Ruthenium catalysed one-pot synthesis of S-allyl and cinnamyl dithiocarbamates using allyl and cinnamyl acetates in water†
Sabir
Ahammed
,
Amit
Saha
and
Brindaban C.
Ranu
*
Department of Organic Chemistry, Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700032, India. E-mail: ocbcr@iacs.res.in; Fax: (+91) 33-24732805; Tel: (+91) 33-24734971
First published on 14th June 2012
Abstract
A convenient and efficient procedure for the synthesis of S-allyl/cinnamyl dithiocarbamates has been developed by a one-pot reaction of allyl/cinnamyl acetate, carbon disulfide and amine in presence of Ru(acac)3 in water. A variety of functionalized dithiocarbamates have been obtained by this procedure in high yields. The reaction proceeds via a catalytic Ru(II) species, generated in situ during the reaction.
1 Introduction
Because of the growing concern on the use of toxic organic solvents and catalysts in chemical processes, the design of chemical reactions using more environmentally acceptable reaction media, reagents and catalysts has received considerable attention in recent years.1 As a part of this drive, the use of water as the reaction medium has gained top priority because of its unique reactivity and selectivity which is different from those in organic solvents, in addition to its environmental acceptability and low cost.2Metals play very important role in organic reactions.3 However; many of them are expensive and toxic too. Thus the use of no metal if possible, or the minimum use of a benign metal complex for reactions which are usually catalyzed by stoichiometric or near stoichiometric quantity of metals, is of much significance. Ruthenium-catalyzed reactions have received considerable interest as they can accommodate a wide range of oxidation states and tolerate many functional groups.4 Ru(III) salts are relatively inexpensive and benign. Moreover, Ru compounds are able to activate allylic substrates through facile oxidative addition.5
Organic dithiocarbamates are of much importance as versatile synthetic intermediates,6 protecting groups in peptide synthesis,7 linkers in solid phase organic synthesis,8 agrochemicals,9 and biologically active compounds.10 Conventional methods for their synthesis involve reactions of amines with thiophosgenes which are highly toxic.11 Later, several one-pot procedures reacting amines with carbon disulfide and alkyl/aryl halides in absence or presence of metals have been developed to produce simple alkyl and aryl dithiocarbamates.12 Functionalization of the carbamate moiety is a useful process as it offers newer derivatives to explore interesting biological properties.10 However, only a few methods are available to access functionalized dithiocarbamates.13 These include reactions of amines and carbon disulfide with conjugated alkenes to provide β-functionalized alkyl dithiocarbamates,13a styrenyl bromides/iodides to give S-styrenyl dithiocarbamates,13b and allyl chlorides to produce S-allyl dithiocarbamates.13c The allyl group is a useful functionality as it provides much scope for further manipulation and thus allyl and substituted allyl dithiocarbamates are of great potential. In the reported procedures13 allylation was carried out using expensive allyl halides as they are more reactive. However, easily accessible, relatively inexpensive, moderately active and configurationally stable14 allyl acetates are more attractive. Nevertheless, the reactions with them are more challenging. We report here a simple condensation reaction of allyl and cinnamyl acetate with carbon disulfide and secondary amines catalyzed by Ru(acac)3 in water (Scheme 1) to produce S-allyl/cinnamyl dithiocarbamates. Although reactions of highly activated allyl acetates from Baylis–Hillman adducts, with carbon disulfide and amines was reported in water in the absence of any metal catalyst15 this procedure was not effective for simple allyl and cinnamyl acetates. To the best of our knowledge we are the first to report such reaction using non-activated allyl and cinnamyl acetates in presence of Ru-catalyst.
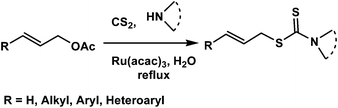 | ||
| Scheme 1 | ||
2 Results and discussion
To standardize the reaction conditions, several experiments were performed for a representative reaction of cinnamyl acetate, carbon disulfide and pyrrolidine under varying reaction parameters such as catalyst, solvent, reaction temperature and time (Table 1). It was found that the best result in terms of yield was obtained using 5 mol% of Ru(acac)3 in water at 100 °C for 14 h (Table 1, entry 9). No reaction was initiated in absence of Ru-catalyst (Table 1, entry 16). No reaction occurred in THF and dioxane using Ru(acac)3 although DMF and NMP furnished better yields (45% and 76% respectively). The yields in neat and in an ionic liquid [pmIm]Br under similar conditions were also not remarkable. The reaction using RuCl3 and Ru nanoparticles in DMF as well as in water did not produce any encouraging results (Table 1, entries 1–4).|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst (loading) | Solvent | Temp. (°C) | Time (h) | Yielda (%) |
| a Yields refer to those of purified products characterized by IR and 1H and 13C NMR spectroscopic data. | |||||
| 1 | RuCl3 (5 mol%) | DMF | 120 | 16 | — |
| 2 | RuCl3 (5 mol%) | H2O | 120 | 16 | — |
| 3 | Ru NP (5 mol%) | DMF | 120 | 16 | — |
| 4 | Ru NP (5 mol%) | H2O | 120 | 16 | — |
| 5 | Ru(acac)3 (5 mol%) | THF | 65 | 15 | — |
| 6 | Ru(acac)3 (5 mol%) | DMF | 120 | 16 | 45 |
| 7 | Ru(acac)3 (5 mol%) | NMP | 120 | 14 | 76 |
| 8 | Ru(acac)3 (5 mol%) | Dioxane | 110 | 15 | — |
| 9 | Ru(acac)3 (5 mol%) | H2O | 100 | 14 | 81 |
| 10 | Ru(acac)3 (5 mol%) | H2O | 70 | 14 | 25 |
| 11 | Ru(acac)3 (8 mol%) | H2O | 100 | 14 | 81 |
| 12 | Ru(acac)3 (3 mol%) | H2O | 100 | 14 | 52 |
| 13 | Ru(acac)3 (1 mol%) | H2O | 100 | 14 | 34 |
| 14 | Ru(acac)3 (5 mol%) | Neat | 80 | 12 | 16 |
| 15 | Ru(acac)3 (5 mol%) | [pmlm]Br | 80 | 12 | 33 |
| 16 | — | H2O | 100 | 14 | — |
| 17 | RuCl2(PPh3)3 (5 mol%) | DMF | 120 | 14 | 73 |
| 18 | [Ru(CO)3Cl2]2 (5 mol%) | DMF | 120 | 14 | 71 |
| 19 | [(C6H6)RuCl2]2 (5 mol%) | DMF | 120 | 14 | 65 |

Thus, in a typical experimental procedure, the amine was added to a stirred mixture of allyl/cinnamyl acetate and carbon disulfide in water at 0–5 °C followed by a catalytic amount of Ru(acac)3 (ruthenium acetylacetonate). The reaction mixture was then heated under reflux for a certain period of time as required for completion (TLC). Standard workup and purification by column chromatography provided the pure products.
Allyl acetates (substituted and unsubstituted) underwent facile reactions with carbon disulfide and various amines (pyrrolidine, morpholine, piperidine, N,N-dimethyl and N,N-diallyl amine) by this procedure to produce the corresponding allyl dithiocarbamates. Interestingly crotyl acetate provided a mixture (77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :23) of (E) and (Z)-dithiocarbamates (Table 2, entry 3) whereas trans-long chain alkyl substituted allyl acetate furnished only the (E)- product (Table 2, entry 4). The branched allyl acetates (Table 2, entries 5, 21 and 22) produced the corresponding (E)-dithiocarbamates. The highly activated allyl acetate with an electron withdrawing group at the β-position underwent clean reaction in the absence of the Ru-catalyst (Table 2, entry 23).15 The reaction of cinnamyl acetate and several substituted cinnamyl acetates by this procedure also produced the corresponding dithiocarbamates efficiently. The trans-cinnamyl acetate provided the (E)-product (Table 2, entry 6) whereas cis-cinnamyl acetate led to a mixture of stereoisomers (Table 2, entry 14). The heteroaryl-substituted trans-allyl acetate also underwent reaction successfully giving an (E)-isomer (Table 2, entry 16). It was observed that the reactions of electron-donating group substituted cinnamyl acetates are more facile than those with electron-withdrawing groups. Significantly, the reaction is uniform with ortho-, meta- and para-substituted allyl acetates.
:23) of (E) and (Z)-dithiocarbamates (Table 2, entry 3) whereas trans-long chain alkyl substituted allyl acetate furnished only the (E)- product (Table 2, entry 4). The branched allyl acetates (Table 2, entries 5, 21 and 22) produced the corresponding (E)-dithiocarbamates. The highly activated allyl acetate with an electron withdrawing group at the β-position underwent clean reaction in the absence of the Ru-catalyst (Table 2, entry 23).15 The reaction of cinnamyl acetate and several substituted cinnamyl acetates by this procedure also produced the corresponding dithiocarbamates efficiently. The trans-cinnamyl acetate provided the (E)-product (Table 2, entry 6) whereas cis-cinnamyl acetate led to a mixture of stereoisomers (Table 2, entry 14). The heteroaryl-substituted trans-allyl acetate also underwent reaction successfully giving an (E)-isomer (Table 2, entry 16). It was observed that the reactions of electron-donating group substituted cinnamyl acetates are more facile than those with electron-withdrawing groups. Significantly, the reaction is uniform with ortho-, meta- and para-substituted allyl acetates.
In general the reactions are clean and high yielding. Several functional groups such as OMe, Br, F, COMe and heteroaryl moieties are compatible with the reaction conditions. A Br-group on the aryl ring of the dithiocarbamates offers scope for further manipulation. It was found that cinnamyl bromides in place of cinnamyl acetates underwent reactions with carbon disulfide and amines under these conditions in the absence of any Ru-catalyst. However, as mentioned earlier, no reaction was initiated without the Ru-catalyst in case of cinnamyl acetate. Primary and aromatic amines did not participate in this reaction. The stereochemistry (E and Z) of the product was determined by the coupling constant of the olefinic protons in the 1H NMR spectra. For confirmation, the stereochemistry of one of the products (Table 2, entry 16) was established by X-ray studies (Fig. 1).16
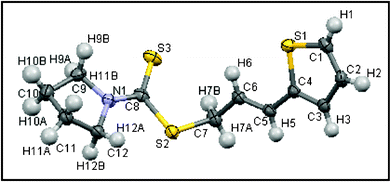 | ||
| Fig. 1 ORTEP diagram of (E)-3-(thiophen-2-yl)allyl pyrrolidine-1-carbodithioate (Table 2, entry 16). | ||
| Entry | Substrate | Amine | Time (h) | Product | Yielda (%) |
|---|---|---|---|---|---|
| a Yields refer to those of purified products characterized by IR and 1H and 13C NMR spectroscopic data. | |||||
| 1 |

|
|
21 |

|
81 |
| 2 |

|

|
23 |

|
78 |
| 3 |

|

|
25 |
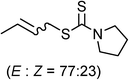
|
69 |
| 4 |

|
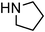
|
28 |

|
65 |
| 5 |
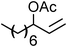
|
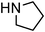
|
28 |

|
67 |
| 6 |

|

|
21 |

|
81 |
| 7 |

|
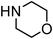
|
23 |

|
78 |
| 8 |

|

|
21 |
|
83 |
| 9 |
|
|
21 |
|
80 |
| 10 |

|

|
22 |

|
77 |
| 11 |

|

|
22 |

|
73 |
| 12 |
|

|
12 |
|
87 |
| 13 |

|

|
12 |

|
81 |
| 14 |

|

|
24 |

|
76 |
| 15 |

|
|
18 |

|
81 |
| 16 |

|
|
16 |
|
85 |
| 17 |

|
|
27 |

|
70 |
| 18 |
|

|
23 |
|
78 |
| 19 |

|
|
13 |
|
86 |
| 20 |

|
|
22 |

|
83 |
| 21 |

|

|
19 |

|
82 |
| 22 |
|
|
22 |
|
81 |
| 23 |
|

|
8 |

|
88 |
We suggest that the reaction proceeds through an oxidative addition of Ru(II), generated in situ by the reduction of Ru(III) by dithiocarbamate ion, with allyl acetate resulting a η3-π-allyl complex, [B] or [C] which undergoes reductive elimination to give the product (Scheme 2). The catalytic cycle involving Ru(II) to Ru(IV) is not unusual in Ru catalyzed allylation reactions.17
 | ||
| Scheme 2 | ||
The involvement of in situ generated Ru(II) in the reaction gains support from the observation that three commercially available Ru(II) catalysts catalyze this reaction producing the corresponding products in 65–73% yields (Table 1, entries 17–19) under identical reaction conditions. The reduction of Ru(III) to Ru(II) by the thiocarbamate anion in the reaction mixture is evidenced by the isolation of a trace amount of the dimer of the dithiocarbamate anion (Scheme 2). The structure of this dimeric compound was established by comparison of its 1H NMR, 13C NMR and HRMS data with those reported.18 As Ru(II) complexes are quite expensive, we considered the use of Ru(acac)3 for this reaction as more cost effective. In case of trans-allyl acetate, it is predicted that the direct interaction of π-allyl complex [B]via a transient intermediate [A] (path-a) with a dithiocarbamate ion, followed by reductive elimination gave the the trans product. However in case of cis-allyl acetate, the complex [C], formed after the oxidative addition via intermediate [A] (path-b), having steric interactions between the α-H and γ-R group is likely to equilibrate with less congested [B]. Thus both the intermediates [B] and [C] participate in the catalytic cycle to give the product as a mixture of cis- and trans-allyl/cinnamyl dithiocarbamates. The transition state for the formation of a Ru-π-allyl complex does not get enough stabilization due to absence of an aromatic ring and the reactions become extremely slow. Thus, the reaction of formation of the Ru-π-allyl complex is likely to become more stable by the π-electrons of the aromatic nucleus of aryl substituted allyl acetates due to stabilization of the electron deficient Ru centre. Thus, the reaction proceeds faster in presence of an electron donating group (Me, OMe) on the aromatic ring of cinnamyl acetate, while rate of the reaction becomes slow in presence of an electron withdrawing group (COMe). In case of branched allyl acetate, exclusively the linear allyl dithiocarbamate was obtained as the sole product indicating a pathway via a η3-π-allyl complex [B] followed by the reductive elimination from the less hindered side of the allyl complex. In case of R = alkyl group, the Ru-π-allyl complex does not get enough stabilization due to absence of the aromatic ring and the reaction becomes extremely slow. Thus, the reaction of alkyl substituted allyl acetates generally takes a longer reaction time (25–28 h) compared to the aromatic allyl acetates.
From all these observations it may be concluded that the formation of η3-π-allyl complex is the rate determining step. In case of trans-crotyl acetate (R = Me) the product was obtained as a mixture of cis and trans-allyl dithiocarbamate. This may be due to presence of a less bulky Me group which forces complex [B] to equilibrate with [C] producing a mixture of cis and trans-allyl dithiocarbamate.
3 Conclusion
In conclusion, we have developed an efficient protocol for the synthesis of allyl and cinnamyl dithiocarbamates by a one pot ruthenium-catalyzed reaction of allyl/cinnamyl acetate, carbon disulfide and an amine in water. To the best of our knowledge we are not aware of any report using non-activated allyl acetates and cinnamyl acetates for this condensation reaction. The operational simplicity, use of inexpensive Ru(acac)3 as catalyst, relatively low catalyst loading (5 mol%), excellent stereoselectivity in the reactions of trans-cinnamyl acetate, high yields, and use of water as reaction medium make this procedure more attractive. More significantly, a wide range of cinnamyl and allyl dithiocarbamates, prepared by this procedure are reported for the first time.4 Experimental Section
General experimental procedure for the synthesis of dithiocarbamates. Representative experimental procedure for condensation of amine, CS2 and cinnamyl acetate (Table 2, entry 6).
Pyrrolidine (84 mg, 1.2 mmol) was added drop by drop at 0–5 °C to a well stirred mixture of cinnamyl acetate (176 mg, 1 mmol) and carbon disulfide (190 mg, 2.5 mmol) in water (3 mL). After stirring for 10 min, ruthenium acetylacetonate (20 mg, 0.05 mmol) was added and the reaction mixture was heated under reflux for 21 h (monitoring via TLC). The reaction mixture was extracted with ethyl acetate (3 × 7 mL) and the extract was washed with brine and dried (Na2SO4). Evaporation of solvent left the crude product which was purified by column chromatography over silica gel (hexane:ether 85:15) to provide the corresponding cinnamyl pyrrolidine-1-carbodithioate as a viscous liquid; IR (neat): νmax = 3026, 2970, 2947, 2870, 1597, 1489, 1435, 1329, 1249, 1182, 1161, 1007, 957, 752, 694 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.94–1.99 (m, 2H), 2.03–2.08 (m, 2H), 3.65 (t, J = 6.5 Hz, 2H), 3.94 (t, J = 7 Hz, 2H), 4.18 (d, J = 7 Hz, 2H), 6.27–6.34 (m, 1H), 6.62 (d, J = 15.5 Hz, 1H), 7.21 (t, J = 7 Hz, 1H), 7.25–7.30 (m, 2H), 7.35 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.3, 26.1, 39.4, 50.6, 55.1, 124.4, 126.5 (2C), 127.7, 128.6 (2C), 133.5, 136.8, 192.3; HRMS m/z calcd for C14H17NS2 [M + H]+ = 264.0881, found 264.0875.
This procedure was followed for all the reactions listed in Table 2. Although this procedure was described with a 1 mmol scale, 10 mmol scale reactions also provided uniform results.
All of these products listed in Table 2 are new except three (Table 2, entries 1, 2 and 23). These known compounds were identified by comparison of their spectral data (1H NMR and 13C NMR) with those reported (entries 1 and 2,12a 2315). The new compounds were properly characterized by their IR, 1H NMR, 13C NMR, and HRMS spectroscopic data which were provided below.
But-2-enyl pyrrolidine-1-carbodithioate (mixture of E and Z) (Table 2, entry 3)
Yellowish viscous liquid; IR (neat): νmax = 3022, 2968, 2936, 2872, 1510, 1433, 1329, 1249, 1219, 1182, 1163, 1009, 957 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.68 (d, J = 6.5 Hz, 3H), 1.72 (d, J = 6.5 Hz, 3H), 1.94–1.99 (m, 2H), 2.03–2.09 (m, 2H), 3.63 (t, J = 7 Hz, 2H), 3.91–3.14 (m, 4H), 3.98 (d, J = 7.5 Hz, 2H), 5.56–5.60 (m, 1H), 5.72–5.77 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 17.9, 24.4, 26.2, 39.3, 50.6, 55, 124.1, 125.3, 129, 130.1, 192.8; HRMS m/z calcd for C9H15NS2 [M + H]+ = 202.0724, found 202.0718.(E)-Dec-2-enyl pyrrolidine-1-carbodithioate (Table 2, entry 4 and 5)
Yellowish viscous liquid; IR (neat): νmax = 2955, 2926, 2854, 1738, 1655, 1460, 1429, 1339, 1250, 1184, 1163, 1009, 957 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.98 (t, J = 7 Hz, 3H), 1.34–1.58 (m,10H), 1.95–2.04 (m, 3H), 2.05–2.13 (m, 3H), 3.63 (t, J = 7 Hz, 2H), 3.91–3.99 (m, 4H), 5.51–5.58 (m, 1H), 5.69–5.75 (m, 1H); 13C NMR (125 MHz, CDCl3) δ 14.2, 22.8, 24.4, 26.1, 26.2, 28.7, 29.3, 29.4, 31.9, 32.5, 34.3, 39.5, 124, 135.7, 192.9; HRMS m/z calcd for C15H27NS2 [M + H]+ = 286.1663, found 286.1657.(E)-Cinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 6 and 21)
Yellowish viscous liquid; IR (neat): νmax = 3026, 2970, 2947, 2870, 1597, 1489, 1435, 1329, 1249, 1182, 1161, 1007, 957, 752, 694 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.94–1.99 (m, 2H), 2.03–2.08 (m, 2H), 3.65 (t, J = 6.5 Hz, 2H), 3.94 (t, J = 7 Hz, 2H), 4.18 (d, J = 7 Hz, 2H), 6.27–6.34 (m, 1H), 6.62 (d, J = 15.5 Hz, 1H), 7.21 (t, J = 7 Hz, 1H), 7.25–7.30 (m, 2H), 7.35 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.3, 26.1, 39.4, 50.6, 55.1, 124.4, 126.5 (2C), 127.7, 128.6 (2C), 133.5, 136.8, 192.3; HRMS m/z calcd for C14H17NS2 [M + H]+ = 264.0881, found 264.0875.(E)-Cinnamyl morpholine-4-carbodithioate (Table 2, entry 7)
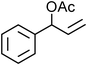
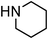
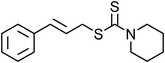
Yellowish viscous liquid; IR (neat): νmax = 2961, 2924, 2853, 1728, 1462, 1418, 1269, 1229, 1115, 977, 752 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.77 (bs, 4H), 3.98 (bs, 2H), 4.21 (d, J = 7.5 Hz, 2H), 4.34 (bs, 2H), 6.28–6.34 (m, 1H), 6.65 (d, J = 15.5 Hz, 1H), 7.22–7.27 (m, 1H), 7.29–7.32 (m, 2H), 7.38 (t, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 40, 50.8, 65.9, 66.3, 123.8, 126.5 (2C), 127.9, 128.6 (2C), 134, 136.7, 197.1; HRMS m/z calcd for C14H17NOS2 [M + H]+ = 280.0830, found 280.0823.(E)-Cinnamyl piperidine-1-carbodithioate (Table 2, entry 8 and 22)
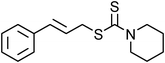
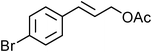
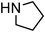
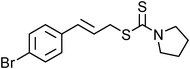
Yellowish viscous liquid; IR (neat): νmax = 3026, 2938, 2855, 1474, 1427, 1281, 1244, 1227, 1132, 1115, 1005, 978, 752 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.69 (bs, 6H), 3.88 (s, 2H), 4.19 (d, J = 7.5 Hz, 2H), 4.29 (s, 2H), 6.29–6.35 (m, 1H), 6.63 (d, J = 15.5 Hz, 1H), 7.20–7.23 (m, 1H), 7.29 (t, J = 7 Hz, 2H), 7.36 (d, J = 8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.3, 25.5, 25.9, 40.2, 51.4, 52.9, 124.2, 126.4 (2C), 127.6, 128.5 (2C), 133.6, 136.7, 195; HRMS m/z calcd for C15H19NS2 [M + H]+ = 278.1037, found 278.1033.(E)-4-Bromocinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 9)
Yellowish viscous liquid; IR (neat): νmax = 3047, 3028, 2976, 2943, 2866, 1904, 1738, 1584, 1487, 1433, 1329, 1292, 1248, 1157, 1070, 1005, 962, 818, 518 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.95–2.0 (m, 2H), 2.04–2.10 (m, 2H), 3.65 (t, J = 6.5 Hz, 2H), 3.94 (t, J = 7 Hz, 2H), 4.17 (d, J = 7 Hz, 2H), 6.28–6.34 (m, 1H), 6.56 (d, J = 15.5 Hz, 1H), 7.22 (d, J = 8 Hz, 2H), 7.40 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.4, 26.2, 39.2, 50.7, 55.2, 121.5, 125.5, 128 (2C), 131.7 (2C), 132.2, 135.8, 192.1; HRMS m/z calcd for C14H16BrNS2 [M + H]+ = 341.9986, found 341.9980.(E)-4-Bromocinnamyl piperidine-1-carbodithioate (Table 2, entry 10)
Yellowish viscous liquid; IR (neat): νmax = 3030, 3003, 2943, 2856, 1647, 1604, 1573, 1510, 1467, 1425, 1284, 1246, 1113, 1032, 978, 806 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.71 (bs, 6H), 3.89 (bs, 2H), 4.17 (d, J = 7 Hz, 2H), 4.3 (bs, 2H), 6.28–6.34 (m, 1H), 6.56 (d, J = 16 Hz, 1H), 7.22 (d, J = 8 Hz, 2H), 7.41 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.4, 25.7 (2C), 40, 51.5, 53.1, 121.5, 125.4, 128 (2C), 131.7 (2C), 132.4, 135.8, 194.9; HRMS m/z calcd for C15H18BrNS2 [M + H]+ = 356.0142, found 356.0137.(E)-4-Bromocinnamyl morpholine-4-carbodithioate (Table 2, entry 11)
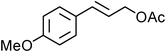
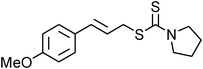
Yellowish viscous liquid; IR (neat): νmax = 2965, 2922, 2856, 2810, 1722, 1585, 1487, 1462, 1267, 1115, 1072, 1009, 814 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.77 (bs, 4H), 3.98 (bs, 2H), 4.18 (d, J = 7.5 Hz, 2H), 4.34 (bs, 2H), 6.27–6.33 (m, 1H), 6.58 (d, J = 16 Hz, 1H), 7.23 (d, J = 8 Hz, 2H), 7.42 (d, J = 7.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 39.8, 51.0, 51.3, 66.0, 66.4, 121.7, 124.9, 128.1 (2C), 131.8 (2C), 132.8, 135.7, 196.9; HRMS m/z calcd for C14H16BrNOS2 [M + H]+ = 357.9935, found 357.9930.(E)-4-Methoxycinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 12)
White solid (mp 68 °C); IR (neat): νmax = 3028, 3001, 2963, 2831, 1643, 1603, 1576, 1212, 1439, 1329, 1246, 1178, 1028, 957, 810, 580 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.92–1.97 (m, 2H), 2.0–2.04 (m, 2H), 3.61 (t, J = 6.5 Hz, 2H), 3.76 (s, 3H), 3.91 (t, J = 7 Hz, 2H), 4.15 (d, J = 7 Hz, 2H), 6.13–6.19 (m, 1H), 6.56 (d, J = 15.5 Hz, 1H), 6.81 (d, J = 9 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.2, 26, 39.5, 50.5, 54.9, 55.2, 113.9, 121.8, 127.5 (2C), 129.5, 132.9 (2C), 159.2, 192.2; HRMS m/z calcd for C15H19NOS2 [M + H]+ = 294.0986, found 294.0981.(E)-4-Methoxycinnamyl piperidine-1-carbodithioate (Table 2, entry 13)
Yellow solid (mp 65 °C); IR (neat): νmax = 3026, 2995, 2933, 2852, 1607, 1510, 1470, 1427, 1281, 1248, 1175, 1115, 1030, 970 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.65 (bs, 6H), 3.75 (s, 3H), 3.84 (s, 2H), 4.14 (d, J = 7.5 Hz, 2H), 4.26 (s, 2H), 6.13–6.19 (m, 1H), 6.55 (d, J = 16 Hz, 1H), 6.80 (d, J = 8.5 Hz, 2H), 7.27 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 24.4, 25.7, 26, 40.6, 51.5, 53, 55.4, 114.1, 121.9, 127.7 (2C), 129.6, 133.3 (2C), 159.4, 195.2; HRMS m/z calcd for C16H21NOS2 [M + H]+ = 308.1143, found 308.1137.(E)-Cinnamyl dimethylcarbamodithioate (Table 2, entry 15)
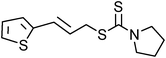
Yellowish viscous liquid; IR (neat): νmax = 3057, 3026, 2924, 1738, 1495, 1373, 1256, 1145, 1053, 982, 752 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.36 (s, 3H), 3.56 (s, 3H), 4.16 (d, J = 7.5 Hz, 2H), 6.27–6.34 (m, 1H), 6.63 (d, J = 15.5 Hz, 1H), 7.20–7.32 (m, 3H), 7.34 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 40.7, 41.5, 45.5, 124.1, 126.5 (2C), 127.7, 128.6 (2C), 133.7, 136.8, 196.8; HRMS m/z calcd for C12H15NS2 [M + H]+ = 238.0724, found 238.0719.(E)-3-(Thiophen-2-yl)allylpyrrolidine-1-carbodithioate (Table 2, entry 16)
Yellow solid (mp 71 °C); IR (KBr): νmax = 3099, 3074, 3020, 2972, 2897, 2864, 1620, 1433, 1381, 1330, 1249, 1157, 1005, 955, 860, 700 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.92–1.97 (m, 2H), 2.01–2.07 (m, 2H), 3.62 (t, J = 7 Hz, 2H), 3.92 (t, J = 7 Hz, 2H), 4.14 (d, J = 7.5 Hz, 2H), 6.09–6.15 (m, 1H), 6.75 (d, J = 15.5 Hz, 1H), 6.91–6.92 (m, 2H), 7.11 (t, J = 3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 24.2, 26, 39.1, 50.6, 55, 123.8, 124.3, 125.7, 126.5, 127.2, 141.7, 191.9; HRMS m/z calcd for C12H15NS3 [M + H]+ = 270.0445, found 270.0438.(E)-3-Acetylcinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 17)
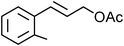
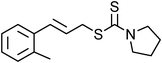
Yellowish viscous liquid; IR (neat): νmax = 3395, 2924, 1684, 1435, 1163, 1007 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.89–1.96 (m, 2H), 1.99–2.05 (m, 2H), 2.54 (s, 3H), 3.6 (bs, 2H), 3.89 (bs, 2H), 4.14 (d, J = 7 Hz, 2H), 6.29–6.36 (m, 1H), 6.61 (d, J = 16 Hz, 1H), 7.32 (t, J = 7.5 Hz, 1H), 7.49 (d, J = 8 Hz, 1H), 7.73 (d, J = 7 Hz, 1H), 7.86 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 24.4, 26.3, 26.8, 39.2, 50.7, 55.3, 126.2, 126.4, 127.6, 128.9, 131, 132.5, 137.4, 137.6, 192.1, 198.2; HRMS m/z calcd for C16H19NOS2 [M + H]+ = 306.0986, found 306.0981.(E)-2-Methylcinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 18)
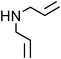
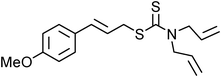
Yellowish viscous liquid; IR (neat): νmax = 3019, 2951, 2924, 2870, 1732, 1460, 1435, 1331, 1290, 1277, 1250, 1219, 1182, 1161, 1006, 957 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.97–2.01 (m, 2H), 2.05–2.09 (m, 2H), 2.34 (s, 3H), 3.66 (t, J = 7 Hz, 2H), 3.95 (t, J = 7 Hz, 2H), 4.2 (d, J = 7.5 Hz, 2H), 6.17–6.22 (m, 1H), 6.86 (d, J = 15.5 Hz, 1H), 7.13–7.15 (m, 3H), 7.43 (t, J = 8.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 19.9, 24.4, 26.2, 39.8, 50.7, 55.1, 125.5, 125.8, 126.1, 127.6, 130.3, 131.5, 135.5, 135.9, 192.3; HRMS m/z calcd for C15H19NS2 [M + H]+ = 278.1037, found 278.1033.(E)-4-Methoxycinnamyl diallylcarbamodithioate (Table 2, entry 19).
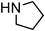
Yellowish viscous liquid; IR (neat): νmax = 3080, 3003, 2955, 2926, 2837, 1606, 1510, 1466, 1398, 1290, 1250, 1175, 1034 cm−1; 1H NMR (500 MHz, CDCl3) δ 3.79 (s, 3H), 4.15 (d, J = 7 Hz, 2H), 4.32 (s, 2H), 4.67 (s, 2H), 5.19–5.29 (m, 4H), 5.86 (bs, 2H), 6.13–6.19 (m, 1H) 6.57 (d, J = 15.5 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 40.9, 53.7, 55.3, 56.5, 114 (2C), 118.5, 118.7, 121.4, 127.7 (2C), 129.5, 130.5, 131.2, 133.4, 159.4, 197.7; HRMS m/z calcd for C17H21NOS2 [M + H]+ = 320.1143, found 320.1136.(E)-4-Fluorocinnamyl pyrrolidine-1-carbodithioate (Table 2, entry 20)
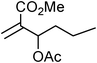
Yellowish viscous liquid; IR (neat): νmax = 2972, 2949, 2924, 2872, 1738, 1601, 1508, 1433, 1228, 1159, 1007 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.95–2.01 (m, 2H), 2.05–2.10 (m, 2H), 3.65 (t, J = 7 Hz, 2H), 3.95 (t, J = 7 Hz, 2H), 4.17 (d, J = 7 Hz, 2H), 6.20–6.26 (m, 1H), 6.62 (d, J = 15.5 Hz, 1H), 6.98 (t, J = 8.5 Hz, 2H), 7.30–7.34 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 24.4, 26.2, 36.6, 50.7, 55.2, 115.5 (d, J = 21 Hz) (2C), 124.3, 128 (d, J = 8.7 Hz) (2C), 132.3, 133, 162.5 (d, J = 245 Hz), 192.2; HRMS m/z calcd for C14H16FNS2 [M + H]+ = 282.0786, found 282.0781.(E)-Methyl-2-((pyrrolidine-1-carbodithioyl)methyl)hex-2-enoate (Table 2, entry 23)
Yellowish viscous liquid; IR (neat): νmax = 2972, 2949, 2872, 1601, 1508, 1433, 1329, 1227, 1157, 1007, 957, 825, 760 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.93 (t, J = 7.5 Hz, 3H), 1.46–1.5 (m, 2H), 1.95 (t, J = 7 Hz, 2H), 2.04 (t, J = 7 Hz, 2H), 2.3–2.35 (m, 2H), 3.59 (t, J = 6.5 Hz, 2H), 3.74 (s, 3H), 3.91 (t, J = 6.85 Hz, 2H), 4.26 (s, 2H), 6.96 (t, J = 7.75 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 13.8, 21.8, 24.1, 26, 31.1, 33.5, 50.4, 51.9, 54.8, 126.7, 147.7, 167, 192.4; HRMS m/z calcd for C13H21NO2S2 [M + H]+ = 288.1092, found 288.1090.Acknowledgements
We are pleased to acknowledge the financial support from DST, New Delhi, in the form of the award J.C. Bose National Fellowship to B.C.R. (Grant No. SR/S2/JCB-11/2008). S.A. and A.S. thank CSIR, New Delhi, for their fellowships.References
- (a) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC; (b) B. C. Ranu, A. Saha and R. Dey, Curr. Opin. Drug. Discov. Devel., 2010, 13, 658 CAS; (c) T. Erdmenger, C. G. Sanchez, J. Vitz, R. Hoogenboom and U. S. Scheubert, Chem. Soc. Rev., 2010, 39, 3317 RSC.
- (a) P. A. Grieco, Organic Synthesis in Water; Blackie Academic and Professional: London, 1998. Search PubMed; (b) Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS; (c) C. J. Lee, Chem. Rev., 2005, 105, 3095 CrossRef.
- J. M. Brown, P. H. Dixneuf, A. Furstner, L. S. Hegedns, P. Hofmann, P. Knochel, G. Vankoten, S. Murai and M. Reetz, Top. Organomet. Chem., 2006, 19, 1 CrossRef.
- C. Bruneau, J.-L. Renaud and B. Demerseman, Pure Appl. Chem., 2008, 80, 861 CrossRef CAS.
- (a) T. Kondo and T. Mitsudo, Curr. Org. Chem., 2002, 6, 1163 CrossRef CAS; (b) T. Kondo, T. Mitsudo, Ruthenium in Organic Synthesis, ed, S.-I. Murahashi, Wiley-VCH, Weinheim, 2004, 129 Search PubMed.
- (a) A. K. Mukherjee and R. Ashare, Chem. Rev., 1991, 91, 1 CrossRef; (b) U. Boas, H. Gertz, J. B. Christensen and P. M. H. Heegaard, Tetrahedron Lett., 2004, 45, 269 CrossRef CAS.
- T. W. Greene, P.G.M. Wuts, Protecting Groups in Organic Synthesis, Wiley Interscience, New York, 3rd edn, 1999, 484 Search PubMed.
- P. Morf, F. Raimondi, H.-G. Nothofex, B. Schnyder, A. Yasuda, J. M. Wessels and T. A. Jung, Langmuir, 2006, 22, 658 CrossRef CAS.
- C. Rafin, E. Veignie, M. Sancholle, D. Postal, C. Len, P. Villa and G. Ronco, J. Agric. Food Chem., 2000, 48, 5283 CrossRef CAS.
- M. Dhooghe and N. De Kime, Tetrahedron, 2006, 62, 513 CrossRef CAS.
- (a) H. Tilles, J. Am. Chem. Soc., 1959, 81, 714 CrossRef CAS; (b) W. Chin-Hsien, Synthesis, 1981, 622 CrossRef.
- (a) N. Azizi, F. Aryanasab and M. R. Saidi, Org. Lett., 2006, 8, 5275 CrossRef CAS; (b) D. Chaturvedi and S. Ray, Tetrahedron Lett., 2006, 47, 1307 CrossRef CAS; (c) B. C. Ranu, A. Saha and S. Banerjee, Eur. J. Org. Chem., 2008, 519 CrossRef CAS; (d) T. Chatterjee, S. Bhadra and B. C. Ranu, Green Chem., 2011, 13, 1837 RSC.
- (a) N. Azizi, F. Aryanasab, L. Torkiyan, A. Ziyaei and M. R. Saidi, J. Org. Chem., 2006, 71, 3634 CrossRef CAS; (b) S. Bhadra, A. Saha and B. C. Ranu, Green Chem., 2008, 10, 1224 RSC; (c) A. Z. Halimehjani, H. Maleki and M. R. Saidi, Tetrahedron Lett., 2009, 50, 2747 CrossRef CAS.
- B. M. Trost and T. R. Verhoeven, J. Am. Chem. Soc., 1980, 102, 4730 CrossRef CAS.
- L. D. S. Yadav, R. Patel and V. P. Srivastava, Tetrahedron Lett., 2009, 50, 1335 CrossRef CAS.
- CCDC No. 861311 for the product in entry 16 of Table 2.
- (a) J. A. Van Rijn, M. L. Lutz, S. V. Chrzanowski, A. L. Spek, E. Bouwman and E. Drent, Adv. Synth. Catal., 2009, 351, 1637 CrossRef CAS; (b) J. A. Van Rijn, M. A. Siegler, A. L. Spek, E. Bouwman and E. Drent, Organometallics, 2009, 28, 7006 CrossRef CAS; (c) H. Kondo, Y. Yamaguchi and H. Nagashima, Chem. Commun., 2000, 1075 RSC.
- C. N. Kapanda, G. G. Muccioli, G. Labar, J. H. Poupaert and D. M. Lambert, J. Med. Chem., 2009, 52, 7310 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all products in Table 2. CCDC reference number 861311. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20856c |
| This journal is © The Royal Society of Chemistry 2012 |