Compressibility and structural stability of nanoparticulate goethite
Sandra
Fernando†
,
Meredith
Baynes
,
Bin
Chen
,
Jillian F.
Banfield
and
Hengzhong
Zhang
*
Department of Earth and Planetary Science, University of California, Berkeley, California 94720, USA. E-mail: heng@eps.berkeley.edu
First published on 21st May 2012
Abstract
Synchrotron high-pressure X-ray diffraction was used to study the compressibility and structural stability of synthetic 17 nm nanoparticulate goethite at pressures up to ∼30 GPa. The lattice parameters and the unit cell volume were determined. We observed much higher contraction along c than a and b (Pnma space group), which we attribute to relatively weak hydrogen bonding along the c axis. In the hydrostatic pressure regime (pressures up to ∼10 GPa) the bulk modulus, K0, of 17 nm goethite was determined to be 109.4 ± 2.2 GPa. The compressibility of the 17 nm goethite is comparable to that of the bulk goethite. Amorphization of the nano-goethite began at ∼17 GPa, an effect not observed in bulk goethite, even at much higher pressures. The findings indicate that for 17 nm particles, the size-dependence of goethite compressibility is small at low to moderate pressures, but at very high pressure, surface effects on nanoparticle structure become very significant.
1. Introduction
Nanoparticles are abundant in the environment, formed via both inorganic and biologic processes. They are nanometer-scale assemblies of atoms with dimensions in between ca. 1–100 nm and can differ in their atomic structure, reactivity, and electronic and magnetic properties relative to larger crystallites.1–4Interest in nanoparticle stability, surface phenomena and interactions in natural systems and materials technology motivates research on both the bulk (e.g., compressibility) and the surface properties (e.g., the surface free energy), as they combine to determine the thermodynamic phase stability and affect kinetic processes such as nucleation and crystal growth. The total surface free energy is expected to increase as particle size decreases because the fraction of ions exposed at the surface increases and these sites have less optimal coordination geometries. Thus, nanoparticles should be substantially more reactive than the related bulk material.
Under compression, nanoparticles may undergo volume contraction, phase change, and/or amorphization. The bulk modulus measures the resistance of a structure to compression. Its dependence on particle size stems in part from the same changes in the surface site coordination environment (e.g., in coordination number, protonation state, or displacement of atoms relative to their position in the bulk) as those that influence surface free energy.3 Measurements of the bulk modulus are useful, as they can be used to derive the surface free energy of nanoparticles.5
Goethite (α-FeOOH, space group Pnma, a = 9.9134 Å, b = 3.0128 Å, and c = 4.5800 Å at room temperature; see ref. 6) is a common nanoparticulate Fe-bearing mineral found in water, soils, sediments, and altered rocks near the Earth's surface. It also forms by phase transformation from ferrihydrite and during early crystallization of the intracellular magnetite crystals in magnetotactic bacteria7,8 and is often associated with cell surfaces and natural organic compounds (e.g.ref. 9). Currently, little is known about the bulk modulus and the surface free energy of goethite nanoparticles, although the bulk modulus of bulk goethite (micron-sized or larger) has been determined previously.10–12 Acquisition of these fundamental quantities for nano-goethite will help achieving a comprehensive understanding of size-dependent phenomena in the iron oxyhydroxide system. Here, we studied the structural response of nano-goethite to compression and decompression in order to quantify the effect of particle size on the bulk modulus. We also evaluated the impact of small particle size on pressure-induced amorphization.
2. Experimental
Goethite nanoparticles were synthesized by titrating 0.5 M 30 mL Fe(NO3)3 with 2.5 M 125 mL KOH at a rate of ∼10 mL min−1 under magnetic stirring.13 The formed suspension was aged at 60 °C for 100 h and then cooled down to room temperature. The precipitates were separated from the solution via six cycles of centrifugation at 8000 rpm and washing using deionized water. The precipitates were dried at 40 °C overnight and stored in a vial.The phase of the synthesized sample was determined using X-ray diffraction (XRD). XRD was conducted in a PANalytical X'Pert Pro diffractometer operated at 40 keV and 40 mA with a Co Kα radiation (wavelength 1.7903 Å). The XRD pattern was collected in the 2θ range 20–90° at a scanning rate of 1° min−1.
High-pressure X-ray diffraction (HPXRD) of the sample was performed at Beamline 12.2.2, Advanced Light Source (ALS), Berkeley, California. High pressure (up to tens of GPa) was achieved in a diamond anvil cell (DAC). A spring steel gasket with a drilled hole ∼100 μm in diameter was placed between a pair of ∼300 μm diameter culet diamonds, forming a micro sample chamber. A small amount of the sample was loaded into the cell with several grains of ruby as the internal pressure calibrator. A mixture of methanol and ethanol in a volume ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used as the pressure medium. Pressure was measured using pressure-dependent ruby fluorescence spectra under laser irradiation. Sample pressure was increased stepwise to a maximum value of ∼30 GPa. Decompression was done at larger pressure intervals. XRD patterns were collected at 30 keV X-ray beam energy (X-ray wavelength 0.4132 Å) and a beam-spot size of 25 × 25 μm2 with a MAR345 image plate. Exposure time was 5 min per XRD pattern. The collected diffraction images were integrated along the Debye–Scherrer rings and converted to the two-dimensional intensity vs. scattering angle data using the FIT2D program.14
:1 was used as the pressure medium. Pressure was measured using pressure-dependent ruby fluorescence spectra under laser irradiation. Sample pressure was increased stepwise to a maximum value of ∼30 GPa. Decompression was done at larger pressure intervals. XRD patterns were collected at 30 keV X-ray beam energy (X-ray wavelength 0.4132 Å) and a beam-spot size of 25 × 25 μm2 with a MAR345 image plate. Exposure time was 5 min per XRD pattern. The collected diffraction images were integrated along the Debye–Scherrer rings and converted to the two-dimensional intensity vs. scattering angle data using the FIT2D program.14
3. Results and discussion
Comparison of the peak positions in the XRD pattern with those predicted for bulk goethite showed that the synthesized sample is goethite (Fig. 1). Significant broadening of the diffraction peaks indicates that the goethite is nanoparticulate. Rietveld fitting (Fig. 1) of the XRD pattern using the program Maud15 indicated that the average particle size of the goethite nanoparticles is 16.6 nm in diameter (the sample is referred to as 17 nm goethite hereafter). The Rietveld fitting assumes that the crystal shape is spherical, yet goethite nanoparticles are often acicular.16 Some peaks in Fig. 1 (e.g., 210) are sharper than predicted by the fitting, indicating that particles are much larger normal to these planes. Previous analysis has shown that the Rietveld size for goethite corresponds to the short particle dimension (lath width).17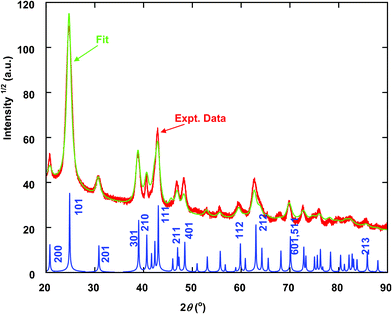 | ||
| Fig. 1 XRD pattern (thick red line) and Rietveld fit (thin green line) of synthesized goethite sample. Calculated XRD pattern of bulk goethite is shown at the bottom for comparison. X-ray wavelength 1.7903 Å. | ||
Fig. 2 shows experimental HPXRD patterns. The diffraction peaks (e.g., (111)) of nano-goethite shift right (lower d spacing) with increasing pressure in compression, and shift left with decreasing pressure in decompression. Peaks that do not shift with pressure (dashed vertical lines in Fig. 2) come from the gasket and/or the instrument: the 2θ = 11.7° peak was from BCC Fe (110), and the 2θ = 5.2 and 6.0° peaks from scattering of the beam pinholes and/or the beam stop. The latter was identified from several weak and low-angle diffraction spots in the original 2D diffraction images. These peaks can be well resolved from sample peaks in fitting (below).
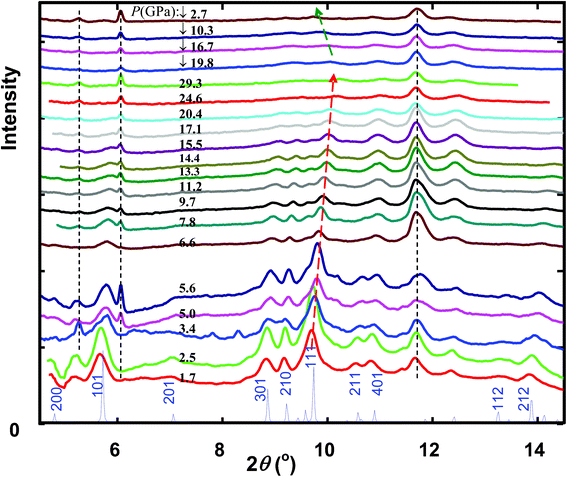 | ||
| Fig. 2 XRD patterns of nano-goethite under compression (1.7–29.3 GPa) and decompression (19.8–2.7 GPa, indicated by downward arrow). Calculated XRD pattern of bulk goethite at ambient pressure is shown at the bottom for comparison. The dashed vertical lines indicate diffraction peaks from the gasket/instrument (see text); the dashed and arrowed lines indicate the shift of the (111) peak with the pressure. X-ray wavelength 0.4132 Å. | ||
Fig. 2 shows that with increased pressure, some prominent peaks such as (101), (301), (210) and (111) decrease in intensity and broaden. This may be due to introduction of strain in the nanoparticles under compression. Decrease in peak intensity is more obvious at pressures above 17.1 GPa. For instance, at 20.4 GPa, the (301) and (210) peaks are barely recognizable. These peaks could not be recovered with the original intensity in decompression (Fig. 2). We interpret this to indicate the onset of amorphization at around 17 GPa. In contrast, in bulk goethite samples, amorphization was not observed even at ∼24 GPa (ref. 10) or ∼30 GPa (ref. 11). In small particles, surface strain causes loss of periodic structure at and near nanoparticle surfaces through bonding distortion and defect formation.18,19 Our results indicate that high pressure causes radial propagation of strain into the nanoparticles until they appear amorphous, an effect not observed in the absence of high surface area.
Diffraction peaks were fitted using Pearson VII functions in the PeakFit Program (Jandel, San Rafael, CA) and the 2θ values were obtained (see an example in Fig. 3). The corresponding d-values of the diffraction peaks were calculated from Bragg's law (using λ = 0.4132 Å for the synchrotron radiation). Lattice parameters were calculated from the prominent goethite diffraction peaks (101), (301), (210), (111), (401), and (212) using linear regression. The derived unit cell parameters are shown in Fig. 4a as a function of the pressure. In regression for the first three lower pressures the (201) d-value was also used, which reduced the regression errors (see Fig. 4a). This peak was excluded from calculations at higher pressures because it was substantially reduced in intensity. The maximum relative errors in the determined d-values are all ∼1%. The lattice parameters at ambient pressure were not determined, since a low pressure (usually ∼1–2 GPa) was needed to securely retain the pressure medium inside the DAC sample chamber once a sample was loaded. These parameters were obtained by linear extrapolation to 0 GPa of the a, b, and c at pressures less than 7 GPa: a0 = 10.038 Å, b0 = 3.0331 Å and c0 = 4.6130 Å.
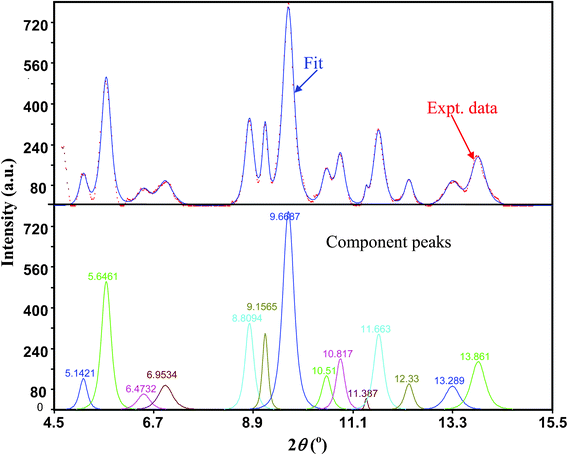 | ||
| Fig. 3 Top panel: fitting (blue solid line) of the XRD pattern (red dotted line) using Pearson VII functions at 1.7 GPa after multi-section background subtraction. Bottom panel: component peaks and the corresponding positions from the fit. X-ray wavelength 0.4132 Å. | ||
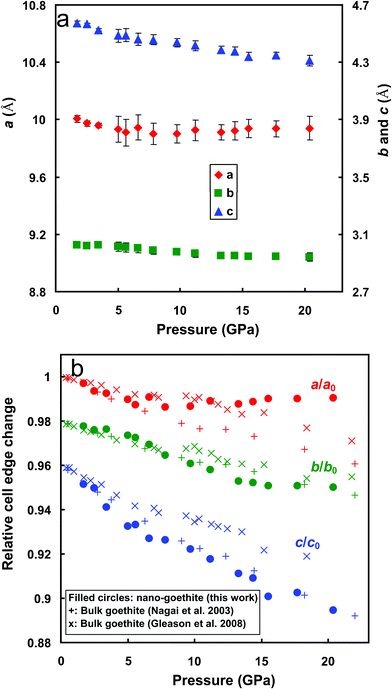 | ||
| Fig. 4 Lattice parameters (a) and their normalized values (b) of nano-goethite as a function of pressure (data points for lattice parameters b and c were shifted down 0.02 and 0.04 units, respectively, for clarity). Vertical bars in (a) indicate standard errors. Data from ref. 10 and 11 are included for comparison in (b). | ||
Lattice parameters normalized using the room pressure values reveal the relative magnitude of lattice contraction along each axis (Fig. 4b). Results show that c is most compressible. Contractions along a and b are similar, as observed in bulk goethite,10,11 though the magnitudes observed by Gleason et al.11 are generally smaller than those by Nagai et al.10 The discrepancies in the degrees of contractions from the three works (Fig. 4b) may be due to the differences in the particle sizes and/or grain sizes of the samples. The higher contraction along c is expected,20,21 given that goethite is made up of double chains of edge-sharing FeO6 octahedra with channels between the chains that run parallel to the b-axis (see Fig. 5a). H-bonds across the channels are approximately parallel to the c-axis (see Fig. 5b). Unlike in layered structures, the overall compression of bulk goethite occurs by shortening of both the ionic bonds between iron and oxygen and the hydroxyl bonds.10,21 Since hydrogen bonds are weaker than Fe–O bonds, most compression occurs along the c axis (Fig. 4b).
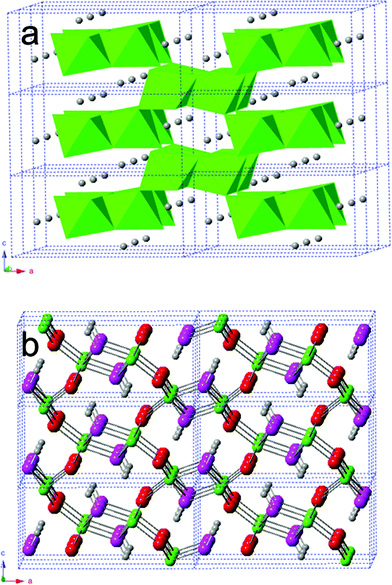 | ||
| Fig. 5 Structure of a 2 × 3 × 3 super cell of goethite in the polyhedral model (a) and in the ball-and-stick model (b). O1: red balls; O2: pink balls; Fe: green balls; H: gray balls. | ||
Fig. 4b shows deflection in the variation of lattice parameters with pressure above ∼10 GPa, especially for a. This was explained by non-hydrostatic compression due to solidification of the methanol–ethanol pressure medium.10,22 Medium solidification shields compression, thus the unit cell contracts less than expected based on the applied pressure. This effect differs in strength in different crystallographic directions, and is most obvious along [100] for the nano sample (Fig. 4b).
The bulk modulus (K0) was derived using the unit cell volume–pressure data according to the second order Birch–Murnaghan equation of state (EOS):23
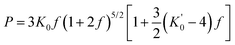 | (1) |
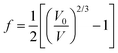 is the volumetric strain with V0 being the unit cell volume at room pressure (V0 = a0b0c0 = 140.45 Å3) and V the unit cell volume at pressure P (V = abc). Commonly, K0′ is set to 4.0.10,20 With this assumption, K0 = P/[3f(1 + 2f)5/2]. Thus, for the 17 nm goethite, the bulk modulus was obtained by averaging all the K0 derived in the pressure range 0–10 GPa: K0 = 109.4 ± 2.2 GPa (standard deviation). Experimental data above ∼10 GPa were not used in deriving K0 due to evidence for non-hydrostatic compression (as seen from the apparent deflection in the unit cell volume vs. pressure plot for both nano and bulk samples in Fig. 6).10
is the volumetric strain with V0 being the unit cell volume at room pressure (V0 = a0b0c0 = 140.45 Å3) and V the unit cell volume at pressure P (V = abc). Commonly, K0′ is set to 4.0.10,20 With this assumption, K0 = P/[3f(1 + 2f)5/2]. Thus, for the 17 nm goethite, the bulk modulus was obtained by averaging all the K0 derived in the pressure range 0–10 GPa: K0 = 109.4 ± 2.2 GPa (standard deviation). Experimental data above ∼10 GPa were not used in deriving K0 due to evidence for non-hydrostatic compression (as seen from the apparent deflection in the unit cell volume vs. pressure plot for both nano and bulk samples in Fig. 6).10
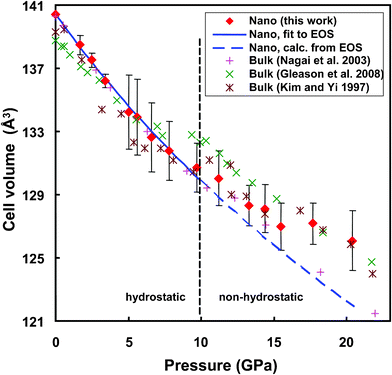 | ||
| Fig. 6 Variation of the unit cell volume of nano-goethite with pressure. Diamonds: experimental data; solid line: calculated using EOS (eqn (1)) at P < 10 GPa; dashed line: extrapolation of the EOS at P > 10 GPa; vertical bars indicate standard errors. Data from ref. 10–12 for bulk goethite are included for comparison. | ||
Fig. 6 compares the calculated and measured unit cell volumes of 17 nm goethite as a function of pressure. The calculation was based on eqn (1) using the determined K0. It is seen that the EOS fits the data well in the hydrostatic pressure region (<∼10 GPa). Above ∼10 GPa, the measured unit cell volume is above the EOS value, as expected if compression is non-hydrostatic.
For bulk goethite, Nagai et al. obtained K0 = 108.5 GPa with data below 11 GPa and K0 = 111 GPa with data in P up to 25 GPa (K0′ = 4).10 Using data at pressures greater than ∼10 GPa, the bulk modulus would be greater21,24 due to non-hydrostatic compression. In two other studies,11,12K0 of bulk goethite was reported as 140.3 GPa at K0′ = 4.6 (ref. 11) and 147.9 GPa (ref. 12), both of which were based on data measured at P up to ∼28 GPa. Non-hydrostatic compression is apparent in their reported data at P > ∼8 GP for the room temperature data set in ref. 11 and at P > ∼10 GPa in ref. 12, as seen from the obvious shifts up in the unit cell volume (see Fig. 6). This explains their higher K0 values than that of Nagai et al.10
The bulk modulus (109.4 GPa) of 17 nm goethite is comparable to that (108.5 GPa) of bulk goethite by Nagai et al.10 This result indicates the size effect on the bulk modulus of goethite is minimal. Prior determinations of the bulk modulus of nano-titania showed that with decreasing particle size, the bulk modulus increases gradually; reaches a maximum at ∼15 nm; then decreases dramatically.25 Thus, further HPXRD study of a series of goethite nanoparticles is needed (and underway) in order to investigate the full size-dependence of the compressibility of nano-goethite.
4. Conclusions
Compression of 17 nm goethite is anisotropic and occurs preferentially in the direction parallel to H-bonds and normal to the double octahedral chains. Non-hydrostaticity arising from solidification of methanol–ethanol pressure medium at above ∼10 GPa causes apparent stiffening of nanocrystals, which reduces the degree of lattice contraction at higher pressures. The bulk modulus of 17 nm goethite is 109.4 ± 2.2 GPa at fixed K0′ = 4 in the second order Birch–Murnaghan equation, slightly higher than that of bulk goethite. The compressibility of 17 nm goethite is comparable to that of bulk goethite at this particular size. Nonetheless, amorphization of 17 nm goethite began above ∼17 GPa, a phenomenon not observed in bulk goethite, even at ∼24 GPa10 or 30 GPa.11Acknowledgements
Financial support was provided by the National Science Foundation (Grant no. EAR-0920921) and U.S. Department of Energy (Grant no. DE-AC02-05CH11231). Synchrotron HPXRD was done at Beamline Station 12.2.2, Advanced Light Source, Berkeley, California. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We thank Dr Jinyuan Yan and Mr. Jason Knight for assistance in the beamline experiments.References
- H. Gleiter, Prog. Mater. Sci., 1989, 33, 223 CrossRef CAS.
- K. Lu, Materials Science and Engineering, 1996, R16, 161 Search PubMed.
- J. F. Banfield and H. Zhang, In Reviews in Mineralogy and Geochemistry, vol. 44, 2001, Mineralogical Society of America and Geochemical Society, Washington, D.C., p. 1 Search PubMed.
- G. A. Waychunas, C. S. Kim and J. F. Banfield, J. Nanopart. Res., 2005, 7, 409 CrossRef CAS.
- H. Zhang, B. Chen and J. F. Banfield, Phys. Chem. Chem. Phys., 2009, 11, 2553 RSC.
- A. F. Gualtieri and P. Venturelli, Am. Mineralogist, 1999, 84, 895 CAS.
- A. M. T. Bell, V. S. Coker, C. I. Pearce, R. A. Pattrick, G. van der Laan and J. R. Lloyd, Zeitschrift für Kristallographie Supplements, 2007, 26, 426 Search PubMed.
- V. S. Coker, A. M. T. Bell, C. I. Pearce, R. A. D. Pattrick, G. van der Laan and J. R. Lloyd, Am. Mineral., 2008, 93, 540 CrossRef CAS.
- C. S. Chan, G. De Stasio, S. A. Welch, M. Girasole, B. H. Frazer, M. Nesterova, S. Fakra and J. F. Banfield, Science, 2004, 303, 1656 CrossRef CAS.
- T. Nagai, H. Kagi and T. Yamanaka, Am. Mineralogist, 2003, 88, 1423 CAS.
- A. E. Gleason, R. Jeanloz and M. Kunz, Am. Mineral., 2008, 93, 1882 CrossRef CAS.
- Y. H. Kim and Z. Yi, J. Miner. Soc. Korea, 1997, 10, 75 Search PubMed.
- L. Mazeina and A. Navrotsky, Clays Clay Miner., 2005, 53, 113 CrossRef CAS.
- A. P. Hammersley, S. O. Svensson, M. Hanfland, A. N. Fitch and D. Häusermann, High Pressure Res., 1996, 14, 235 CrossRef.
- L. Lutterotti and M. Bortolotti, IUCr: Compcomm Newsletter, 2003, 1, 43 Search PubMed Available at http://www.ing.unitn.it/~maud/.
- D. J. Burleson and R. L. Penn, Langmuir, 2006, 22, 402 CrossRef CAS.
- H. Zhang, M. Bayne, S. Fernado, B. Legg, M. Zhu, R. L. Penn and J. F. Banfield, J. Phys. Chem. C, 2011, 115, 17704 CAS.
- D. Machon, M. Daniel, P. Bouvier, S. Daniele, S. L. Floch, P. Melinon and V. Pischedda, J. Phys. Chem. C, 2011, 115, 22286 CAS.
- V. Pischedda, G. R. Hearne, A. M. Dawe and J. E. Lowther, Phys. Rev. Lett., 2006, 96, 035509 CrossRef CAS.
- K. D. Grevel, M. Burchard and D. W. Fasshauer, J. Geophys. Res., 2000, 105, 27877 CrossRef CAS.
- J. Xu, J. Hu, L. Ming, E. Huang and X. Hongsen, Geophys. Res. Lett., 1994, 21, 161 CrossRef CAS.
- R. J. Angel, M. Bujak, J. Zhao, G. D. Gatta and S. D. Jacobson, J. Appl. Crystallogr., 2007, 40, 26 CrossRef CAS.
- F. Birch, J. Geophys. Res., 1978, 83(B3), 1257 CrossRef CAS.
- S. E. Bradbury and Q. Williams, Am. Mineralogist, 2003, 88, 1460 CAS.
- B. Chen, H. Zhang, K. A. Dunohy-Guzman, D. Spagnoli, M. B. Kruger, D. V. S. Muthu, M. Kunz, S. Fakra, J. Z. Hu, Z. Guo and J. F. Banfield, Phys. Rev. B, 2009, 79, 125406 CrossRef.
Footnote |
| † Current address: Department of Earth and Environmental Sciences, University of Michigan Ann Arbor, 2534 C.C. Little Bldg., 1100 N. University Avenue, Ann Arbor, MI 48109-1005, USA |
| This journal is © The Royal Society of Chemistry 2012 |