DOI:
10.1039/C2RA20608K
(Paper)
RSC Adv., 2012,
2, 8201-8208
Superior photocatalytic behaviour of novel 1D nanobraid and nanoporous α-Fe2O3 structures†
Received
2nd April 2012
, Accepted 3rd July 2012
First published on 4th July 2012
Abstract
We have produced novel nanostructures of pure and ceramic α-Fe2O3 using electrospinning, followed by annealing at 500 °C for 5 h with ramp rate of 5 °C min−1. Electron microscopy clearly reveals the novel morphologies, namely nanobraids and nanoporous α-Fe2O3, suggesting that the precursor, (iron(III) acetylacetonate, (Fe(acac)3)) to polyvinylpyrrolidone (PVP) ratio greatly influences structural transformations of Fe2O3. 4 wt% of Fe(acac)3/PVP solution used for electrospinning at 15 kV a potential produced nanobraid-like ceramic α-Fe2O3, indicating that binodal phase separation is predominant at this ratio. On the other hand, the electrospinning of 6 wt% of Fe(acac)3/PVP solution induces spinodal phase separation that results in the formation of nanoporous ceramic α-Fe2O3 fibers. The nanobraids and nanoporous ceramic α-Fe2O3 exhibit superior photocatalytic performances of up to 91.2% and 90.2% for the organic dye, Congo red (CR) in the shorter time of 140 min under photoirradiation. It is concluded that the presence of the porous surface and smaller crystallite size in the α-Fe2O3 nanostructures act as active catalytic centers and play a key role in allowing effective interaction between organic dye and α-Fe2O3, in turn enhance photocatalytic degradation performance.
1 Introduction
Novel nanostructures in materials, especially metal oxides have recently received more attention due to the rapidly evolving fields of nanoenergetics and nanoelectronics. Ferric oxide, α-Fe2O3 is a promising photovoltaic, photocatalytic, and electrochemical sensing material due to its ability to harvest the solar energy, catalyze reactions and act as rapid responsive material. The size and shape of the α-Fe2O3 nanostructure plays a dominant role in its catalytic, magnetic, electrical and optical properties.1–6 Specifically, 1 dimensional (1D) nanostructures of α-Fe2O3 that possess large surface-to-volume ratios per unit volume and offer unique chemical and optical properties are preferred for their broad range of applications such as field effect transistors,7 biosensors,8 magnetic resonance imaging (MRI),9 bio-probes,10 batteries,11 and photo-electrochemical cells.12,13 A sensor based on a 1D α-Fe2O3 nanotubular structure exhibited rapid response and good stability,11 attributed to the large surface area of the nanotubes that effectively absorbs the solute on the surface of α-Fe2O3. Recently, α-Fe2O3 nanostructures that allows metals or metal oxides to be embedded within α-Fe2O3 has received interest for energy and environmental related applications, especially catalysis.13–16 Ultrathin platinum layered on the surface of α-Fe2O3 film was reported to improve the photocatalytic degradation activity of α-Fe2O3 against methanol.15 The presence of tin oxide (SnO2) on α-Fe2O3 in a core–shell arrangement markedly enhanced the photodegradation efficiency of α-Fe2O3 against rhodamine B dye, owing to the synergistic effects of α-Fe2O3 and SnO2.14 1D α-Fe2O3 morphologies with a characteristic structure of interconnected nanometer-sized pores have been very encouraging. The porous structure within α-Fe2O3 will allows for faster diffusion of organic pollutants and better interaction that would eventually improve the photocatalytic performance.
Various methods including chemical vapor deposition,17,18 sol–gel,19 hydrothermal,20 electrochemical anodization,21 template,22 and electrospinning23,24 were explored to produce α-Fe2O3 nanostructures. Formation of nanostructures by a template-directed approach was well proven, but the extent to which this method could be applied is currently limited to relatively shorter structures; long and flexible templates could cause interconnections or entanglements between nanotubes. Fan et al. employed an activated carbon template based the supercritical fluid (CO2) method to prepare porous α-Fe2O3 film.25 However, this method did not yield 1D nanostructures with fine pores, and even a small change in the temperature or pressure could impact on the purity of α-Fe2O3 and percentage of yield. McKenzie et al. obtained a nanoporous α-Fe2O3 film by a layer-by-layer method using phytic acid, a linker molecule, followed by calcination at 500 °C in air.26 This approach however imposed numerous repeating process sequences to form the layers, which impacted on the process cost.
Our work in this report demonstrates an effective electrospinning method that can produce novel 1D α-Fe2O3 with unique and characteristic morphologies. Our study revealed that the combination of polymer induced phase separation (thermodynamic) and electrospinning (electro-hydrodynamic) is the key to produce the novel nanostructures: 1D nanoporous structures and nanobraids α-Fe2O3. The potential application of the as-prepared α-Fe2O3 nanostructures was investigated for its photocatalytic performance. In this work, Congo red (CR) has been considered as an organic model pollutant because of its common usage in textile and biomedical industries. Congo red is a water soluble, azo type dye widely used as a biological stain and to color the textiles. It is carcinogenic in nature.27 The results showed that the unique ceramic 1D nanoporous and nanobraid morphologies of α-Fe2O3 exhibited superior photocatalytic behaviour against Congo red dye upon photoirradiation compared to the currently available literature.
2 Experimental sections
2.1 Materials
Polyvinylpyrrolidone (PVP; Mw = 1,300,000) and iron(III) acetylacetonate (Fe(acac)3) were purchased from Sigma-Aldrich and Fluka, respectively. Ethanol (HPLC grade) and glacial acetic acid were purchased from Tedia, Singapore and used as received.
2.2 Synthesis of Fe(acac)3/PVP composite fibers and α-Fe2O3 fibers
In a typical procedure, 1 g of PVP was added to 10 ml of ethanol and stirred at room temperature for 1 h. After PVP completely dissolved in ethanol, the precursor, Fe(acac)3 was added in the solution with different weight ratios of 4 wt% and 6 wt% and continuously stirred for 6 h. Finally, the mixed solution was loaded into a plastic syringe with internal diameter of pinhead around 0.859 mm. Later, the pinhead was connected to a high-voltage supply that is capable of generating direct current (DC) voltage of up to 30 kV (ELECTROSPUNRA). The applied potential was varied from 15 kV to 20 kV. An aluminum foil was used as the counter electrode, and the distance between the pinhead and the collector was maintained at 15 cm. Fig. 1 shows the simple schematic representation of electrospinning set-up. The as-electrospun Fe(acac)3/PVP composite fibers were placed in a vacuum oven for 12 h at room temperature to remove the residual solvent. Then, electrospun fibers were calcined at 500 °C for 5 h in air at a heating rate of 5 °C min−1, and finally α-Fe2O3 fibers were obtained.
 |
| | Fig. 1 Schematic representation of a typical electrospinning set-up. | |
2.3 Characterization
The crystallographic information of the as-prepared α-Fe2O3 nanostructures was analyzed by powder X-ray diffraction (XRD, Shimadzu XRD-6000, Cu-Kα radiation operating at 30 kV/40 mA). The surface morphology of the structures was characterized by a JEOL JSM-6700F field emission scanning electron microscope (FESEM). Transmission electron microscopy (JEM-2010, JEOL USA Inc.) was employed to study the detailed surface morphologies of the α-Fe2O3 nanostructures. Thermal decomposition of Fe(acac)3/PVP composite nanofibers were measured by a thermo-gravimetric analyzer (TA Instruments, TGA 2050) up to 800 °C with a heating rate of 5 °C min−1 under ambient atmosphere. Fourier transform infra-red (FTIR) was used to study the fundamental vibrations of chemical bonds and functional groups present in the composite. FTIR spectra of Fe(acac)3/PVP composite fibers before and after calcination were obtained from an Avatar 380 FTIR (Thermo Nicolet, Waltham, MA) over a range of 400 to 4000 cm−1 at a resolution of 2 cm−1. The specific surface area of as-prepared samples was determined using Brunauer–Emmett–Teller (BET) (NOVA 4200E Surface Area and Pore Size Analyzer, Quantachrome, USA). BET surface area has been averaged for three measurements and had an error average well within 3 m2 g−1.
2.4 Evaluation of photocatalytic activity
The photocatalytic performance of α-Fe2O3 nanostructures for photodegradation of Congo red (CR) dye was evaluated as follows. In a typical process, 20 ppm of CR dye was dispersed in 100 ml aqueous solution and 25 mg of α-Fe2O3 photocatalyst was added. Later, the mixture was mixed thoroughly in a shaker for 30 min to attain adsorption equilibrium. A light source of 400 W metal halide lamp (λ ≥ 365 nm) was used to initiate the reaction. At a regular interval of 10 min irradiation, 3 ml of dye solution was taken out and centrifuged to remove the catalysts. The photocatalysis experiment was carried out under static condition and the photodegradation of CR dye was monitored by measuring the maximum absorption peak at 498 nm using UV-Visible spectrometer (Shimadzu UV 3600 UV-Vis-NIR spectrophotometer).
3 Results and discussions
3.1 Morphology and crystallinity study of composite fibers
The size and distribution of the Fe(acac)3/PVP composite fibers were found to be changed upon varying the potential. The average fiber diameter of 328 nm ± 130 nm and 269 nm ± 110 nm were observed for the 4 wt% and 6 wt% Fe(acac)3/PVP composite green fibers at 15 kV and 18 kV, respectively (Fig. S1, ESI†). The decrease in the diameter of the nanofibers with the increase in the applied potential was observed, which was due to the dominance of the Coulombic repulsive forces. In electrospinning, a longer flight time of the jet will allow more time for the fibers to stretch and elongate with solvent evaporation before they randomly collect on grounded aluminum foil. At lower potentials, the acceleration of the jet is lower and the weaker potential favors the formation of finer fibers with broad distribution for 15 kV. The as-electrospun composite fibers were vacuum dried overnight to remove the residual solvent and calcined at 500 °C in a tubular furnace.
XRD analysis was carried out on the annealed sample to investigate phase formation. Fig. 2(a & b) shows the XRD pattern of calcined Fe(acac)3/PVP fibers obtained from 4 wt% and 6 wt% at 15 kV and 18 kV respectively. The diffraction peaks observed at 24.15°, 33.3°, 35.8°, 41.0°, 49.6°, 54.8°, 62.6° and 64.2° correspond to the (012), (104), (110), (113), (024), (116), (214) and (300) planes of the rhombohedral α-Fe2O3 phase. All the peaks in XRD pattern were well indexed to hematite (JCPDS: 33–0664, a = 5.035 Å, c = 13.749 Å), indicating the formation of the pure and spinel phase of α-Fe2O3.20,28 The strong and sharp diffraction peaks showed the high degree of crystallization of α-Fe2O3 nanostructures. The average crystallite sizes of these nanostructures were calculated from the peak widths using the Scherrer formula.29 The crystallite size was found to be 35 nm. XRD patterns of samples for 4 wt% and 6 wt% Fe(acac)3 precursor concentrations at 15 kV and 18 kV respectively were similar, confirming that the varying the precursor concentration had no influence on the crystalline nature of the α-Fe2O3.
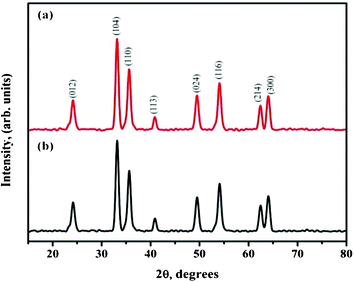 |
| | Fig. 2 XRD pattern of calcined (a) 4 wt% and (b) 6 wt% Fe(acac)3/PVP nanofibers at 15 kV and 18 kV power supply. | |
Thermogravimetric analysis (TGA) on Fe(acac)3 precursor and PVP electrospun composite nanofibers were carried out to determine the change in weight with temperature and to investigate the decomposition temperature of the PVP polymer and Fe(acac)3 precursor in the nanofiber. Fig. 3a shows the TGA plot of the 6 wt% Fe(acac)3/PVP composite nanofiber after calcining up to 800 °C. Overall fiber weight loss of about 90% was observed after calcining to 800 °C. The weight loss occurring between 70 °C to 120 °C was due to residual ethanol solvent evaporation and water vaporization. Furthermore, decrease in weight loss occurred from above 150 °C which was due to the fractional decomposition of the acetylacetonate group in the precursor and the PVP polymer decomposition. The transition temperature of Fe(III) acetylacetonate to the formation of α-Fe2O3 phase was noted to occur in the temperature ranges from 230 °C to 365 °C.30 The complete decomposition of the PVP and Fe(acac)3 to α-Fe2O3 phase were observed after 450 °C. The calcination of composite fibers at 500 °C for 5 h formed pure α-Fe2O3 phase.
 |
| | Fig. 3 TGA analysis of 6 wt% Fe(acac)3/PVP composite electrospun fibers at 18 kV potential, and (b) FTIR spectra of 6 wt% Fe(acac)3/PVP composite electrospun fibers and after calcining at 500 °C in air. | |
3.2 FTIR spectral analysis
Fig. 3b shows the IR spectra of 6 wt% Fe(acac)3/PVP composite fibers and calcined nanofibers at 500 °C for 5 h at 5 °C min−1. The bands at 3441 cm−1 and 3213 cm−1 were assigned to the hydroxyl stretching mode of residual ethanol and water, whose intensity decreased after calcination. Similarly, the intensity of the bending mode of water at 1611 cm−1 also disappeared after calcining at 500 °C. The two bands at 2954 cm−1 and 2880 cm−1 were the symmetric and asymmetric stretching vibrations of –CH and –CH2 bands in the PVP. In the region 1250–1750 cm−1, the band at 1657 cm−1 was due to symmetric stretching of non hydrogen bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N stretching, and that at 1437 cm−1 and 1373 cm−1, –CH deformation of cyclic CH2 groups occurred. The peak at 1279 cm−1 was assigned to C–N stretching and the peaks appearing between 1018 cm−1 and 931 cm−1 were attributed to the acetonate precursor.19 The bands below 700 cm−1 (663 cm−1) were due to Fe–O stretching associated with acetylacetonate group. After calcining at 500 °C, there were only two adsorptions in the range of 400–700 cm−1, which indicates that 500 °C was good enough for complete organic decomposition. Around 530 cm−1 and 448 cm−1 two peaks appeared which were ascribed to the metal-oxygen vibration (Fe–O) of the α-Fe2O3 nanostructures.1
O and C–N stretching, and that at 1437 cm−1 and 1373 cm−1, –CH deformation of cyclic CH2 groups occurred. The peak at 1279 cm−1 was assigned to C–N stretching and the peaks appearing between 1018 cm−1 and 931 cm−1 were attributed to the acetonate precursor.19 The bands below 700 cm−1 (663 cm−1) were due to Fe–O stretching associated with acetylacetonate group. After calcining at 500 °C, there were only two adsorptions in the range of 400–700 cm−1, which indicates that 500 °C was good enough for complete organic decomposition. Around 530 cm−1 and 448 cm−1 two peaks appeared which were ascribed to the metal-oxygen vibration (Fe–O) of the α-Fe2O3 nanostructures.1
3.3 FESEM and TEM analysis
In order to investigate the formation mechanism and nature of α-Fe2O3 nanostructures, microscopy imaging techniques (FESEM and TEM) were employed. A microscopy image (Fig. 4a) indicated that the discontinuous form of α-Fe2O3 fibers were produced for the composite fibers obtained by electrospinning of 4 wt% precursor and annealed at 500 °C. Closer examination with FESEM revealed the formation of nanobraid like structures, suggested that they were composed of nanorods (ellipsoidal shape of nanoparticles) which were uniformly plaited along the direction of fibers. It is believed that the PVP and Fe(acac)3 solutions undergo binodal phase separation at 4 wt% precursor ratio. The phase separation process involving polymer/precursor forms “islands” that account for the generation of Fe2O3 nanorods. The stretching of the jet was increased during electrospinning, Fe2O3 nanorods in the solution jet were coerced to plait together via spinning and whirling effects at an applied potential of 15 kV. Upon sintering, the PVP phase present all over the fibrous structure decomposed and yielded nanobraid like structures. Fig. 4 confirmed that the annealed Fe2O3 fiber consisted of the nanorods in its structure whose diameter was about 20–40 nm. The adhesion between nanorods was found to be extremely stable and the Fe2O3 nanorods remain attached even after rigorous sonication. Eid et al. and Zhan et al. synthesized hollow and tubular α-Fe2O3 fibers using electrospinning.23,31 However, they obtained fiber diameters in the range of 450 nm to 5 μm, whereas our approach not only yielded nanofibers with diameters below 100 nm but also distinct morphologies with more surface active sites.
 |
| | Fig. 4 FESEM images of a) sintered Fe2O3 discontinuous fibers obtained using 4 wt% Fe(acac)3/PVP nanofibers at 15 kV, and b) magnified image revealing the nano-braid like structure of α-Fe2O3. | |
FESEM image of calcined Fe2O3 sample obtained by electrospinning of 6 wt% Fe(acac)3/PVP at 18 kV is shown in Fig. 5. The FESEM image disclosed the formation of continuous, uniform and a very fine porous structure within each α-Fe2O3 fiber. Appearance of nanopores within the fibrous structure after sintering indicates that the phase separation played a key role. A 6 wt% Fe(acac)3/PVP composition resulted in better dispersion and an improved interpenetrating network of Fe(acac)3 in PVP.
 |
| | Fig. 5 FESEM images of a) sintered Fe2O3 continuous fibers obtained using 6 wt% Fe(acac)3/PVP nanofibers at 18 kV, and b) sintered fibers revealing the mesoporous structure upon magnification. | |
In the electrospinning process, kinetic effects determine the morphology of the fibers. Kinetic factors are mainly the diffusion rates of the polymer and precursor solutions. Hence, the molecular weight of PVP, PVP/precursor concentration ratio, and the solvent decide the type of phase separation: binodal and spinodal for the formation of specific nanostructures (Fig. 6). In the binodal type, nucleation from the precursor propagates to form ‘islands’ which then separate out from the polymer rich phase, whereas in spinodal type phase separation, concentration fluctuations occur in a wave-like fashion that result in the uniform dispersion of polymer rich and polymer poor areas throughout the solution. Spinodal phase separation thus yields fine pores after the thermal process.
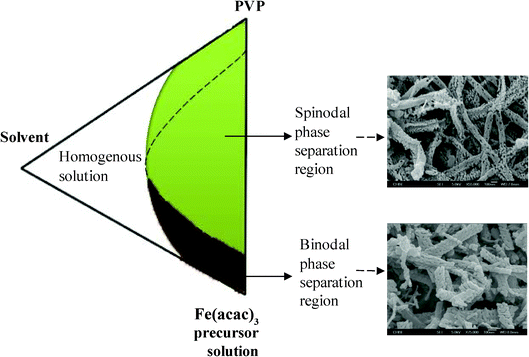 |
| | Fig. 6 Schematic of main components involved in phase separation and the types of phase separation in the formation of α-Fe2O3 nanostructures. | |
Spinodal type phase separation was evidenced by the presence of 15–30 nm size pores in each fiber, formed by electrospinning of 6 wt% Fe(acac)3/PVP solution. At higher concentrations of the precursor, the diffusion process is expected to allow for more phase coarsening, in turn a more porous structure. It is believed that the applied potential of 18 kV during electrospinning of 6 wt% Fe(acac)3/PVP solution caused a stronger whipping motion along the path of the jet, which favored spinodal type phase separation and led to the formation of nanoporous structures in α-Fe2O3 fibers at 500 °C. The synthesized α-Fe2O3 nanobraids and nanoporous structures were quite unique on their own and the average surface area was measured to be 22 ± 3 m2 g−1 and 24 ± 3 m2 g−1. These results convey that through electrospinning, 1D novel structures with high porosity can be produced, which is beneficial in many potential applications, and especially ideal for batteries and photocatalytic applications.
TEM analysis also confirmed the conformational changes in the morphology of Fe(acac)3/PVP fibers that occurred after calcination. Fig. 7 shows the TEM images and corresponding lattice-resolved TEM images of α-Fe2O3 nanostructures obtained at 15 kV and 18 kV for 4 wt% and 6 wt% Fe(acac)3/PVP fibers. The average diameter of α-Fe2O3 nanostructures was 94.1 nm for 4 wt% and 85.8 nm for 6 wt% Fe(acac)3/PVP. The lattice spacing of 4 wt%, and 6 wt% Fe(acac)3/PVP α-Fe2O3 nanostructures were found to be 0.274 nm, which was in good agreement with the literature corresponding to the rhombohedral α-Fe2O3 (104) plane.20
 |
| | Fig. 7 TEM images of (a) 4 wt% and (b) 6 wt% Fe(acac)3/PVP fibers after calcination at 500 °C at 15 kV and 18 kV power supply. (c) & (d) correspond to the lattice-resolved image of 4 wt% and 6 wt% Fe(acac)3/PVP fibers after calcination. | |
3.4 Photocatalytic performance analysis
Fig. 8(a&b) shows the absorbance spectra of Congo red (CR) dye obtained upon photoirradiation in the presence of nanobraid and nanoporous α-Fe2O3 respectively. The UV-Vis absorbance spectrum of synthesized α-Fe2O3 is available in the ESI† (Fig. S2). Fig. 8c shows the percentage of CR dye degradation with respect to irradiation time. The rate of decolourization of CR dye due to the effect of α-Fe2O3 was expressed as photocatalytic performance of α-Fe2O3, which was determined in terms of change in the intensity of the absorption maximum of CR dye over irradiation time. The higher photocatalytic performances of 91.2% and 90.2% were achieved within 140 min of photoirradiation for nanobraids and nanoporous structures of α-Fe2O3, respectively. A negligible change in the absorbance was observed for CR dye solution when irradiated in the absence of α-Fe2O3 catalyst (Fig. S3, ESI†). Similarly, the absorbance change was insignificant when the CR dye solution was kept in the dark with α-Fe2O3, suggesting that the dye degradation was mainly due to the effect of photoirradiated α-Fe2O3 nanostructures.
The photodegradation pathway generally follows either by semiconductor initiated photocatalysis or photosensitized oxidation of the organic molecules.32–35 We observed the enhanced photodegradation of CR dye in the presence of the α-Fe2O3 photocatalyst, which could be the result of photosensitization of the dye molecules followed by semiconductor-initiated photocatalysis. The mechanism is proposed as follows: photosensitization of the CR dye takes place firstly where the charge transfer occurs from the LUMO level of the dye to the conduction band of the photocatalyst (eqn. 1 & 2)33,35 followed by the semiconductor-initiated photocatalytic reaction where α-Fe2O3 generates an electron and hole pair upon photoirradiation. The electron ejects from the valence band of α-Fe2O3 to the conduction band, leaving behind a hole in the valence band (eqn. 3). The photogenerated electron of α-Fe2O3 generates intermediate superoxide radicals (O2•−) by reacting with the chemisorbed oxygen on the catalyst surface and oxygen in the aqueous solution (eqn. 5). The superoxide radicals (O2•−) along with dissociated water (H+) form hydroperoxyl radicals (˙HO2) (eqn. 6) and hydrogen peroxide (H2O2) is formed from the hydroperoxyl radicals (eqn. 7). The hydroxyl radicals (OH•−) are formed on the surface of the catalyst by the reaction with photogenerated holes (h+) with adsorbed water (OH−) (eqn. 4 & 8). These reactive radicals and intermediate species react with CR dye, and degrade them into non-toxic organic compounds (eqn. 9).
| | | CR* + α-Fe2O3 → CR*+ + α-Fe2O3(e−CB) | (2) |
| | | α-Fe2O3 + hν → α-Fe2O3(e−CB + h+VB) | (3) |
| | | ˙HO2 + ˙HO2 ↔ H2O2 + O2 | (7) |
| | | CR*/CR*+ + (O2*− , ˙HO2, H2O2, OH*−) → Degradation products | (9) |
The catalytic performance of various nanostructures of α-Fe2O3 such as nanorods, nanoparticles reported in the literature was found to be inferior compared to the nanobraids and nanoporous structures obtained in this study. For example, Pradhan et al. showed the photocatalytic degradation performance of α-Fe2O3 nanorods only up to 27% against Congo red dye and increased the maximum up to 63% by doping with Pt and upon exposure to sunlight over the period of 240 min.1 The porous surface morphology of as-synthesized nanobraid and nanoporous α-Fe2O3 lead dye diffusion onto the surface. The active surface sites enabled better interaction of the CR dye molecules with α-Fe2O3, and degraded the dye quickly.
The photodegradation ability of a catalytic material is influenced by its physicochemical properties like crystallite size, crystallinity, surface morphology, and surface area. Our study on the α-Fe2O3 revealed that the crystallite size and surface morphology played important role for the higher photocatalytic activity. The presence of smaller crystallite sized nanobraids and nanoporous α-Fe2O3 provided more active sites along these fiber surface than regular nanofibers. The nanobraids and nanoporous α-Fe2O3 showed superior photocatalytic performances of up to 91.2% and 90.2%, respectively, under irradiation for 140 min. The superior photocatalytic performance of nanobraids and nanoporous α-Fe2O3 can be extended to remove organic pollutants from waste water.
4 Conclusions
We prepared nanostructured α-Fe2O3 with 1D nanobraids and nanoporous morphologies via electrospinning, and applied them to the photocatalytic decomposition of dye under light irradiation. The synthesized materials were characterized using SEM/TEM/XRD/FTIR methods. The photocatalytic performance of 1D nanobraids and nanoporous morphologies was found to be superior to that of other nanostructures of α-Fe2O3 reported in the literatures. The photocatalysis study revealed that the maximum removal capacity of the α-Fe2O3 was up to 91.2% within 140 min. The superior performance was due to the high crystallinity, smaller crystallite size and porous surface morphology. The study conveyed that nanoporous structures offer the advantage of having more sites and thus are useful for the improvement of catalytic behavior against chemical contaminants and also in water splitting. The low-cost and easy fabrication methodologies of α-Fe2O3 with such unique morphologies should be of great interest in terms of potential applications such as adsorbents, catalysts and catalyst supports. We are currently investigating the possibilities and performances of these novel structures in energy applications.
Acknowledgements
This work was partially supported by Singapore NRF-CRP grant number NRF-CRP-4-2008-03 on ‘Nanonets: New Materials, Devices for Integrated Energy Harnessing and Storage’ and also NUS and NTU for providing facilities to carry out the research. Dr V. Thavasi acknowledges NUSNNI for support.
References
- G. K. Pradhan and K. M. Parida, ACS Appl. Mater. Interfaces, 2011, 3, 317–323 CAS.
- X.-L. Fang, C. Chen, M.-S. Jin, Q. Kuang, Z.-X. Xie, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, J. Mater. Chem., 2009, 19, 6154–6160 RSC.
- J. Ma, J. Lian, X. Duan, X. Liu and W. Zheng, J. Phys. Chem. C, 2010, 114, 10671–10676 CAS.
- L. Liu, H.-Z. Kou, W. Mo, H. Liu and Y. Wang, J. Phys. Chem. B, 2006, 110, 15218–15223 CrossRef CAS.
- C. Wu, P. Yin, X. Zhu, C. OuYang and Y. Xie, J. Phys. Chem. B, 2006, 110, 17806–17812 CrossRef CAS.
- S. Boumaza, A. Boudjemaa, S. Omeiri, R. Bouarab, A. Bouguelia and M. Trari, Sol. Energy, 2010, 84, 715–721 CrossRef CAS.
- Z. Fan, X. Wen, S. Yang and J. G. Lu, Appl. Phys. Lett., 2005, 87, 013113–013113 CrossRef.
- W. Zhang, T. Yang, X. Li, D. Wang and K. Jiao, Biosens. Bioelectron., 2009, 25, 428–434 CrossRef CAS.
-
Y. Li, Y. Lu, H. Hong, Y. Chen, X. Ma, L. Guo, Z. Wang, J. Chen, M. Zhu, J. Ni, H. Gu, J. Lu and J. Y. Ying, Chem. Commun. (Cambridge, U. K.), 2011, 47, 6320–6322. Search PubMed.
- L. Sun, J. Wang and Z. Wang, Nanoscale, 2010, 2, 269–276 RSC.
- J. Chen, L. Xu, W. Li and X. Gou, Adv. Mater., 2005, 17, 582–586 CrossRef CAS.
- A. Boudjemaa, S. Boumaza, M. Trari, R. Bouarab and A. Bouguelia, Int. J. Hydrogen Energy, 2009, 34, 4268–4274 CrossRef CAS.
- E. Thimsen, F. Le Formal, M. Grätzel and S. C. Warren, Nano Lett., 2010, 11, 35–43 CrossRef.
- L.-P. Zhu, N.-C. Bing, D.-D. Yang, Y. Yang, G.-H. Liao and L.-J. Wang, CrystEngComm, 2011, 13, 4486–4490 RSC.
- Z. Zhang, M. F. Hossain, T. Miyazaki and T. Takahashi, Environ. Sci. Technol., 2010, 44, 4741–4746 CrossRef CAS.
- E. Sánchez Mora, E. Gómez Barojas, E. Rojas Rojas and R. Silva González, Sol. Energy Mater. Sol. Cells, 2007, 91, 1412–1415 CrossRef.
- A. A. Tahir, K. G. U. Wijayantha, S. Saremi-Yarahmadi, M. Mazhar and V. McKee, Chem. Mater., 2009, 21, 3763–3772 CrossRef CAS.
- J. R. Morber, Y. Ding, M. S. Haluska, Y. Li, J. P. Liu, Z. L. Wang and R. L. Snyder, J. Phys. Chem. B, 2006, 110, 21672–21679 CrossRef CAS.
- P. Zhifa, Nanotechnology, 2006, 17, 799 CrossRef.
- W. Wu, X. Xiao, S. Zhang, J. Zhou, L. Fan, F. Ren and C. Jiang, J. Phys. Chem. C, 2010, 114, 16092–16103 CAS.
- E. P. Haripriya, Nanotechnology, 2006, 17, 4285 CrossRef.
- A. Mao, G. Y. Han and J. H. Park, J. Mater. Chem., 2010, 20, 2247–2250 RSC.
- C. Eid, A. Brioude, V. Salles, J.-C. Plenet, R. Asmar, Y. Monteil, R. Khoury and P. Miele, Nanotechnology, 2010, 21, 125701 CrossRef.
- W. Zheng, Z. Li, H. Zhang, W. Wang, Y. Wang and C. Wang, Mater. Res. Bull., 2009, 44, 1432–1436 CrossRef CAS.
- H. Fan, Q. Xu, Y. Guo, Q. Peng and Z. Hou, Mater. Sci. Eng., A, 2006, 422, 272–277 CrossRef.
- K. J. McKenzie, F. Marken, M. Hyde and R. G. Compton, New J. Chem., 2002, 26, 625–629 RSC.
- S. Chatterjee, S. Chatterjee, B. P. Chatterjee and A. K. Guha, Colloids Surf., A, 2007, 299, 146–152 CrossRef CAS.
- J. Lian, X. Duan, J. Ma, P. Peng, T. Kim and W. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS.
- P. S. Kumar, S. A. S. Nizar, J. Sundaramurthy, P. Ragupathy, V. Thavasi, S. G. Mhaisalkar and S. Ramakrishna, J. Mater. Chem., 2011, 21, 9784–9790 RSC.
- B. Pal and M. Sharon, Thin Solid Films, 2000, 379, 83–88 CrossRef CAS.
- S. Zhan, D. Chen, X. Jiao and S. Liu, J. Colloid Interface Sci., 2007, 308, 265–270 CrossRef CAS.
-
C. Chen, Z. Wang, W. Ma and J. Zhao, Photocatalytic degradation of organic contaminants on mineral surface. In: Biophysico-Chemical Processes of Anthropogenic Organic Compounds in Environmental Systems, ed. Xing, B., Senesi, N., Huang, P. M. (Hoboken: John Wiley & Sons, Inc.), 2011, 91–111 Search PubMed.
- I. K. Konstantinou and T. A. Albanis, Appl. Catal., B, 2004, 49, 1–14 CrossRef CAS.
- C. Baumanis, J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2011, 115, 25442–25450 CAS.
- J. Bandara, J. A. Mielczarski and J. Kiwi, Langmuir, 1999, 15, 7680–7687 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 