Electromodulated release of nitric oxide through polymer material from reservoir of inorganic nitrite salt†
Lajos
Höfler
a,
Dipankar
Koley
a,
Jianfeng
Wu
b,
Chuanwu
Xi
b and
Mark E.
Meyerhoff
*a
aDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109-1055, USA. E-mail: mmeyerho@umich.edu
bDepartment of Environmental Health Sciences, University of Michigan, Ann Arbor, MI 48109-1055, USA
First published on 12th July 2012
Abstract
Herein, we report a new approach to electromodulate the release of NO at physiological levels through polymeric materials from a stable nitrite electrolyte reservoir, with potential application in controlling biofilm formation and clotting on intravascular catheters. The NO flux can be turned ‘on’ and ‘off’ electrochemically, on demand.
Nitric oxide (NO) has been shown to have several important physiological functions. It is an important vasodilator,1 it inhibits platelet adsorption and activation,2 and it is a mediator in antimicrobial activities.3 Indeed, nitric oxide production from NO donors doped within plastic materials has also been shown to control infection, prevent biofilm formation, and minimize inflammation and fibrosis.4 Implantable devices that can deliver physiologically relevant fluxes of NO in a controllable fashion have significant medical potential. For example, infection and blood clotting are important risk factors associated with the use of various intravascular catheters.5 Utilization of NO releasing catheters can provide the solution for both of these problems by actively protecting against bacterial biofilm formation and attenuating platelet activation and adhesion. The inhibition of platelet adhesion by NO has been achieved previously by employing NO releasing polymeric materials and coatings based on diazeniumdiolate chemistry.6–13 Although these reports have demonstrated continuous NO release (at fluxes above those produced by endothelial cells) for up to 2–3 weeks, longer-term release profiles (> 1 month) are needed for certain implantable devices. Further, the instability of diazeniumdiolates and other NO donors (e.g., S-nitrosothiols) employed to create NO emitting polymeric coatings makes the commercialization of these technologies challenging and costly. Herein, we describe a new approach to create implantable catheters and potentially other biomedical devices that release NO via electrochemical generation of NO from a reservoir of low cost and stable sodium nitrite salt.
The most efficient NO generation from nitrite in solution phase is obtained by using a copper working electrode (e.g., 1 mm diameter in glass housing) and generating NO during an anodic pulse in which a low concentration of Cu(I) ions is produced locally at the electrode surface. It is known that Cu(I) reacts directly with nitrite to form NO(g).14 However, simply applying an anodic voltage to a copper electrode that is not pre-cleaned first via an applied cathodic voltage pulse for a fixed time period does not usually generate significant levels of NO from nitrite. Indeed, fresh Cu° wires are generally passivated by an oxide layer, and even after cleaning via application of a cathodic voltage, subsequent application of an anodic voltage generates NO, but only for a limited time interval, as Cu2O/CuO/CuOH forms (at physiological pH) quickly on the electrode to foul its surface.15–21 Therefore, to observe consistent reduction of nitrite to NO, as measured by chemiluminescence detection (see Fig. 1S in SI file†), requires a two-step applied potential sequence to the working Cu° electrode, with a cathodic pulse to refresh the Cu° electrode surface.
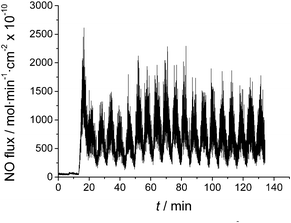 | ||
| Fig. 1 Nitric oxide generation with 0.785 mm2 Cu electrode placed in 0.1 M NaNO2, 50 μM EDTA in 10 mM PBS (pH 6.8). Sample purged with N2(g) continuously into nitric oxide chemiluminescence analyzer. Applied voltage step changes initiated at 10 min time point. | ||
In practice, following the cathodic step (at −0.7 V vs. normal hydrogen electrode (NHE)), a small positive potential step (to +0.2 V vs. NHE) is required to produce Cu(I) ions that can react with nitrite to form NO, with concomitant surface oxidation and passivation of the copper. Fig. 1 illustrates production of NO from the surface of the copper electrode with surface area of 0.785 mm2 in a solution of 0.1 M NaNO2 made in phosphate buffered saline (PBS) (pH 6.8) containing 50 μM EDTA. EDTA was added to help drive the reduction reaction of Cu(I) with nitrite (Cu(I) + NO2− + 2H+ → Cu(II) + NO + H2O) to the product side via the stronger chelation of Cu(II) (than Cu(I)) by EDTA.22,23 Under these conditions, continuous application of either only the reduction potential (−0.7 V) or only the NO liberation potential (+0.2 V) to the copper electrode does not result in any significant long-term NO generation. Note that the applied voltage cycle employed in Fig. 1 is −0.7 V for 3 min, then +0.2 V for 3 min, and this sequence is continuously repeated. Further, a two electrode system was employed, using a commercial Ag/AgCl reference electrode. For consistency, we report potential values vs. NHE reference, so that if the potential of Ag/AgCl differs owing to different chloride activity present, the actual applied potential can be altered to keep it the same vs. NHE (see below). During the +0.2 V portion of the cycle, the highest fluxes of NO are obtained, but the average flux during the cycling period is ca. 800 × 10−10 mol cm−2 min−1, based on the geometric surface area of the copper electrode employed in these preliminary experiments. In separate experiments, we proved that exposure to nitrite when the copper electrode is polarized by a cathodic applied voltage pulse is not required to generate NO once the potential is switched to the more positive anodic value (see Fig. 2S in SI file;† note: a three electrode setup was used in these experiments to examine the effect of different applied voltages on NO generation to avoid any slight potential shifts).
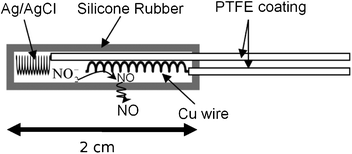 | ||
| Fig. 2 Schematic of the electrochemical NO generating catheter. | ||
To prepare a more biomedically relevant catheter configuration to investigate the prevention of biofilm formation using electrochemically generated NO, 2 cm lengths of small diameter silicone rubber tubing (0.51 mm i.d.; 0.94 mm o.d.) were sealed at one end with silicone rubber sealant. As shown in Fig. 2, a polytetrafluoroethylene (PTFE)-coated silver/silver chloride wire was employed as the reference electrode and a PTFE-coated Cu wire (bare wire o.d. 0.127 mm; PTFE coated wire o.d. 0.152 mm) served as the working electrode. The reservoir solution within the tubing was 1 M NaNO2, 0.138 M NaCl, 0.02 M EDTA, 0.5 M Na2HPO4, 0.5 M NaH2PO4, and the pH was adjusted to 6.8 with NaOH. Again, a high concentration of EDTA was used to guarantee there is enough free EDTA to shift the reaction towards providing a reproducible amount of NO during each pulse cycle. The PTFE was removed from the ends of Ag and Cu wires in 20 and 10 mm lengths, respectively. Both wires were coiled and were inserted and sealed in the silicone rubber sleeve in a manner that avoids direct metallic connection. Since the chloride activity in the internal solution differs from that used in Fig. 1 experiments, the applied voltages used in the cycle were altered to keep the potential of the copper working electrode at the desired values with respect to a NHE. The outer silicone rubber tubing plays a dual role by providing a barrier against the leaching of the nitrite and copper ions24 from the catheter, but allowing for the electrochemically generated NO gas to readily diffuse out. The NO flux from the surface of the SR tubing is dependent on the surface area of the copper wire and could be modulated by changing the length of the copper wire and/or concentration of nitrite electrolyte. As shown in Fig. 3, a catheter with a 1 cm long thin copper wire within was able to generate an average NO flux of 0.6 × 10−10 mol cm−2 min−1 when the voltage cycle is implemented, and zero flux in the absence of any applied voltage. The average flux reported in this case is based on surface area of the entire silicone rubber tubing, not the copper wire alone.
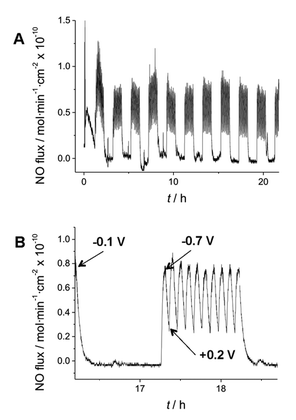 | ||
| Fig. 3 Electromodulated NO generation in silicone rubber catheter filled with 1 M NaNO2, 20 mM EDTA, 0.138 M NaCl in 1 M PBS (pH 6.8). (A) NO flux from surface of SR tubing over 24 h period, with the applied −0.7 V (3 min)/+0.2 V (3 min) pulse sequence initiated for 1 h every 2 h. (B) Expanded view of 2 h segment when voltage cycle is “off” and then turned “on” (oscillating between −0.7 V and 0.2 V vs. NHE) to create flux of NO from SR catheter surface. | ||
Higher NO flux can also be achieved by modulating the positive pulse potential as well as increasing the surface area of the copper electrode (data not shown). Further, measurement of copper ions in the solution in which an active catheter is placed for 7 d yields no detectable increase in trace copper ion content (as determined by ICP-ES) above that found in the solution to start. As shown in Fig. 3, control of the potential applied to the copper electrode can “turn on” and “turn off” the release of NO through the walls of the 0.94 mm diameter SR catheter tubing. An important advantage of this concept is that nitrite salts can be dissolved in high concentrations as reservoirs in the lumen of the catheter. Indeed, a 150 μm thick layer of 1 M nitrite solution is, in theory, enough to produce a 1 × 10−10 mol min−1 cm−2 NO flux continuously for at least 100 d in a planar or cylindrical arrangement.
To assess potential applications of this new approach, we performed bacterial biofilm prevention studies using the new electrochemical NO generation concept in complex biological environment. Two strains of bacteria were used, the widely adopted E. coli, and A. baumannii. The latter is of special importance, since it is known to show resistance to a majority of antibiotics.25 The experiment was performed over a one week period (see SI file, Fig. 3S, for experimental details†) with continuous flow of media using a CDC bioreactor. Four NO generating catheters were immersed in the media containing either E. coli or A. baumannii. Two of the electrodes were connected to the amperometric workstation and a continuous potential program was applied, 3 min at −0.97 V vs. Ag/AgCl (−0.7 vs. NHE) for reduction and −0.07 V (+0.2 vs. NHE) vs. Ag/AgCl for liberation of NO. The other two catheters were used as controls (containing the same nitrite salt solution, but not linked to the potentiostats). After 7 days incubation, four catheters were taken out. One NO generating catheter and one control catheter were used for detection of viable bacteria on the catheter surfaces by plate counting method. The other NO generating and control catheters were used for imaging. As shown in Fig. 4, the bacterium counting results after 7 d show that the NO generating catheter that releases NO had 97% less viable E. coli on its surfaces. A larger decrease, 98%, in the number of A. baumannii could be observed, even in the presence of low NO flux (0.6 × 10−10 mol cm−2 min−1) from catheter surface. Micrographs also confirmed this observation (see Figure 3S in SI file for fluorescence images of catheters).
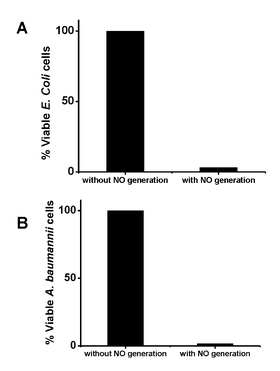 | ||
| Fig. 4 Effect of electrogeneration of NO from catheter surface on biofilm formation after 7 d for E. coli (A) and A. baumannii (B) using CDC bioreactor with continuous flow of media. | ||
In summary, the results indicate that it is possible to generate NO efficiently from nitrite using copper electrodes using a two-step voltage cycle involving reductive cleaning of the copper working electrode, followed by anodic liberation of Cu(I) ions by applying a positive potential pulse to the copper electrode. This electrochemical process can be potentially employed to modulate the amount of NO release from polymer surfaces used to prepare indwelling catheters and should extend the longevity of potential catheter placement with a given supply of NO precursors (nitrite). However the biggest advantage of this electrochemical NO generation scheme is its inherent ability to produce the desired amount of NO on demand for a given period of time, which may also yield extended use lifetimes for catheters and other biomedical devices. Indeed, when the NO is produced at high enough levels in short pulses at a defined frequency to achieve desired biological effects (e.g., inhibit adhesion of bacteria and platelets to polymer surfaces, disperse microbial biofilms, etc.), the total moles of NO required for delivery over a given time period can be much less than with continuous release NO donor chemistry employed to date.4–13 Experiments to test dual lumen catheters with electrochemical NO generation in one lumen for their ability to prevent both biofilm formation and platelet activation/adhesion when implanted in the blood vessels of animals are currently in progress. In addition, fluorescence-based mapping of the NO flux distribution across the dual-lumen catheter is also under investigation.
Acknowledgements
This research was supported by the National Institutes of Health (EB-00783 and EB-004527).References
- L. J. Ignarro, G. M. Buga, K. S. Wood, R. E. Byrns and G. Chaudhuri, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 9265–9265 CrossRef CAS.
- M. W. Radomski, R. M. Palmer and S. Moncada, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5193–5197 CrossRef CAS.
- C. F. Nathan and J. B. Hibbs, Curr. Opin. Immunol., 1991, 3, 65–70 CrossRef CAS.
- M. H. Schoenfisch and E. M. Hetrick, Chem. Soc. Rev., 2006, 35, 780–789 RSC.
- M. E. Meyerhoff and M. C. Frost, Anal. Chem., 2006, 78, 7370–7377 CrossRef.
- G. M. Annich, J. P. Meinhardt, K. A. Mowery, B. A. Ashton, S. I. Merz, R. B. Hirschl, M. E. Meyerhoff and R. H. Bartlett, Crit. Care Med., 2000, 28, 915–920 CrossRef CAS.
- H. A. Zhang, G. M. Annich, J. Miskulin, K. Stankiewicz, K. Osterholzer, S. I. Merz, R. H. Bartlett and M. E. Meyerhoff, J. Am. Chem. Soc., 2003, 125, 5015–5024 CrossRef CAS.
- T. C. Major, D. O. Brant, M. M. Reynolds, R. H. Bartlett, M. E. Meyerhoff, H. Handa and G. M. Annich, Biomaterials, 2010, 31, 2736–2745 CrossRef CAS.
- P. S. Fleser, V. K. Nuthakki, L. E. Malinzak, R. E. Callahan, M. L. Seymour, M. M. Reynolds, S. I. Merz, M. E. Meyerhoff, P. Bendick, G. Zelenock and C. Shanley, J. Vasc. Surg., 2004, 40, 803–811 CrossRef.
- P. G. Parzuchowski, M. C. Frost and M. E. Meyerhoff, J. Am. Chem. Soc., 2002, 124, 12182–12191 CrossRef CAS.
- M. C. Frost, M. M. Reynolds and M. E. Meyerhoff, Biomaterials, 2005, 26, 1685–1693 CrossRef CAS.
- M. M. Reynolds, J. A. Hrabie, B. K. Oh, J. K. Politis, M. L. Citro, L. K. Keefer and M. E. Meyerhoff, Biomacromolecules, 2006, 7, 987–994 CrossRef CAS.
- Z. Zhou and M. E. Meyerhoff, Biomacromolecules, 2005, 6, 780–789 CrossRef CAS.
- B. K. Oh and M. E. Meyerhoff, Biomaterials, 2004, 25, 283–93 CrossRef CAS.
- M. J. A. Shiddiky, A. P. O'Mullane, J. Zhang, L. D. Burke and A. M. Bond, Langmuir, 2011, 27, 10302–11 CrossRef CAS.
- M. Štulíková and F. Vydra, J. Electroanal. Chem., 1973, 44, 117–127 CrossRef.
- H. H. Strehblow and B. Titze, Electrochim. Acta, 1980, 25, 839–850 CrossRef CAS.
- S. Fletcher, J. Electrochem. Soc., 1978, 125, 1960 CrossRef CAS.
- R. L. Deutscher and R. Woods, J. Appl. Electrochem., 1986, 16, 413–421 CrossRef CAS.
- H. Leckie, J. Electrochem. Soc., 1970, 117, 1478 CrossRef CAS.
- J. M. M. Droog, C. A. Alderliesten, P. T. Alderliesten and G. A. Bootsma, J. Electroanal. Chem., 1980, 111, 61–70 CrossRef CAS.
- K. Srinivasan and R. S. Subrahmanya, J. Electroanal. Chem., 1971, 31, 257–263 CrossRef CAS.
- R. Belcher, D. Gibbons and T. S. West, Anal. Chim. Acta, 1955, 12, 107–114 CrossRef CAS.
- N. Donaldson, P. Baviskar, J. Cunningham and D. Wilson, J. Biomed. Mater. Res. A, 2012, 100, 588–598 CrossRef.
- J. H. Shin, H. W. Lee, S. M. Kim and J. Kim, J. Microbiol., 2009, 47, 728–735 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details on how NO fluxes were determined from bare electrodes and SR catheter devices, as well as methods to assess biofilm formation over a 7 d period. See DOI: 10.1039/c2ra20853a |
| This journal is © The Royal Society of Chemistry 2012 |