Ionic liquid-stabilized graphene and its use in immobilizing a metal nanocatalyst
Wenjing
Xiao†
,
Zhenyu
Sun†
,
Sha
Chen
,
Hongye
Zhang
,
Yanfei
Zhao
,
Changliang
Huang
and
Zhimin
Liu
*
Beijing National Laboratory for Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: liuzm@iccas.ac.cn
First published on 7th August 2012
Abstract
A new ionic liquid (IL), 1-butyl-3-methylimidazolium cholate, was first synthesized through an ion exchange reaction of 1-butyl-3-methylimidazolium chloride with sodium cholate. Stable aqueous dispersions of graphene were achieved by exfoliating graphite in the presence of the IL under ultrasonication. Both transmission electron microscopy and Raman measurements showed that the IL-stabilized graphene (IL–G) sheets existed with only a few (<5) layers. Furthermore, the IL–G was used to immobilize noble metal nanoparticles (Pt, Pd, Ru, Rh, etc), and a series of graphene–metal (G–M) composites with metal size ≤2 nm and very narrow size distributions were obtained. The resulting G–M exhibited superior catalytic performance with respect to hydrogenation of arenes. In particular, the as-prepared G–Ru with Ru content of 5% was very active for the hydrogenation of benzene to hexane with a turnover frequency as high as 6000 h−1. The catalysts could be reused without detectable loss of activity, a result of their stable structure.
1. Introduction
Graphene and graphene-based 2D nanomaterials have attracted much attention due to their unique mechanical, electronic, and thermal properties,1 showing potential applications in nanoelectronics,2,3 capacitors,4 fuel cells,5 catalysis,6 and sensors.7,8 Methods to prepare graphene mainly include mechanical exfoliation,9,10 thermal expansion,11 epitaxial growth,12 chemical vapor deposition13 as well as reduction of exfoliated graphite oxide (GO).14–17 Recently direct exfoliation of pristine graphite to produce graphene in liquid media has been reported,18–22 and the surfactant-assisted exfoliation is of particular interest as the solvent used is water. For example, high-yield aqueous dispersions of graphene were obtained in the presence of sodium cholate (SC) under long-lasting bath sonication,23 and the thickness of SC-stabilized graphene can be controlled via ultrasonication and density gradient ultracentrifugation.24In recent years, ionic liquids (ILs), a kind of organic salt with melting points below 100 °C, have been paid much attention owing to their unique properties, such as high charge density, high polarity, high dielectric constant, excellent solvent power for organic and inorganic compounds and supramolecular network formation, very low vapor pressure, etc. More importantly, the properties of ILs can be designed through judicious combination of anions and cations, and thereby ILs with unique functions can be obtained. Imidazolium cation-based ILs have been widely investigated, showing potential applications in many areas. In a recent work, dispersion of graphene oxide in IL, 1-butyl-3-methylimidazolium hexafluorophosphate, was successfully achieved with the aid of a polymerized ionic liquid (PIL).25 The surfactant-like property of some functional ILs may make the IL suitable to be applied in the production of graphene via exfoliating graphite. However, exfoliation of pristine graphite into graphene in ILs has not been found in a literature survey. On the other hand, very few studies have been carried out concerning the combination of ILs with graphene in catalysis.
Graphene as a 2D carbon material is expected to be an ideal support for metal and/or metal oxide nanocatalysts because of its excellent physical and chemical properties, including high mechanical strength, high thermal conductivity, outstanding electron mobility, and chemical inertness. Indeed graphene-based catalysts have shown promising applications in catalysis. For example, graphene-supported Pd particles displayed extraordinary high activities for Suzuki–Miyaura coupling reactions.26 Pt particles deposited on graphene exhibited good catalytic activities for both methanol oxidation and hydrogen conversion reactions.17 Graphene-supported Ru and Rh nanocomposites showed high efficiency in hydrogenations of cyclohexene and benzene.27 Despite recent advances in this regard, synthesis of graphene supported catalysts remains an ongoing challenge in terms of control of particle size, size distribution, and their thermal and/or electrochemical stability under different conditions.
In this work, a new functional IL, 1-butyl-3-methylimidazolium cholate, was designed and used to exfoliate pristine graphite to make graphene. The resulting IL-stabilized graphene sheets were further applied in the immobilization of noble metal particles including Ru, Pt and Rh. The as-obtained graphene and graphene-supported metal composites were characterized by IR and Raman spectroscopy, transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA). Moreover, the catalytic performances of the metal/graphene nanocomposites for the arene hydrogenation were investigated as well.
2. Experimental section
2.1 Materials
Graphite powder was purchased from Sigma-Aldrich, and sodium cholate hydrate was obtained from ABCR. All other chemicals were purchased from Sinopharm Chemical Reagent Beijing Co. Ltd., and used as supplied.2.2 Synthesis of ionic liquid
The IL, 1-butyl-3-methylimidazolium cholate, was synthesized via an ion exchange reaction of 1-butyl-3-methylimidazolium chloride with sodium cholate hydrate. Typically, 1 mol of 1-butyl-3-methylimidazolium chloride and an equivalent amount of sodium cholate hydrate were initially dissolved in 30 mL of methanol, and then stirred overnight; subsequently, the mixture was filtered to remove the resultant salt, and the solvent was removed via distillation under reduced pressure. The obtained white solid product was rinsed by absolute ethanol twice, and dried under vacuum for further characterization and application.2.3 Preparation of graphene-noble metal (G–M) composites
The IL-stabilized graphene was first prepared via exfoliating graphite in the presence of IL under sonication irradiation. Typically, 100 mg of graphite was dispersed in 20 mL of an aqueous solution of the as-synthesized IL with a concentration of 5 mg mL−1. The mixture was subjected to bath sonication (KQ100DB, Kunshan Ultrasonic Instruments Co., Ltd., 40 kHz, 100 W) for 24 h. Subsequently, the suspension was centrifuged at 5000 rpm for 90 min, and the supernatant was collected to give aqueous graphene dispersion. The IL-stabilized graphene, denoted as IL–G, was obtained via removing solvent from the supernatant, which was used to immobilize metal particles and for characterization.The IL–G was used to prepare G–M nanocomposites. In a typical experiment to prepare the G–Ru composite, 5 mL of RuCl3 ethanol solution at a designated concentration was mixed with 20 mL ethanol suspension of IL–G (1 mg mL−1), followed by dropwise addition of 5 mL of an NaBH4 ethanol solution (its concentration being 5 times higher than that of the metal precursor) under tip ultrasonication (Vibra Cell CVX, 500 W, 20 kHz, 25% of amplitude). The mixture was then centrifuged, and the collected precipitate was washed repeatedly with absolute ethanol and distilled water, and subsequently vacuum-dried at 60 °C. Using similar procedures, other G–M (including Rh, Pt, etc) composites with metal loading at 5 wt% were prepared as well by varying metal precursors.
2.4 Characterization
MALDI-TOF mass spectra were taken on a Bruker BIFLEX III ultrahigh resolution Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer with α-cyano-4-hydroxycinnamic acid as matrix. 1H NMR spectra were recorded on a Bruker DPX 400 spectrometer in D2O. Raman spectroscopy was performed using a Renishaw inVia plus spectrometer with 514 nm laser excitation. Fourier-transform infrared (FT-IR) spectra were recorded on a Bruker Tensor 27 spectrophotometer using KBr pellets. Thermogravimetric analysis (TGA) was carried out on a PERKIN-ELMER 7 Series Thermal Analysis System with a heating rate of 10 °C min−1 in air with a flow rate of 20 mL min−1. Transmission electron microscopy (TEM) observation was performed on a transmission electron microscope (JEOL JEM-2010) equipped with an energy-dispersive X-ray spectrometer (EDS), operated at 200 kV. TEM samples were prepared by depositing droplets of sample on copper grids coated with lacey carbon film. The particle size distribution was calculated by counting over 100 particles. X-Ray photoelectron spectroscopy data were obtained with an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. The base pressure was about 3 × 10−9 mbar. The binding energies were referenced to the C1s line at 284.8 eV from adventitious carbon. X-Ray diffraction (XRD) was carried out on a D/MAX-RC diffractometer, operated at 40 kV and 200 mA with Cu-Kα radiation.2.5 Catalytic activity test of G–M composites
The catalytic performances of the resultant G–M catalysts for arene hydrogenation were examined. The G–Ru composite was used to catalyze the hydrogenation of benzene under different conditions. In a typical experiment, 5 mg of the catalyst with a Ru loading of 5 wt% and 10 mmol of benzene were loaded into a high-pressure stainless steel reactor, and the reactor was then sealed and flushed with H2 three times to remove the air inside. Subsequently, the reactor was moved to an air bath set at the desired temperature, and H2 was charged into the reactor up to the pressure of interest. The hydrogenation reaction started with magnetic stirring. The H2 pressure was kept constant by replenishing H2 as the reaction proceeded. After the reaction, the mixture after removal of the catalyst was analyzed by gas chromatography (GC) (Agilent 4890D) with a capillary column and a flame ionization detector (FID). The isolated catalyst was reused in the next hydrogenation run to explore its stability. Alternatively, hydrogenation of toluene and nitrobenzene were investigated using G–Rh and G–Pt as catalysts, respectively.3. Results and discussion
3.1 Preparation of IL–G
The IL, 1-butyl-3-methylimidazolium cholate, was synthesized via the reaction of 1-butyl-3-methylimidazolium chloride with sodium cholate hydrate. The NMR and ESI-MS spectral characteristics of the as-synthesized IL are shown as follows: 1H NMR (400 MHz, D2O): δ 7.335–7.331 (d, 1H), δ 7.285–7.281 (d, 1H), δ 4.073–4.037 (t, 2H), δ 3.949 (s, 1H), δ 3.748 (s, 3H), δ 3.772 (s, 1H), δ 0.596 (s, 1H). ESI-MS: m/z 548.8, m/z 409.6. These data indicate the formation of the IL with the chemical structure as shown in Scheme 1.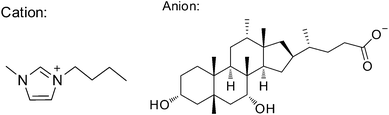 | ||
| Scheme 1 The chemical structure of the as-synthesized IL. | ||
The efficiency of the as-synthesized IL for graphite exfoliation was investigated under sonicating irradiation. It was found that stable graphene dispersions were achieved after being subjected to sonication in the IL aqueous solution for 24 h. The dispersion remained highly stable for several months. Fig. 1 shows the Raman spectra of graphite and IL–G. Compared to the spectrum of the used graphite, the spectrum of IL–G exhibited characteristics of graphene sheets with three peaks at 1350 cm−1, 1580 cm−1, and 2700 cm−1, which are ascribed to the G, D, and 2D peaks of graphene, respectively. The Raman analysis for IL–D confirms the formation of graphene sheets. Moreover, the shape of the 2D peak (∼2700 cm−1) was consistent with that of graphene sheets with fewer than 5 layers;23 especially, the symmetric 2D band further confirms the formation of single-layer graphene. The formation of graphene was also supported by the observation that a shift (∼29 cm−1) towards lower wavenumbers occurred for the 2D band of IL–G accompanied by an enhancement of its intensity when compared to that of graphite.
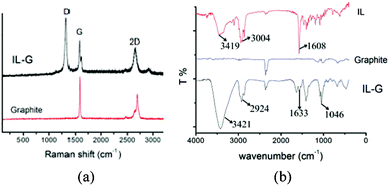 | ||
| Fig. 1 (a) Raman spectra of graphite and IL–G; (b) IR spectra of graphite, IL–G and IL. | ||
Fig. 1b shows the IR spectra of the used graphite, IL–G and the IL. In comparison with the spectra of the IL and the graphite, the spectrum of IL–G displayed the characteristic peaks assigned to the IL, suggesting the presence of IL in this sample. This also suggests that the IL played important roles in exfoliating graphite and stabilizing the resultant graphene sheets.
To determine the amount of IL in IL–G, TGA analysis was carried out for both IL–G and IL. As shown in Fig. 2, the IL started to decompose at 270 °C and completely decomposed at about 570 °C. In contrast, there are three weight loss peaks in the TGA curve of IL–G. The weight loss of about 3% below 250 °C was ascribed to the removal of adsorbed water. The sharp weight loss between 250 and 570 °C was assigned to the decomposition of the IL, from which the amount of IL in IL–G was estimated to be approximately 52%. This is indicative of strong interactions between the graphene and the IL, resulting in stabilization of exfoliated graphene against reaggregation. The further weight loss in the range of 570–850 °C was attributed to the oxidation of the graphene sheets.
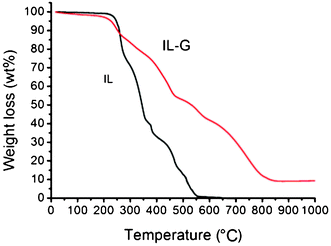 | ||
| Fig. 2 TGA curves of IL and IL–G. | ||
A typical TEM image of IL-stabilized graphene flakes is illustrated in Fig. 3a, which clearly showed a large number of thin graphene sheets. Monolayer and bilayer graphene were observed as well, as displayed in Fig. 3b, indicating the successful exfoliation of graphene in the IL. The mechanism for the formation of stable graphene dispersion in the IL aqueous solution under sonicating irradiation can be explained as follows. The mechanical energy due to intensive sonication is sufficient to overcome the van der Waals attraction between graphite layers, thus the graphite was exfoliated into single- and few-layered sheets under the sonication irradiation. The attraction between the graphene surface and the hydrophobic groups of the IL molecules led to the adsorption of a large amount of IL molecules onto the graphene sheets, preventing them from restacking. The stabilization of the graphene dispersion in the IL aqueous solution may result from the electrostatic repulsion between the hydrophilic head ions coating adjacent graphene. Depending on the exfoliating extent of graphite, graphene sheets with different layers can be obtained. It should be pointed out that the dried IL–G can be redispersed in aqueous solutions to form stable suspension without aggregation.
 | ||
| Fig. 3 TEM images of IL–G sheets (a) and 2-layer graphene (b). | ||
3.2 Immobilization of metal particles on the IL–G sheets
The as-prepared IL enabled effective stabilization of metal particles. In this work, a series of G–M (M referring to Pt, Ru, Rh, etc) composites were prepared using IL–G as the support. From the TEM image (Fig. 4), it was found that all individual graphene sheets were uniformly decorated with ultrafine nanoparticles, and no free-standing metal aggregates or agglomerates from the graphene sheets were observed. The particles are characteristically of a small size (≤2.0 nm) and very narrow size distributions. Energy-dispersive spectroscopy (EDS) analysis during TEM observations from different regions suggested the presence of noble metal elements in the particles. The elemental composition of the NPs would be in the metallic state since the particles were produced via reduction of the metal precursor by excessive NaBH4. The deposited particles remained strongly immobilized on the graphene sheets, regardless of intense sonication treatment for several hours.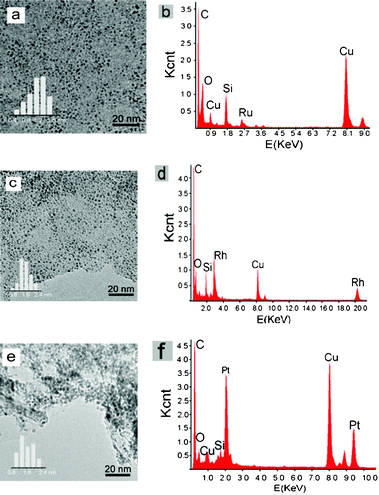 | ||
| Fig. 4 TEM images, particle size distribution and EDS spectra of G–M: (a) and (b) G–Ru (5 wt.%, 1.1 nm), (c) and (d) G–Rh (5 wt.%, 1.6 nm), (e) and (f) G–Pt (5 wt.%, 1.7 nm). | ||
Fig. 5 illustrates the XRD patterns of IL–G and G–Ru in the wide range of 2θ = 10–80°. The diffraction peak at 26.7° in both cases was assigned to the (111) reflection of the graphitic structure from graphene. Note that no diffraction peaks attributable to metal nanoparticles was observed for G–Ru with a Ru loading of 5 wt.% in the XRD patterns. This may be an indication that the Ru particles were very small.
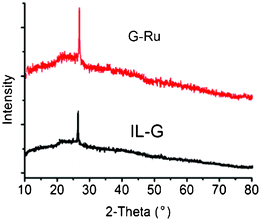 | ||
| Fig. 5 XRD patterns of G–Ru and IL–G. | ||
XPS measurements were employed to analyze the surface composition and oxidation states of species in the G–M composites. Fig. 6a shows the wide survey XPS pattern of G–Ru, which indicates the coexistence of C, Ru, N and O in the catalyst, and no Cl was detectable, suggesting its high purity without the presence of the metal precursor. The element N was originated from the IL. The deconvoluted Ru 3d XPS profile shows a doublet with peak binding energies (BEs) at 281.5 (Ru 3d5/2) and 285.5 eV (Ru 3d3/2), which were ascribed to a Ru–O species. Two peaks appearing at BEs of 485.5eV (Ru 3p1/2) and 463.4eV (Ru 3p3/2) were also attributed to a Ru–O species. These results suggest that the Ru species were present in the form of an oxide state in the composite. As expected, Ru species should be present in the form of a Ru(0) state because they were obtained via the reduction of RuCl3 by NaBH4. The presence of Ru oxide in G–Ru may result from the subsequent oxidation of Ru(0) particles as they were exposed to air. Moreover, the absence of Ru(0) in the G–Ru composite indicates that the initially obtained Ru(0) particles were completely converted into oxide, probably due to their tiny size. In addition, the other peaks at BEs of 285.4, 286.5 and 288.6 eV were ascribed to C–C, C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, respectively, which were originated from the IL.
O, respectively, which were originated from the IL.
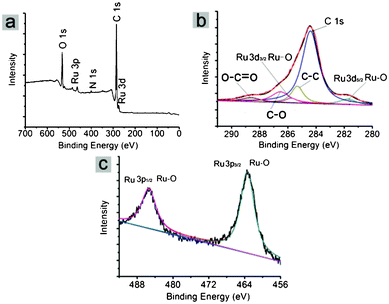 | ||
| Fig. 6 XPS spectra of G–Ru: (a) survey spectrum, (b) Ru 3d and C 1s, (c) Ru 3p. | ||
G–Pt and G–Rh were also examined by XPS analysis, and the metal oxides were detectable in these G–M composites as well, suggesting the oxidation of metal particles after the composites were exposed to air.
3.2 Catalytic performances of the G–M composites
The as-prepared ultrafine noble metal NPs supported on graphene sheets are expected to have promising applications in catalysis. In such a scenario, they were used to catalyze the hydrogenation of some arenes, and the results are listed in Table 1. It should be pointed out that these catalysts can be well dispersed in the arenes forming stable suspensions. They can also be easily separated from the final products.| Entry | Catalyst | Substrate | T/°C | PH2/MPa | t/min | Conv./% | TOFa |
|---|---|---|---|---|---|---|---|
| a Turnover frequency (TOF) of substrate was defined as moles of substrate converted per moles of Ru per hour. b The catalyst was reused for the sixth run for benzene hydrogenation. | |||||||
| 1 | G–Ru | Benzene | 110 | 8 | 40 | >99.9 | 6060 |
| 2 | G–Rub | Benzene | 110 | 8 | 40 | >99.9 | 6060 |
| 3 | G–Ru | Benzene | 110 | 4 | 65 | >99.9 | 3730 |
| 4 | G–Ru | Benzene | 110 | 2 | 60 | 10 | 420 |
| 5 | G–Ru | Benzene | 60 | 4 | 170 | 68.5 | 990 |
| 6 | G–Ru | Benzene | 25 | 4 | 300 | 32.9 | 270 |
| 7 | G–Rh | Toluene | 110 | 8 | 68 | >99.9 | 3630 |
| 8 | G–Pt | Nitrobenzene | 60 | 2 | 13 | >99.9 | 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
| 9 | TMG-MMT/Ru28 | Benzene | 110 | 8 | 150 | >99.9 | 4000 |
The G–Ru catalyst was very active for benzene hydrogenation to hexane, and the turnover frequency (TOF) reached over 6000 h−1 at 110 °C and H2 pressure at 8 MPa, which was much higher than that of most of the reported Ru catalysts.28 We note that the catalyst exhibited high activity even at room temperature (entry 6), and its activity increased with temperature and H2 pressure. It is known that Ru(0) is the active component for catalyzing hydrogenations. As analyzed above, the Ru particles were present in the form of oxide in the as-prepared G–Ru catalyst. Therefore, it can be deduced that the Ru oxide with sizes less than 2.0 nm can be easily reduced to the Ru(0) state during the hydrogenation process, which served as the actual active species for catalyzing benzene hydrogenation.
Moreover, the G–Ru catalyst could be reused 6 times without detectable loss of activity, suggesting the high stability of the catalyst. To explore the reason for its high stability, the recovered catalyst after 6 times was examined by TEM. It was shown that the metal particles remained highly distributed on the graphene surfaces without aggregation, and the particle size was kept unchanged, compared to the fresh G–Ru catalyst. The final product after reaction was examined by ICP, and it was found that the metal species were undetectable, indicating that there was no leaching of the metal species during the reaction process. As discussed above, the metal particles were immobilized onto the graphene sheets via the IL. The high activity and good stability of the catalyst indicates that the moderate interaction existed between the particles and the IL, which prevented particles from leaching into the reaction mixture and/or aggregating into larger ones.
Similarly, the as-prepared G–Rh and G–Pt catalysts were applied to the hydrogenation of toluene and nitrobenzene, respectively, which showed remarkably high activities (entries 7 and 8) and good stabilities without activity loss even after 5 times recovered use. The high performance of the as-synthesized nanocatalysts may result from their unique structures, in which small metal particles with high monodispersibility were strongly immobilized on the graphene sheets with the assistance of the IL.
4. Summary
In summary, the as-synthesized IL, 1-butyl-3-methylimidazolium cholate, resulted in the exfoliation of graphite into graphene sheets with few layers (<5), and further mediated the immobilization of metal particles onto the graphene sheets, producing G–M composites. The metal particles were uniformly distributed on the graphene sheets with sizes less than 2.0 nm and very narrow size distribution. The as-prepared G–M composites exhibited superior activity and high stability for the solvent-free hydrogenation of arenes. These catalysts may find promising applications in catalysis.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos.: 20903105, 21073202, 21021003) and the Chinese Academy of Sciences (KJXC2-YW-H30).References
- (a) X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC; (b) A. A. Balandin, Nat. Mater., 2011, 10, 569 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- X. Li, X. Wang, L. Zhang, S. Lee and H. Dai, Science, 2008, 319, 1229 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, A. Velamakanni, R. D. Piner and R. S. Ruoff, Carbon, 2010, 48, 2118 CrossRef CAS.
- G. Lee, J. H. Shim, H. Kang, K. M. Nam, H. Song and J. T. Park, Chem. Commun., 2009, 5036 RSC.
- L. Dong, R. R. S. Gari, Z. Li, M. M. Craig and S. Hou, Carbon, 2010, 48, 781 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, A. K. Mishra, N. H. Kim and J. H. Lee, Biosens. Bioelectron., 2011, 26, 4637 CrossRef CAS.
- S. Guo, D. Wen, Y. Zhai, S. Dong and E. Wang, ACS Nano, 2010, 4, 3959 CrossRef CAS.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451 CrossRef CAS.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Naud, D. Mayou, T. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191 CrossRef CAS.
- M. J. McAllister, J.-L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud'homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396 CrossRef CAS.
- P. W. Sutter, J. Flege and E. A. Sutter, Nat. Mater., 2008, 7, 406 CrossRef CAS.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J.-H. Ahn, P. Kim, J.-Y. Choi and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS.
- S. Moussa, V. Abdelsayed and M. S. El-Shall, Chem. Phys. Lett., 2011, 510, 179 CrossRef CAS.
- S. Chandra, S. Bag, R. Bhar and P. Pramanik, J. Nanopart. Res., 2011, 13, 2769 CrossRef CAS.
- J. J. Guo, S. M. Zhu and Z. X. Chen, Ultrason. Sonochem., 2011, 18, 1082 CrossRef CAS.
- P. Kundu, C. Nethravathi and P. A. Deshpande, Chem. Mater., 2011, 23, 2772 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704 CrossRef.
- V. C. Tung, M. J. Allen, Y. Yang and R. B. Kaner, Nat. Nanotechnol., 2009, 4, 25 CrossRef CAS.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun and S. De, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS.
- Y. Hernandez, M. Lotya, D. Rickard, S. D. Bergin and J. N. Coleman, Langmuir, 2009, 26, 3208 CrossRef.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155 CrossRef CAS.
- A. A. Green and M. C. Hersam, Nano Lett., 2009, 9, 4031 CrossRef CAS.
- X. S. Zhou, T. B. Wu, K. L. Ding, B. J. Hu, M. Q. Hou and B. X. Han, Chem. Commun., 2010, 46, 386 RSC.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS.
- D. Marquardt, C. Vollmer, R. Thomann, P. Steurer, R. Mülhaupt, E. Redel and C. Janiak, Carbon, 2011, 49, 1326 CrossRef CAS.
- S. D. Miao, Z. M. Liu, B. X. Han, J. Huang, Z. Y. Sun, J. L. Zhang and T. Jiang, Angew. Chem., Int. Ed., 2006, 45, 266 CrossRef CAS.
Footnote |
| † W. J. Xiao and Z. Y. Sun equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2012 |