the unexpected new emission band located at a longer wavelength of 500 nm. A separate experiment indicated that the FL spectrum of the aq. SECPS solution (Fig. S9, ESI†) at pH = 2 also contained the same emission band at 500 nm. At highly acidic conditions, the sulfonate terminal of SECPS was protonated and converted into sulfonic acid, which was suggested to be responsible for the emission at 500 nm.
![FL emission spectra of the aq. TP-PNIPAM solutions in the presence of (A) HCl and (B) NaOH. ([TP-PNIPAM] = 6.676 × 10−5 M; λex = 350 nm).](/image/article/2012/RA/c2ra20857a/c2ra20857a-f5.gif) | ||
| Fig. 5 FL emission spectra of the aq. TP-PNIPAM solutions in the presence of (A) HCl and (B) NaOH. ([TP-PNIPAM] = 6.676 × 10−5 M; λex = 350 nm). | ||
The gradual emission weakening is also expected at high pH conditions since the ionic bond will be ruptured by the alkaline NaOH. As illustrated in Fig. 5B, the disruption of the ionic bonds upon addition of aq. NaOH solution caused a continuous weakening of the FL emission intensity from pH = 8 to 13. Upon the attack of alkaline NaOH, the dissociation of the ionic Am-Sul bonds should lead to the release of SECPS and TP2NH2. In this case, the observed precipitate was the organic TP2NH2 molecule due to its insolubility in water.
Conclusions
Organic compound TP2NH3+ and polymer SECPS were prepared and mixed together to generate the amphiphilic polymer complex TP-PNIPAM. The solution emission in solvent/non-solvent mixtures suggest that the organic TP2NH3+ was indeed an AIEE-active materials and can be used to inherit the AIEE-operative FL property into TP-PNIPAM when complexed to SECPS. In water, TP-PNIPAM formed micelles with an architecture where the aggregated, hydrophobic TP core was surrounded by the hydrophilic PNIPAM shell.The ionic Am-Sul bonds connecting the core and shell segments acted as a trigger to launch the multiple responses toward metal ions, acid and base. Under the operation of metal ions, acid and base, the ionic Am-Sul bonds dissociated to result in the collapse of the micelles and the release of the AIEE-active fluorophores, thus leading to weakening of the FL emission. A novel fluorogenic sensor capable of responding to the stimuli of metal ions, acid and base was therefore generated.
References
- (a) C. Schatz, S. Louguet, J. -F. Le Meins and S. Lecommandoux, Angew. Chem., Int. Ed., 2009, 48, 2572–2575 CrossRef CAS; (b) G. Riess, Prog. Polym. Sci., 2003, 28, 1107–1170 CrossRef CAS; (c) C. L. McCormick, B. S. Sumerlin, B. S. Lokitz and J. E. Stempka, Soft Matter, 2008, 4, 1760–1773 RSC; (d) A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS; (e) J.-F. Lutz, Polym. Int., 2006, 55, 979–993 CrossRef CAS.
- (a) S. Ganta, H. Devalapally, A. Shahiwala and M. Amiji, J. Controlled Release, 2008, 126, 187–204 CrossRef CAS; (b) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS; (c) N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS; (d) F. Meng, Z. Zhong and J. Feijen, Biomacromolecules, 2009, 10, 197–209 CrossRef CAS; (e) M. H. Stenzel, Chem. Commun., 2008, 3486–3503 RSC; (f) H. Wei, S.-X Cheng, X.-Z. Zhang and R.-X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- D. M. Vriezema, M. C. Aragone`s, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS.
- (a) Y. Wang, H. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849–2864 CrossRef CAS; (b) C. Wang, Y. Guo, Y. Wang, H. Xu and X. Zhang, Chem. Commun., 2009, 5380–5382 RSC; (c) A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS.
- (a) K. N. Power-Billard, R. J. Spontak and I. Manners, Angew. Chem., Int. Ed., 2004, 43, 1260–1264 CrossRef CAS; (b) N. Ma, Y. Li, H. Xu, Z. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442–443 CrossRef CAS; (c) A. Napoli, M. Valentini, N. Tirelli, M. Müller and J. A. Hubbell, Nat. Mater., 2004, 3, 183–189 CrossRef CAS; (d) X.-S. Wang, H. Wang, N. Coombs, M. A. Winnik and I. Manners, J. Am. Chem. Soc., 2005, 127, 8924–8925 CrossRef CAS; (e) S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS; (f) W.-F. Dong, A. Kishimura, Y. Anraku, S. Chuanoi and K. Kataoka, J. Am. Chem. Soc., 2009, 131, 3804–3805 CrossRef CAS; (g) J. Bigot, B. Charleux, G. Cooke, F. Delattre, D. Fournier, J. Lyskawa, L. Sambe, F. Stoffelbach and P. Woisel, J. Am. Chem. Soc., 2010, 132, 10796–10801 CrossRef CAS.
- Y. Morishama, Angew. Chem., Int. Ed., 2007, 46, 1370–1372 CrossRef.
- A. Klaikherd, C. Nagamani and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 4830–4838 CrossRef CAS.
- A. P. Goodwin, J. L. Mynar, Y. Ma, G. R. Fleming and J. M. J. Fre'chet, J. Am. Chem. Soc., 2005, 127, 9952–9953 CrossRef CAS.
- R. J. Amir, S. Zhong, D. J. Pochan and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 13949–13951 CrossRef CAS.
- (a) H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS; (b) Y. H. Bae, T. Okano, R. Hsu and S. W. Kim, Makromol. Chem. Rapid Commun., 1987, 8, 481–485 CrossRef CAS; (c) A. S. Hoffman, J. Controlled Release, 1987, 6, 297–305 CrossRef CAS; (d) H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, J. Membr. Sci., 1991, 64, 283–294 CrossRef CAS; (e) A. Gustafsson, H. Wennerstrom and F. Tjerneld, Fluid Phase Equilib., 1986, 29, 365–371 CrossRef CAS; (f) S. B. La, T. Okano and K. Kataoka, J. Pharm. Sci., 1996, 85, 85–90 CrossRef CAS; (g) A. Matsuyama and F. Tanaka, J. Chem. Phys., 1991, 94, 781–786 CrossRef CAS; (h) C. Wu and S. Zhou, Macromolecules, 1995, 28, 5388–5390 CrossRef CAS; (i) Y. Okada and F. Tanaka, Macromolecules, 2005, 38, 4465–4471 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC; (b) H. Y. Chen, W. Y. Lam, J. D. Luo, Y. L. Ho, B. Z. Tang, D. B. Zhu, M. Wong and H. S. Kwok, Appl. Phys. Lett., 2002, 81, 574–576 CrossRef CAS; (c) J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS; (d) J. Chen, Z. Xie, J. W. Y. Lam, C. C. W. Law and B. Z. Tang, Macromolecules, 2003, 36, 1108–1117 CrossRef CAS; (e) J. Chen, H. Peng, C. C. W. Law, Y. Dong, J. W. Y. Lam, I. D. Williams and B. Z. Tang, Macromolecules, 2003, 36, 4319–4327 CrossRef CAS.
- (a) S. Dong, Z. Li and J. Qin, J. Phys. Chem. B, 2009, 113, 434–441 CrossRef CAS; (b) C. X. Yuan, X. T. Tao, L. Wang, J. X. Yang and M. H. Jiang, J. Phys. Chem. C, 2009, 113, 6809–6814 CrossRef CAS; (c) Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975–3981 CrossRef CAS; (d) B. Xu, Z. Chi, Z. Yang, J. Chen, S. Deng, H. Li, X. Li, Y. Zhang, N. Xu and J. Xu, J. Mater. Chem., 2010, 20, 4135–4141 RSC; (e) Z. Zhao, S. Chen, J. W. Y. Lam, P. Lu, Y. Zhong, K. S. Wong, H. S. Kwok and B. Z. Tang, Chem. Commun., 2010, 46, 2221–2223 RSC; (f) L. Tang, J. K. Jin, A. Qin, W. Z. Yuan, Y. Mao, J. Mei, J. Z. Sun and B. Z. Tang, Chem. Commun., 2009, 33, 4974–4976 RSC; (g) J. Liu, Y. Zhong, J. W. Y. Lam, P. Lu, Y. Hong, Y. Yu, Y. Yue, M. Faisal, M. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Macromolecules, 2010, 43, 4921–4936 CrossRef CAS; (h) A. Qin, J. W. Y. Lam, L. Tang, C. K. W. Jim, H. Zhao, J. Sun and B. Z. Tang, Macromolecules, 2009, 42, 1421–1424 CrossRef CAS; (i) R. H. Chien, C. T. Lai and J. L. Hong, J. Phys. Chem. C, 2011, 115, 5958–5965 CrossRef CAS; (j) C. T. Lai and J. L. Hong, J. Phys. Chem. B, 2010, 114, 10302–10310 CrossRef CAS; (k) C. A. Chou, R. H. Chien, C. T. Lai and J. L. Hong, Chem. Phys. Lett., 2010, 501, 80–86 CrossRef CAS; (l) C. T. Lai, R. H. Chien, S. W. Kuo and J. L. Hong, Macromolecules, 2011, 44, 6546–6556 CrossRef CAS.
- F. K. Su, J. L. Hong and L. L. Lin, Synth. Met., 2004, 142, 63–69 CrossRef CAS.
- C. H. Yang, L. L. Lin and J. L. Hong, Polym. Int., 2005, 54, 679–685 CrossRef CAS.
- S. Inceoglu, S. C. Olugebefola, M. H. Acar and A. M. Mayes, Des. Monomers Polym., 2004, 7, 181–189 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: 1H NNR, FTIR, MALDI-TOF, UV, DLS, TEM and FL spectra of all orgnaic molecules and polymers. See DOI: 10.1039/c2ra20857a/ |
| This journal is © The Royal Society of Chemistry 2012 |