Peculiarities of quasi-aromatic hydrogen bonding†
Agata
Martyniak
,
Irena
Majerz
and
Aleksander
Filarowski
*
Faculty of Chemistry, University of Wrocław, Joliot-Curie 14, PL 50-383 Wrocław, Poland. E-mail: aleksander.filarowski@chem.uni.wroc.pl
First published on 6th July 2012
Abstract
The analysis of the calculated topological and energetic characteristics is presented for the hydroxyaryl, alkyl Schiff and Mannich bases. The quantum theory of the atoms-in-molecules methodology has been used to explore the topology of the electron density at the bond and ring critical points in the studied compounds with intramolecular hydrogen bonding. The dependencies between the electron-topological, aromatic, and energetic parameters under tautomeric equilibrium have been analysed. The calculated non-adiabatic potential curves show the difference between the unconjugated and π-conjugated compounds. The calculated dependency between the binding energy of hydrogen bonding (ΔEHB) and the aromaticity of the chelate chain (the HOMA aromaticity index) reveals a difference between the two types of compounds. The results obtained have made it possible to figure out a basic difference between unconjugated and π-conjugated compounds.
Introduction
This paper presents a study on the impact of the proton transfer in quasi-aromatic hydrogen bonding on the electron density at the critical points of bonds and rings as well as on the aromaticity of the chelate and aryl formations. The proton transfer process plays an important role in biological activity,1,2 therefore the description of this phenomenon by means of the quantum theory of atoms-in-molecules theory (QTAIM),3,4 aromaticity,5,6 and potential energy surface7–11 is a significant task. It is noteworthy that the terms “hydrogen bond” and “proton transfer” are highly cited in the natural sciences.12 However, no perfect theoretical model describing these phenomena exists so far. The intensive discussion relating to the existence of the resonance assisted hydrogen bond (RAHB)13–42 serves as an example. The pioneer investigations of quasi-aromatic hydrogen bonding belong to Shigorin43 who introduced an additional component (π-electronic energy, ΔEπ) into the general formula for the estimation of hydrogen bond energy.44 The explanation of the strengthening of hydrogen bonding by resonance (based upon X-ray research and valence bond theory13–23) has led to some contradictions in this area over the last decade.30–35 The existence of the RAHB was put into question due to the quantum-mechanical calculations of compounds with (π-conjugated) and without (unconjugated) quasi-aromatic hydrogen bond formation.30–35. In Ref. 30 and 31 the principal difference in the strength of hydrogen bonds between saturated and unsaturated compounds is shown on the basis of calculations of the 2hJO–O and 2hJN–N spin–spin coupling constants.However, quite the opposite conclusion is presented in Ref. 40, where the NMR spectroscopic σ and J parameters of the malonaldehyde, nitromalonaldehyde, and nitromalonamide are analysed. The next stage of computational research into the RAHB was reported in Ref. 32–35, and concluded that there is no difference of principle between the structural parameters of hydrogen bonding in the unconjugated (non-quasi-aromatic) and π-conjugated (quasi-aromatic) compounds. However, despite some criticism towards the RAHB model and the explanation of related facts, there are experimental data proving the impact of π-electronic coupling in the chelate chain on the proton vibrations in intramolecular hydrogen bonding. Spectroscopic experimental research45,46 reveals a visible decrease in the integral intensity of the stretching vibration of the X–H bond (the derivative of a dipole moment on a normal coordinate, ∂μ/∂R) in compounds with quasi-aromatic intramolecular hydrogen bonding (o-hydroxyaryl Schiff bases) compared to those without π-electronic coupling (o-hydroxyaryl Mannich bases).45 This phenomenon is conditioned by the redistribution of a charge in the chelate chain, e.g. π-electron delocalization in an OCCCN chain. In non-π-electronic conjugated hydrogen bonding, the movement of a proton is co-directed with the movement of charge along the X–H bond, whereas in quasi-aromatic hydrogen bond formation, proton movement brings about both the charge reorganization in the chelate chain and the mutual compensation of dipole moments of the hydrogen bridge and the chelate chain. Notably, this explanation does not contradict the conclusions in Ref. 47 and 48. The reduction of the integral intensity of the ν(XH) bond for compounds with quasi-aromatic formation is also presented in Ref. 49–51. However, one should keep in mind that an increase in integral intensity is accompanied by the strengthening of quasi-aromatic bonding52 but not by the weakening.53
The phenomena described above refers to the intramolecular hydrogen bond, whereas in Ref. 15 and 16 it testifies to the existence of RAHB in complexes with hydrogen bonding. The logical grounding of the strength of intermolecular hydrogen bonding has been given by Beck and Mó27 who point out a significant contribution of polarization in hydrogen bonding. However, there is no doubt that polarization in particular fragments of the complex (the proton-donor and the proton-acceptor molecules) provides an additional stabilizing component. Moreover, Bickelhaupt et al.54–57 estimated the resonance contribution toward the strength of intermolecular hydrogen bonding as being mediocre (1.7–4.8 kcal mol−1).
It is noteworthy that a significant shortening of the length of the hydrogen bridge in o-hydroxyaryl ketimines58 and malonaldehydes39,59,60 analysed in the literature does not discard the existence of RAHB. In these compounds, quasi-aromatic hydrogen bond formation undergoes a serious reduction due to a steric effect which surpasses the resonance effect. Remarkably, o-hydroxyaryl ketimines (compounds with a strong steric squeezing) were referred to as quasi-aromatic hydrogen bond formations because of a small integral intensity of the ν(XH) mode.52 The description of quasi-aromatic hydrogen bonding by means of the RAHB model requires a further development. Therefore, the study of quasi-aromatic formations should be developed in order to determine the existence of resonance assisted hydrogen bonding.
To clarify the nature of the hydrogen bond and the impact of the proton transfer process on the electronic state and the aromatic formation of the molecule we have selected hydroxyalkyl and aryl Schiff bases (Scheme 1), which are the samples with quasi-aromatic hydrogen bonding. For the purpose of comparison with the compounds possessing π-electronic conjugation, hydroxyalkyl and aryl Mannich bases are chosen. These compounds are not quasi-aromatic formations due to the isolation of proton-acceptors from proton-donors by a methene bridge (Scheme 1).
 | ||
| Scheme 1 Diagram of studied compounds. | ||
Computational details
The calculations were performed with Gaussian 0961 sets of code using the 6-311++G(d,p) basis set62 at the hybrid Hartree–Fock density functional (B3LYP).63 The use of diffuse functions is the accepted approach for studies of hydrogen bonding.64 The one-dimensional approach was employed to perform the calculations at selected points along the proton transfer reaction path. The approach is based on the stepwise elongation of the XH bond length with full optimisation of the remaining structural parameters. This approach reflects the proton transfer process in the intramolecular hydrogen bridge.10 QTAIM analysis was performed using the AIM2000 program65 with all default options.To describe the aromatic and quasi-aromatic formations, the harmonic oscillator model of aromaticity (HOMA) index was selected as the most reliable parameter in the description of hydrogen bonding and tautomeric equilibrium.66 For this purpose, the HOMA index67 was applied to determine the degree of aromaticity.
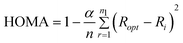
| (1) |
Results and discussion
Interrelation between potential energy function, aromaticity and structural parameters
The presented results are based on the calculated dependencies between the energetic, aromatic, and electron-topological parameters. The analysis of such dependencies makes it possible to elucidate the physicochemical nature of quasi-aromatic hydrogen bonding. The dependence of the potential energy on the length of the OH or HN bond (ΔE = f(d(OH or HN))) (Fig. 1), which describes the change in molecule energy under the proton transfer process, is the curve with two minima for the π-conjugated compounds (S1–S6, S8–S10) and one minimum for the unconjugated compounds (M1–M10, S7, S11). The experimental and theoretical studies in Ref. 68–72 disclose the fact that the intramolecular proton transfer is described by the OH and HN tautomeric forms for Schiff bases. Meanwhile for Mannich bases the proton transfer process is conditioned by the disruption of the intramolecular hydrogen bond and the formation of the dimer.73 The collation of the calculated and experimental data confirms that the ΔE = f(d(XH)) dependence verifies the presence of the tautomeric equilibrium in the experiment. It is noteworthy that the shape (the degree of asymmetry) of the potential curve is a measure of the strength of hydrogen bonding.74–80 An increase of the potential curve asymmetry (with the exception of low barrier hydrogen bonds), for example the transition from the potential curve M4 to the potential curve S4, exposes a stronger interaction in the hydrogen bond (Fig. S1†). Symmetric double-well potential curves are observed for the S2 and S3 compounds (the potential curves with almost equal minima, ΔEPT = 0.41 and 0.45 kcal mol−1, respectively, Table 1). This fact supports the presence of the strongest intramolecular hydrogen bond in the studied compounds. Hydrogen bridges with a small energetic barrier (ΔETS ≤ 2–3 kcal mol−1)81 for proton transfer are known as strong short hydrogen bonds (SSHB), low barrier hydrogen bonds (LBHB),82 and Speackman-Hadži hydrogen bonds.83,84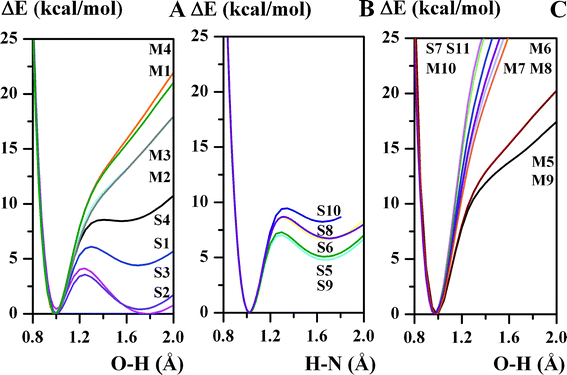 | ||
| Fig. 1 Calculated (B3LYP/6-311++G(d,p)) non-adiabatic potential functions for proton transfer displacement for studied compounds. | ||
| Compound | ΔEPT (kcal mol−1) | ΔETS (kcal mol−1) |
|---|---|---|
| S1 | 4.4 | 6.1 |
| S2 | 0.5 | 4.1 |
| S3 | 0.4 | 3.6 |
| S4 | 8.4 | 8.6 |
| S5 | 5.1 | 7.3 |
| S6 | 6.7 | 8.8 |
| S8 | 6.7 | 11.9 |
| S9 | 4.8 | 10.4 |
| S10 | 8.3 | 9.5 |
The difference between the potential curves for the S2 and S3 compounds (the potential for the S4 compound is more asymmetric, Fig. 1A) is explained by the differences in the pKa values experimentally obtained for α- and β-naphtholic fragments.85 The acidity of the hydroxyl group of α-naphthol is larger than that of β-naphtols. A larger asymmetry of the potential curves is indicated between the phenolic (S1) and naphtholic (S2 and S3) Schiff base derivatives. A smaller asymmetry of the potential curve and the prevailing OH tautomeric form (global minimum is observed in the d(OH) ≅ 1 Å region) for the phenolic derivatives with respect to the naphthol derivatives is caused by a smaller acidity of the hydroxyl group of the phenolic fragment (pKa = 10 for phenol; pKa = 9.57 and 9.34 for β- and α-naphthols, respectively).85 The aforesaid shows an agreement between the experimental and calculated results. Similar trends are also obtained for Mannich bases (M1–M4) but with far smaller changes occurring in the asymmetry of the potential curve (Fig. 1A).
For exhaustive information on the impact of the adjacent (A) aromatic formation on the tautomeric equilibrium of the quasi-aromatic hydrogen bond formation, a number of calculations were accomplished. A complete lack of double bonds in the adjacent formation of unconjugated compounds brings about the single-well potential curve (the OH tautomeric form for S7, M6 and M7). For hydroxy Schiff base derivatives (S5, S6 and S8–S10), the doubling of two bonds in the chelate chain (the creation of a quasi-aromatic hydrogen bond formation)44 triggers the occurrence of the double-well potential curve with the HN form prevailing (Fig. 1B). It should be underlined that this situation (the HN form prevailing) does not change when one bond is doubled into the A formation (transition from S5 to S8–S10, Fig. 1B). However, the situation undergoes changes for the phenolic Schiff base derivative (S1) where the double-well potential curve is observed but with the OH tautomeric form prevailing (Fig. 1A). It is noteworthy that the presence of two double bonds in the chelate chain (the quasi-aromatic hydrogen bond formation) enhancing the π-electronic coupling between the acid and base centers, leads to the occurrence of the double-well potential curve. Meanwhile, the absence of single/double bond alteration in the OCCCN chain shows the single-well potential curve for both aliphatic (S7, S11 and M5–M10) and aromatic (M1–M4) derivatives. Therefore, the existence of the quasi-aromatic hydrogen bond formation in the Schiff bases causes the tautomeric equilibrium. This confirmation exposes the specific character of the compounds possessing quasi-aromatic hydrogen bonding. The impact of the adjacent formation reveals the following tendency: the growing number of double bonds in the adjacent formation in the conjugated compounds (S6 → S5 → S8–S10 → S1) provokes a change of the tautomeric equilibrium from the prevalence of the HN form (S5, S6, S8–S10) to the OH form (S1) (Fig. 1).
For a description of π-electronic delocalization in the chelate chain and aromatic formations, Krygowski's HOMA aromaticity index86 was chosen. In the studies,87–89 it was shown that an increase in aromaticity reflects the rising participation of π-electronic components in formation. Thus, the analysis of the aromaticity of the compounds is accompanied by the π-electronic coupling impact. Despite a significant increase in the number of papers dealing with this phenomenon,90–102 a more complete description of aromaticity under the step-by-step proton transfer in the intramolecular quasi-aromatic hydrogen bond formation was accomplished herein.
The marked differences in the aromaticity behaviour of the chelate chain under the proton transfer process are observed between π-conjugated and unconjugated compounds. Thus, the aromaticity of the chelate chain increases up to the region of the transition state, and then slightly decreases in the region of the HN form under stepwise proton transfer (Fig. 2A) in the S2 and S3 compounds with a symmetrical double-well potential and the compounds with an asymmetrical double-well potential with the OH form prevailing (S1, S4). For the S5, S6, S8–S10 π-conjugated compounds (with a visible prevalence of the HN tautomeric form having the double-well potential curve) the aromaticity of the chelate chain continues to increase up to the region of the transition state (Fig. 2B), where it then decreases rapidly under the proton transfer process. In such a situation, the tautomeric equilibrium undergoes a transformation from a more stable HN form to a less stable one (OH tautomeric form). For the M1–M3, M, and M9 compounds with a single-well of the potential curve, the aromaticity of the chelate chain increases and remains almost stable (Fig. 2C). As for the rest of unconjugated compounds, the behaviour of aromaticity of the chelate chain is not regular. The comparison of the HOMA(ch) = f(d(XH)) dependency for the studied compounds results in the observation that π-electron delocalization increases under the quasi-aromatic hydrogen bond strengthening (TS points), whereas for the unconjugated compounds it is not regular.
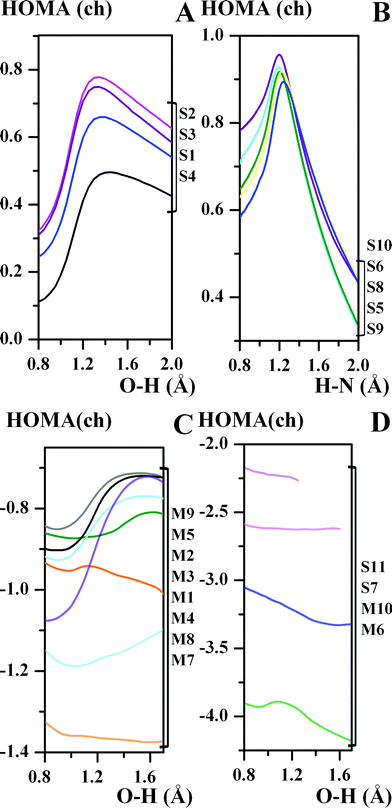 | ||
| Fig. 2 Correlations between the aromaticity of the chelate chain (the HOMA(ch) aromaticity index) and the OH/HN bond length (d(OH/HN), Å) for the studied compounds. | ||
The aromaticity analysis conducted in Ref. 90–92 and 99 makes it possible to define the state of the tautomeric equilibrium of the molecule. In this paper, we have continued the study on the basis of the HOMA(A), HOMA(B) = f(d(XH)) dependencies (Fig. 3). For the aromaticity of the A formation, there is a decrease for the compounds S1–S4 and M1–M4 (Fig. 3A). Moreover, this decline is clearly exposed for the π-conjugated compounds with the double-well potential curve (S1–S4). This effect is conditioned by the formation of the two partially double CO and C2C7 bonds of the π-conjugated compounds, whereas for the unconjugated compounds only one CO bond is partly doubled. It is remarkable that the aromatic balance evoked by proton transfer and described in Ref. 93, 94 and 99 for naphthalene derivatives is assigned to a decrease of aromaticity of the A ring and an increase in the aromaticity of the B ring (Fig. 3). However, such a balance is not traced for the S4 and M4 compounds where a weakening of the aromaticity of both the A and B rings is observed (Fig. 3A and 3D). Therefore, the energy loss under the dearomatisation of the A and B rings does not support the stabilization of the HN form; this explains the high energy barrier of the potential curve (Fig. 1). The aromaticity of the A formation in the S5 and S8–S10 compounds increases up to the transition state region and then remains almost stable under the HN bond elongation. This observation reveals a reverse tendency for the partly aromatic Schiff bases (S5, S8–S10) compared to the aromatic ones (S1–S4). This reverse tendency is also obtained for the potential curves (the prevailing of the OH form for S1–S4 and the HN form for S5, S8–S10, Fig. 1) and provides the following conclusions: A remarkable change in the aromaticity of the A formation affects the proton transfer process. One of the ways to obtain two almost equal minima on the potential curve (the LBHB form stabilization) is to achieve the aromaticity balance between the particular fragments of the molecule.
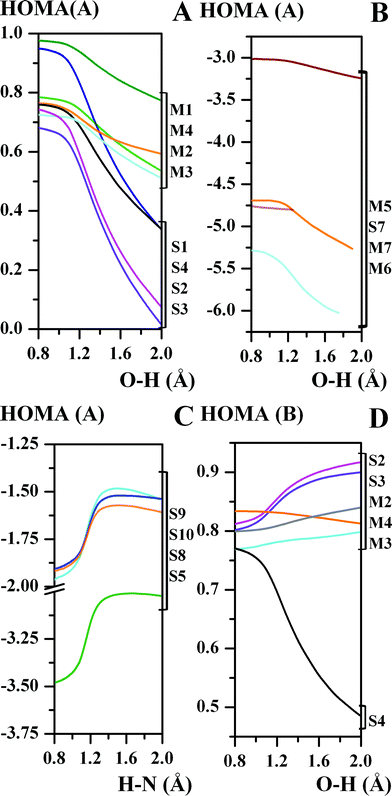 | ||
| Fig. 3 Correlations between the aromaticity of the A formation (the HOMA(A) aromaticity index) (A − C), the B formation (the HOMA(B) aromaticity index) and the OH/HN bond length (d(OH/HN), Å) for the studied compounds. | ||
Electron-topological analysis of intramolecular hydrogen bonding
The description of the dependence between the structural, electron topological, and aromatic parameters of the hydrogen bonding has become a popular subject of discussion in the last decade.103–112 Therefore, our objective here is to show the relation between aromaticity and electron density at critical points. To define the specific feature distinguishing the unconjugated and π-conjugated compounds, a series of dependencies were calculated: ρBCP(XH) = f(d(XH)), ρBCP(C2C7) = f(d(OH/HN)), ρBCP(C1C2) = f(d(OH/HN)), ρBCP(CO) = f(d(OH/HN)), ρBCP(CN) = f(d(OH/HN)) and ρRCP(ch) = f(d(OH/HN)) (Fig. S2†).The series describes the changes in electron density at critical points of the quasi-aromatic formation under the proton transfer process. The ρBCP(CO), ρBCP(C1C2), ρBCP(C2C7) and ρBCP(CN) = f(d(OH/HN)) dependencies for the π-conjugated compounds show a regular alteration of the electron density at the bond critical points of the corresponding bonds according to Scheme 2. The scheme reflects the electron-topological changes of the tautomeric equilibrium. As for the unconjugated compounds, they show irregular changes in electron density at the bond critical points.
 | ||
| Scheme 2 Scheme of the electron-topological changes in a quasi-aromatic formation under the proton transfer process in the π-conjugated compounds. | ||
It is noticeable that the dependence of the HOMA aromaticity index of the A and B formations from the sum of the electron density at the bond critical points for these formations is of a linear character (Fig. 4). The increase in the sum of electron density at the bond critical points brings about the increase of aromaticity. This phenomenon is an example of the reliability of the HOMA aromaticity index. The dependence of the sum of electron density at the OH and NH bonds critical points (ρBCP(HB) = ρBCP(OH) + ρBCP(HN)) under the transfer from one tautomeric form to the transition state initially decreases, and under the following transfer (from TS to the next tautomeric form) it increases (Fig. 5). According to the aforesaid we can state that the chelate chain and hydrogen bonding interact. Thus, for the OH and HN tautomeric forms, the electron density at the bond critical points is more localized on the OH and HN bonds, whereas under the transition state it undergoes a re-distribution and transfers to the chelate chain. A similar correlation was also observed by Albayrak et al.98 for o-hydroxyaryl aldimines. The ρBCP(HB) = f(d(OH)) dependence is supported by the experimental data. Thus, according to NMR studies, the signal of the bridged proton shifts into the direction of a low-field under hydrogen bond strengthening, which is characterised by a decrease of electron density on the bridged hydrogen.113 This picture is also verified by the analysis of the crystallographic and neutronographic structural data.114,115 The results show the movement of electron density in the direction of the proton-donor atom with respect to the bridged proton position.
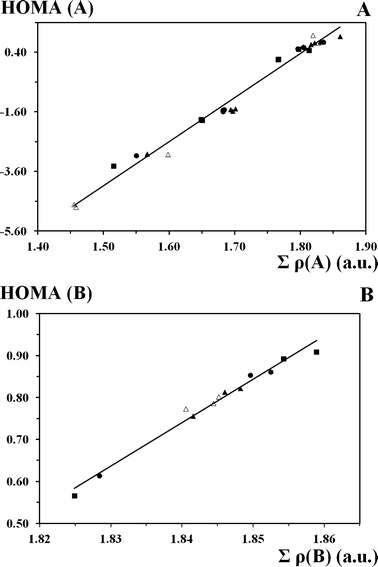 | ||
| Fig. 4 Correlations between the aromaticity of the A (HOMA(A) = 14.81 × Σρ(A) − 26.30; R² = 0.985) and B (HOMA(B) = 10.37 × Σρ(B) − 18.28; R² = 0.981; S = 0.08 and 0.01) formations and the Σρ(A) (Σρ(A) = ρBCP(C1C2) + ρBCP(C2C3) + ρBCP(C3C4) + ρBCP(C4C5) + ρBCP(C5C6) + ρRCP(A), a.u.), and Σρ(B) (Σρ(B) = ρBCP(C10C6) + ρBCP(C5C6) + ρBCP(C5C7) + ρBCP(C7C8) + ρBCP(C8C9) + ρBCP(C9C10) + ρRCP(B), a.u.) sums of the electron density at the critical points, respectively. Filled triangles, squares and circles correspond to the OH and HN forms, and transition state of π-conjugated compounds, respectively; open triangles correspond to the OH form of unconjugated compounds. | ||
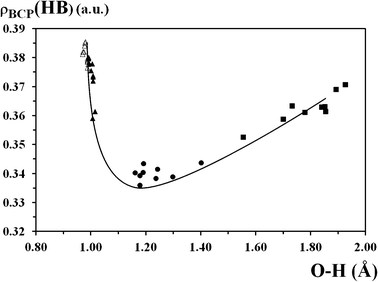 | ||
| Fig. 5 Scatter plot of electron density at the hydrogen bonding critical points (ρBCP(HB) = ρBCP(OH) + ρBCP(HN), a.u.) versus the OH bond length (d(OH), Å); ρBCP(HB) = 1.40 × d(OH)6 − 11.91 × d(OH)5 + 42.41 × d(OH)4 − 81.24 × d(OH)3 + 88.84 × d(OH)2 − 52.77 × d(OH) + 13.66; R² = 0.958; S = 0.02. Filled triangles, squares and circles correspond to the OH and HN forms, and transition state of π-conjugated compounds, respectively; open triangles correspond to the OH form of unconjugated compounds. | ||
It is interesting that the electron density at the chelate ring critical point (ρRCP(ch)) correlates with the main parameters describing the hydrogen bond (d(ON), ΔEHB). Fig. 6 presents good correlations: (A) d(ON) = 0.61 × ρRCP(ch)−0.37; R² = 0.954 and (B) ΔEHB = 9E+07 × ρRCP(ch)4.00; R² = 0.948. These correlations support the reliability of the ρRCP(ch) parameter in the description of the intramolecular hydrogen bonding strength.
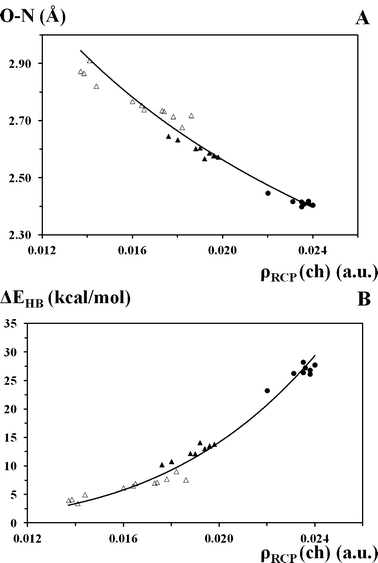 | ||
| Fig. 6 Correlations between (A) hydrogen bonding length (d(ON), Å), (B) the energy of hydrogen bonding (ΔEHB, kcal mol−1) and the electron density at the chelate ring critical point (ρRCP(ch), a.u.). Filled triangles and circles correspond to the OH form and transition state of π-conjugated compounds, respectively; open triangles correspond to the OH form of unconjugated compounds. (A) d(ON) = 0.61 × ρRCP(ch)−0.37; R² = 0.954; S = 0.14 and 0.0013. (B) ΔEHB = 9E+07 × ρRCP(ch)4.00; R² = 0.948. | ||
The resonance assisted hydrogen bond
To elicit the π-electronic conjugation influence on hydrogen bonding, the calculations of flattened unconjugated compounds were carried out (the optimization under the fixed dihedral OCCC = CCCN = 0° angles). This limitation creates similar geometrical conditions both for the unconjugated compounds, where the angle between the OCC and CCN planes is 20–40° for the full optimized structure, and for the π-conjugated compounds, and makes it possible to collate the hydrogen bridges in the two types of compounds. This restriction was applied to infer differences between hydrogen formations in the π-conjugated and flat unconjugated compounds. It is noteworthy that an equal length of hydrogen bridges (d(ON)) for the similar unconjugated and π-conjugated compounds (e.g. S1 and M1) was not fixed during the calculations. In the calculations, this restriction was not employed because the length of a hydrogen bridge serves as a basic parameter describing the strength (energy) of hydrogen bonding.116–118 Thus, it seems unreasonable to fix this parameter on an equal length and perform the analysis, as it was employed in Ref. 32–34.The computed potential curves did not expose any drastic changes for the “restricted” unconjugated compounds (compare the potential curves for the M1 with M1* compounds and the M2 with M2* compounds, Fig. S3†). Unharmonicity of the potential curves shows the strengthening of the hydrogen bridge in the “restricted” unconjugated derivatives corresponding to the unconjugated derivatives (M1–M1* etc). Moreover, this restriction did not cause such results (Fig. S3†, compare the M1* with S1 and M2* with S2 compounds), which showed that the hydrogen bonding in the “restricted” unconjugated derivatives is stronger than in the π-conjugated compounds. The analysed results make it clear that hydrogen bonding is stronger in the π-conjugated compounds than in the corresponding “restricted” unconjugated compounds and the unconjugated compounds.
One of the basic dependencies which reveals the difference between the π-conjugated and unconjugated compounds is ΔEHB = f(HOMA(ch)) (Fig. 7). This correlation reflects the relations between the energy of hydrogen bonding (calculated by equations: ΔEHB = (−5.554 × 105) × exp(−4.12 × d(ON))118 and ΔEHB = −0.37 × V(rcp) + 3.1,119Table 2 and Fig. S4†) and the degree of aromaticity of the chelate chain (the HOMA(ch) aromaticity index) which is a measure of π-electronic conjugation.
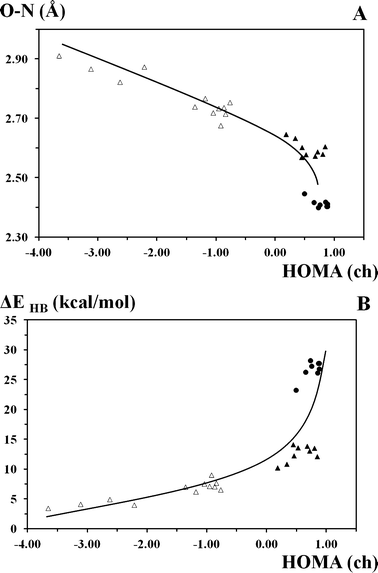 | ||
| Fig. 7 Correlations between (A) the hydrogen bonding length (d(ON), Å), (B) the energy of hydrogen bonding (ΔEHB, kcal mol−1) and the aromaticity of the chelate chain (the HOMA(ch) aromaticity index) for the OH tautomeric form. Filled triangles and circles correspond to the OH tautomeric form and transition state of π-conjugated compounds, respectively; open triangles correspond to the OH tautomeric form of unconjugated compounds. (A) d(ON) = −0.01 × HOMA(ch)3 − 0.06 × HOMA(ch)2 − 0.15 × HOMA(ch) + 2.64; R² = 0.857. (B) ΔEHB = 0.48 × HOMA(ch)4 + 3.74 × HOMA(ch)3 + 7.84 × HOMA(ch)2 + 7.29 × HOMA(ch) + 9.54; R² = 0.747. | ||
| Compound | Form | ΔEHB(I) | ΔEHB(II) | ΔEHB(III) | ΔEHB(IV) |
|---|---|---|---|---|---|
| S1 | OH | 10.8 | 11.4 | 8.9 | 14.3 |
| TS | 26.2 | — | — | — | |
| NH | 13.4 | 11.8 | 9.3 | 14.9 | |
| M1 | OH | 7.1 | 8.7 | 6.7 | 10.7 |
| M1* | OH | 9.3 | 11.5 | 9.0 | 14.5 |
| S2 | OH | 12.2 | 12.9 | 10.2 | 16.4 |
| TS | 27.3 | — | — | — | |
| NH | 11.6 | 9.4 | 7.3 | 11.7 | |
| M2 | OH | 7.7 | 9.4 | 7.3 | 11.7 |
| M2* | OH | 9.8 | 12.2 | 9.6 | 15.4 |
| S3 | OH | 14.1 | 14.6 | 11.6 | 18.7 |
| TS | 28.2 | — | — | — | |
| NH | 13.8 | 11.2 | 8.8 | 14.1 | |
| M3 | OH | 9.0 | 10.7 | 8.4 | 13.4 |
| M3* | OH | 10.7 | 13.0 | 10.3 | 16.5 |
| S4 | OH | 10.2 | 10.9 | 8.5 | 13.7 |
| TS | 23.3 | — | — | — | |
| NH | 18.0 | 18.5 | 14.9 | 23.9 | |
| M4 | OH | 7.1 | 8.9 | 6.8 | 11.0 |
| M4* | OH | 9.8 | 12.2 | 9.6 | 15.4 |
| S8 | OH | 13.0 | 13.7 | 10.9 | 17.5 |
| TS | 26.8 | — | — | — | |
| NH | 8.7 | 6.8 | 5.1 | 8.1 | |
| M5 | OH | 7.6 | 9.1 | 7.0 | 11.2 |
| M5* | OH | 8.6 | 10.5 | 8.2 | 13.2 |
| S7 | OH | 5.0 | 5.3 | 3.5 | 5.6 |
| M6 | OH | 3.5 | 4.2 | 2.9 | 4.6 |
| M6* | OH | 9.6 | 11.0 | 8.6 | 13.8 |
| S9 | OH | 13.5 | 14.1 | 11.2 | 18.0 |
| TS | 27.8 | — | — | — | |
| NH | 10.7 | 8.0 | 6.0 | 9.7 | |
| S5 | OH | 13.6 | 14.2 | 11.3 | 18.1 |
| TS | 27.7 | — | — | — | |
| NH | 10.3 | 7.7 | 5.8 | 9.3 | |
| M7 | OH | 7.0 | 8.9 | 6.9 | 11.0 |
| M7* | OH | 7.4 | 9.6 | 7.4 | 11.9 |
| S10 | OH | 13.9 | 14.9 | 11.9 | 19.1 |
| TS | 26.1 | — | — | — | |
| NH | 8.9 | 6.8 | 5.1 | 8.1 | |
| S6 | OH | 12.1 | 12.7 | 10.0 | 16.1 |
| TS | 26.4 | — | — | — | |
| NH | 7.7 | 6.2 | 4.5 | 7.3 | |
| M8 | OH | 6.2 | 8.0 | 6.1 | 9.8 |
| M8* | OH | 6.2 | 8.2 | 6.2 | 10.0 |
| S11 | OH | 4.0 | 4.1 | 2.8 | 4.5 |
| M10 | OH | 4.1 | 5.4 | 3.9 | 6.2 |
| M10* | OH | 8.3 | 9.1 | 7.0 | 11.3 |
| M9 | OH | 6.6 | 7.8 | 5.9 | 9.5 |
| M9* | OH | 7.2 | 8.7 | 6.6 | 10.6 |
The comparison of the two parameters for the unconjugated and π-conjugated compounds provides a method to estimate the impact of π-conjugation in the chelate chain on the hydrogen bond strength. The calculated ΔEHB = f(HOMA(ch)) dependence (Fig. 7 and S4†) clearly features two sets of points for the unconjugated and π-conjugated compounds, respectively. According to this correlation, the hydrogen bond energy in the unconjugated compounds is 2–9 kcal mol−1 under a lack of π-electronic conjugation, meanwhile the energy of hydrogen bonding in the π-conjugated compounds is over 10 kcal mol−1 under a stronger π-conjugation in the chelate chain. For the question about the existence of RAHB, it is the strengthening of hydrogen bonding in the π-conjugated compounds by means of the increase of π-conjugation in the chelate chain that speaks in favour of the phenomenon. Moreover, as mentioned above, we have fulfilled the calculations for the unconjugated compounds for the plane chelate fragment. The calculations with the θ(OCC/CCN) = 0° restriction show that the hydrogen bridge is slightly stronger for the flattened unconjugated compounds than for the fully optimized unconjugated compounds. However, the strength of hydrogen bonds in the flattened unconjugated compounds is still much weaker than in the π-conjugated compounds (ΔEHB(conjug.) = 10.37–14.12 kcal mol−1 (23.28–28.24 kcal mol−1 for TS) > ΔEHB(unconjug.) = 1.81–9.04 kcal mol−1). The presented analysis does not deny the influence of the σ-skeleton framework, or any steric- or charge-assisted effects on the structural parameters of the hydrogen bonding. However, ΔEπ (ΔEπ ≅ ΔEHB(conjug.) − ΔEHB(unconjug.)) for the presented π-conjugated compounds is larger compared with the unconjugated compounds and reaches up to 4–9 kcal mol−1.
Conclusions
The most complete analysis of the topological vs. aromatic and energetic changes has been obtained for ortho-hydroxyaryl or alkyl Schiff and Mannich bases with intramolecular hydrogen bonding. The changes of aromaticity and electron density have been examined at the critical points of all the bonds and rings in the unconjugated and π-conjugated compounds depending on the tautomeric equilibrium. The specific character of compounds with quasi-aromatic hydrogen bond formation has been shown by way of comparison of the aromaticity and electron density of the unconjugated and π-conjugated systems.This paper presents the principal difference between the potential curves calculated for the π-conjugated and unconjugated compounds under a non-adiabatic approach. The potential curves for the π-conjugated compounds take a double-well, which supports the existence of tautomeric equilibrium, whereas this effect is not observed for the unconjugated compounds. It has been shown that hydrogen bond strengthening increases with a corresponding increase of chelate chain aromaticity. This phenomenon is more significant for the π-conjugated systems. The comparison of the π-conjugated compounds with the unconjugated compounds makes it obvious that π-electronic conjugation in the chelate chain enhances hydrogen bonding by 4–8 kcal mol−1 (30–40%). In summary, the abovementioned facts demonstrate the peculiarity of compounds with quasi-aromatic hydrogen bonding with respect to the lack of π-electronic conjugation in the chelate chain.
Acknowledgements
The authors acknowledge the Wrocław Center for Networking and Supercomputing for generous computer timeReferences
- G. A. Jeffery and W. Saenger, Hydrogen Bonding in Biological Structures, Springer, Berlin, 1991 Search PubMed.
- J. T. Hynes, J. P. Klinman, H.-H. Limbach and R. L. Schowen, Hydrogen-transfer reactions, Wiley-VCH, Weinheim, 2006 Search PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed.
- P. Coppens, X-Ray Charge Densities and Chemical Bonding, Oxford University Press, New York, 1997 Search PubMed.
- T. M. Krygowski and H. Szatyłowicz, Trends Org. Chem., 2006, 11, 37 CAS.
- L. Sobczyk, S. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513 CrossRef CAS.
- D. Hadži, Theoretical Treatments of Hydrogen Bonding, John Wiley & Sons, Chichester, 1997 Search PubMed.
- G. S. Denisov, J. Mavri and L. Sobczyk, in Hydrogen Bonding—New Insights; ed. S. Grabowski,Springer, Berlin, 2006, 377 Search PubMed.
- S. Scheiner, Acc. Chem. Res., 1994, 27, 402 CrossRef CAS.
- A. Filarowski, A. Koll, P.-E. Hansen and M. Kluba, J. Phys. Chem. A, 2008, 112, 3478 CrossRef CAS.
- M. V. Basilevsky and M. V. Vener, Usp. Khim., 2003, 72, 3 Search PubMed.
- ISI Web of Science, apps.webofknowledge.com, retrieved March 2012.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 564 CrossRef.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 51, 1004 CrossRef.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1994, 116, 909 CrossRef CAS.
- P. Gilli, V. Ferretti, V. Bertolasi and G. Gilli, in Advances in Molecular Structure Research, ed. M. Hargittai and I. Hargittai, JAI Press, Greenwich, CT, 1996, Vol. 2, 67 Search PubMed.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Chem. Soc., Faraday Trans., 1997, 945 CAS.
- P. Gilli, V. Bertolasi, L. Pretto, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, A. Lyčka and G. Gilli, J. Am. Chem. Soc., 2002, 124, 13554 CrossRef CAS.
- P. Gilli, V. Bertolasi, L. Pretto, L. Antonov and G. Gilli, J. Am. Chem. Soc., 2005, 127, 4943 CrossRef CAS.
- G. Gilli and P. Gilli, The Nature of the Hydrogen Bond. Outline of a Comprehensive Hydrogen Bond Theory, Oxford University Press, Oxford, 2009 Search PubMed.
- S. J. Grabowski and J. Leszczyński, in Hydrogen Bonding—New Insights; ed. S. J. Grabowski, Springer, Berlin, 2006, 487 Search PubMed.
- G. Buemi, in Hydrogen Bonding—New Insights, ed. S. J. Grabowski, Springer, Berlin, 2006, 51 Search PubMed.
- T. Steiner, Chem. Commun., 1998, 411 RSC.
- J. F. Beck and Y. J. Mó, J. Comput. Chem., 2007, 28, 455 CrossRef CAS.
- R. Kurczab, M. P. Mitoraj, A. Michalak and T. Ziegler, J. Phys. Chem. A, 2010, 114, 8581 CrossRef CAS.
- J. N. Woodford, J. Phys. Chem. A, 2007, 111, 8519 CrossRef CAS.
- I. Alkorta, J. Elguero, O. Mó, M. Yanez and J. E. del Bene, Mol. Phys., 2004, 102, 2563 CrossRef CAS.
- I. Alkorta, J. Elguero, O. Mó, M. Yanez and J. E. del Bene, Chem. Phys. Lett., 2005, 411, 411 CrossRef CAS.
- P. Sanz, O. Mó, M. Yanez and J. Elguero, J. Phys. Chem. A, 2007, 111, 3585 CrossRef CAS.
- P. Sanz, O. Mó, M. Yanez and J. Elguero, ChemPhysChem, 2007, 8, 1950 CrossRef CAS.
- P. Sanz, O. Mó, M. Yanez and J. Elguero, Chem.–Eur. J., 2008, 14, 4225 CrossRef CAS.
- P. Sanz, O. Mó, M. Yanez and J. Elguero, Phys. Chem. Chem. Phys., 2009, 11, 762 RSC.
- M. Jablonski, A. Kaczmarek and A. Sadlej, J. Phys. Chem. A, 2006, 110, 10890 CrossRef CAS.
- T. M. Krygowski, J. Zachara-Horeglad and M. Palusiak, J. Org. Chem., 2010, 75, 4944 CrossRef CAS.
- A. E. Lutskii and T. N. Marchenko, in Vodorodnaya svyaz' (Hydrogen bond), ed. N. D. Sokolov, Nauka, Moscow, 1981, 50 Search PubMed.
- M. B. Piccoli, T. F. Koetzle, A. J. Schultz, E. A. Zhurova, J. Stare, A. A. Pinkerton, J. Eckert and D. Hadži, J. Phys. Chem. A, 2008, 112, 6667 CrossRef.
- N. Zarycz, G. A. Aucar and C. O. D. Vedova, J. Phys. Chem. A, 2010, 114, 7162 CrossRef CAS.
- R. I. Zubatyuk, O. V. Shishkin, L. Gorb and J. Leszczynski, J. Phys. Chem. A, 2009, 113, 2943 CrossRef CAS.
- R. I. Zubatyuk, Y. M. Volovenko, O. V. Shishkin, L. Gorb and J. Leszczyński, J. Org. Chem., 2007, 72, 725 CrossRef CAS.
- D. N. Shigorin, Zh. Fiz. Khim., 1949, 23, 505 CAS.
- D. N. Shigorin, in Vodorodnaya svyaz' (Hydrogen bond), ed. N. D Sokolov and A. D. Chulanovsky, Nauka, Moscow, 1964, 195 Search PubMed.
- A. Filarowski and A. Koll, Vib. Spectrosc., 1998, 17, 123 CrossRef CAS.
- M. Takasuka and Y. Matsui, J. Chem. Soc., Perkin Trans. 2, 1979, 1743 RSC.
- B. Schiøtt, B. B. Iversen, G. K. H. Madsen and T. C. Bruice, J. Am. Chem. Soc., 1998, 120, 12117 CrossRef.
- G. K. H. Madsen, B. B. Iversen, F. K. Larsen, M. Kapon, G. M. Reisner and F. H. Herbstein, J. Am. Chem. Soc., 1998, 120, 10040 CrossRef CAS.
- M. L. Josien and G. Sourisseu, p.129; D. M. Shigorin, p.191; A. Burawoy, p.259; M. D. Cohen, et al., p.293; M. Hunsberger, et al., p.461; Hydrogen Bonding, papers presented at the symposium on hydrogen bonding held at Ljubljana, July 29–August 3, 1957; ed. D. Hadži, Pergamon Press, London, UK., 1959 Search PubMed.
- D. Hadži, J. Chem. Soc., 1956, 2143 RSC.
- H. Tsubomura, J. Chem. Soc., 1956, 24, 927 CAS.
- A. Filarowski, J. Phys. Org. Chem., 2005, 18, 686 CrossRef CAS.
- T. S. Kopteva and D. N. Shigorin, Zh. Fiz. Khim., 1974, 48, 532 CAS.
- C. Fonseca Guerra and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 1999, 38, 2942 CrossRef.
- C. Fonseca Guerra, H. Zijlstra, F. M. Bickelhaupt and G. Paragi, Chem.–Eur. J., 2011, 17, 12612 CrossRef CAS.
- C. Fonseca Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem.–Eur. J., 1999, 5, 3581 CrossRef.
- C. Fonseca Guerra, Z. Szekeres and F. M. Bickelhaupt, Chem.–Eur. J., 2011, 17, 8816 CrossRef CAS.
- A. Filarowski, A. Koll and L. Sobczyk, Curr. Org. Chem., 2009, 13, 172 CrossRef CAS.
- J. A. Belot, J. Clark, J. A. Cowan, G. S. Harbison, A. I. Kolesnikov, Y.-S. Kye, A. J. Schultz, C. Silvernail and X. Zhao, J. Phys. Chem. B, 2004, 108, 6922 CrossRef CAS.
- J. Emsley, Struct. Bonding, 1984, 27, 147 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS; (b) R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS; (c) T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS; (d) M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1993, B37, 785 Search PubMed.
- S. Scheiner, Hydrogen Bonding: A Theoretical Perspective, Oxford University Press, New York, 1997 Search PubMed.
- J. Biegler-König, D. Schönbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545 CrossRef.
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385 CrossRef CAS.
- T. M. Krygowski, J. Chem. Inf. Model., 1993, 33, 70 CrossRef CAS.
- N. Zarycz and G. A. Aucar, J. Phys. Chem. A, 2008, 112, 8767 CrossRef CAS.
- V. I. Minkin, A. V. Tsukanov, A. D. Dubonosov and V. A. Bren, J. Mol. Struct., 2011, 998, 179 CrossRef CAS.
- W. Bruyneel, J. Charette and E. De Hoffmann, J. Am. Chem. Soc., 1966, 88, 3808 CrossRef CAS.
- O. Dominguez, B. Rodriguez-Molina, M. Rodriguez, A. Ariza, N. Farfan and R. Santillan, New J. Chem., 2011, 35, 156 RSC.
- I. Król-Starzomska, M. Rospenk, A. Filarowski and A. Koll, J. Phys. Chem. A, 2004, 108, 2131 CrossRef.
- A. Koll, M. Rospenk and L. Sobczyk, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2309 RSC.
- C. Sandorfy, in The Hydrogen Bond. Recent Developments in Theory and Experiments, ed. P. Schuster, G. Zundel and C. Sandorfy, North-Holland, Amsterdam, 1976, vol. 2, 613 Search PubMed.
- T. Lankau and C.-H. Yu, Phys. Chem. Chem. Phys., 2007, 9, 299 RSC.
- A. Filarowski, A. Koll and T. J. Głowiak, J. Chem. Soc., Perkin Trans. 2, 2002, 835 RSC.
- T. F. Markle, A. L. Tenderholt and J. M. Mayer, J. Phys. Chem. B, 2012, 116, 571 CrossRef CAS.
- I. Majerz and I. Olovsson, Phys. Chem. Chem. Phys., 2009, 11, 1297 RSC.
- M. V. Vener, Chem. Phys., 1992, 166, 311 CrossRef CAS.
- I. Majerz and I. Olovsson, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 650 CrossRef.
- Y. Pan and M. A. McAllister, J. Org. Chem., 1997, 62, 8171 CrossRef CAS.
- W. W. Cleland and M. M. Kreevoy, Science, 1994, 264, 1887 CAS.
- J. C. Speakman, J. Am. Chem. Soc., 1949, 3357 CAS.
- D. Hadži, Pure Appl. Chem., 1965, 11, 435 CrossRef.
- E. P. Serjeant and B. Dempsey, Ionisation Constants of Organic Acids in Aqueous Solution, Pergamon Press, New York, 1979 Search PubMed.
- T. M. Krygowski, M. Cyrański, Z. Czarnoski, G. Hafelinger and A. Katritzky, Tetrahedron, 2000, 56, 1783 CrossRef CAS.
- S. Shaik, P. C. Hiberty, J. M. Lefour and G. Ohanessian, J. Am. Chem. Soc., 1987, 109, 363 CrossRef CAS.
- K. Jug and A. Koester, J. Am. Chem. Soc., 1990, 112, 6772 CrossRef CAS.
- S. Shaik and P. C. Hiberty, J. Am. Chem. Soc., 1985, 107, 3089 CrossRef CAS.
- T. M. Krygowski, K. Woźniak, R. Anulewicz, D. Pawlak, W. Kolodziejski, E. Grech and A. Szady, J. Phys. Chem. A, 1997, 101, 9399 CrossRef CAS.
- A. Filarowski, A. Koll, T. Głowiak, E. Majewski and T. Dziembowska, Ber. Bunsenges. Phys. Chem., 1998, 102, 393 CrossRef CAS.
- A. Filarowski, A. Kochel, K. Cieślik and A. Koll, J. Phys. Org. Chem., 2005, 18, 880 CrossRef.
- M. Palusiak and T. M. Krygowski, Chem.–Eur. J., 2007, 13, 7996 CrossRef CAS.
- M. Palusiak, S. Simon and M. Sola, J. Org. Chem., 2006, 110, 5875 Search PubMed.
- H. Houjou, T. Motoyama, S. Banno, I. Yoshikawa and K. Araki, J. Org. Chem., 2009, 74, 520 CrossRef CAS.
- H. Karabiyik, H. Petek, N. O. Iskeleli and C. Albayrak, Struct. Chem., 2009, 20, 1055 CrossRef CAS.
- H. Petek, C. Albayrak, M. Odabasoglu, I. Senel and O. Buyukgungor, Struct. Chem., 2010, 21, 681 CrossRef CAS.
- C. Albayrak, R. Sevincek, H. Petek and M. Aygun, J. Mol. Model., 2011, 17, 1295 CrossRef.
- A. Filarowski, A. Kochel, M. Kluba and J. Kamounah, J. Phys. Org. Chem., 2008, 21, 939 CrossRef CAS.
- T. M. Krygowski, J. E. Zachara and H. Szatyłowicz, J. Phys. Org. Chem., 2005, 18, 110 CrossRef CAS.
- E. D. Raczyńska, T. M. Krygowski, J. E. Zachara, B. Osmiałowski and R. Gawinecki, J. Phys. Org. Chem., 2005, 18, 892 CrossRef.
- M. Kluba, P. Lipkowski and A. Filarowski, Chem. Phys. Lett., 2008, 463, 426 CrossRef CAS.
- J. Overgaardand and B. B. Iversen, Struct. Bonding, 2011, 146, 53 CrossRef.
- S. J. Grabowski, Chem. Rev., 2011, 11, 2597 CrossRef.
- L. F. Pacios and P. C. Gomez, J. Phys. Chem. A, 2004, 108, 11783 CrossRef CAS.
- R. A. Klein, J. Am. Chem. Soc., 2002, 124, 13931 CrossRef CAS.
- M. V. Vener, A. V. Manaev, A. N. Egorova and V. G. Tsirelson, J. Phys. Chem. A, 2007, 111, 1155 CrossRef CAS.
- M. Palusiak, Wiad. Chem., 2010, 64, 264 Search PubMed.
- A. Filarowski and I. Majerz, J. Phys. Chem. A, 2008, 112, 3119 CrossRef CAS.
- S. J. Grabowski, W. A. Sokalski and J. Leszczyński, J. Phys. Chem. A, 2006, 110, 4772 CrossRef CAS.
- A. Makal, W. Schilf, B. Kamieński, A. Szady-Chełmieniecka, E. Grech and K. Woźniak, Dalton Trans., 2011, 40, 421 RSC.
- P. M. Dominiak, A. Makal, P. R. Mallinson, K. Trzcinska, J. Eilmes, E. Grech, M. Chruszcz, W. Minor and K. Woźniak, Chem.–Eur. J., 2006, 12, 1941 CrossRef CAS.
- H.-H. Limbach, P. M. Tolstoy, N. Perez-Hernandez, J. Guo, I. G. Shenderovich and G. S. Denisov, Isr. J. Chem., 2009, 49, 199 CrossRef CAS.
- W. C. Hamilton and J. A. Ibers, Hydrogen Bonding in Solids; ed. W. A. Benjamin, INC,New York, 1968 Search PubMed.
- I. Majerz and M. J. Gutmann, RSC Adv., 2011, 1, 219 RSC.
- G. A. Jeffery, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- R. N. Musin and Y. H. Mariam, J. Phys. Org. Chem., 2006, 19, 425 CrossRef CAS.
- I. Mata, I. Alkorta, E. Espinosa and E. Molins, Chem. Phys. Lett., 2011, 507, 185 CrossRef CAS.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20846f/ |
| This journal is © The Royal Society of Chemistry 2012 |