DOI:
10.1039/C2RA20841E
(Paper)
RSC Adv., 2012,
2, 7714-7723
9-O-N-aryl/arylalkyl amino carbonyl methyl substituted berberine analogues induce self-structure in polyadenylic acid†
Received
2nd May 2012
, Accepted 13th June 2012
First published on 14th June 2012
Abstract
This manuscript describes the interaction of 9-O-substituted analogues of the plant alkaloid berberine with single stranded (ss) poly(A). Three new analogues of berberine with aryl/arylalkyl amino carbonyl methyl substituents at the 9-position of the isoquinoline chromophore were evaluated for their binding to ss poly(A) by a wide variety of biophysical techniques. The results indicated that these analogues exhibited several-fold higher binding affinities than berberine. The Scatchard binding isotherms revealed that all the analogues retained the cooperative binding mode of berberine. Circular dichroism and FTIR studies unequivocally established the strong interaction potential of the analogues to ss poly(A). Optical melting and the unique dilution experiments revealed that these analogues induced self-structure formation in poly(A) and the nature of the substituent was important for the ease of formation of self-structure. Energetics of the binding suggested an entropy driven binding for the analogues in sharp contrast to the enthalpy driven binding of berberine. The introduction of the aryl/arylalkyl amino carbonyl methyl substituent thus switched the enthalpy driven binding of berberine to entropy dominated binding. Salt and temperature dependent calorimetric studies established the involvement of multiple weak noncovalent interactions in the binding process. The study also revealed that the spacer length at the 9-position had an important role in the self-structure induction.
Introduction
Polyadenylic acid has been the focus of increasing attention recently for its role in mRNA functioning as all eukaryotic mRNAs have the long poly(A) tail at the 3′ end that is added during post transcriptional modification. This single stranded (ss) poly(A) tail is an important factor for nuclear export, in the maturation and stability of mRNA, and in the initiation of translation. Many recent studies have implicated that the neo polyadenylic polymerase, a human PAP enzyme that catalyzes the poly(A) addition, was significantly over expressed in human cancer cells in comparison to its expression in normal or virally transformed cells.1 This relates polyadenylation to neoplasms and the presence of enhanced levels of poly(A) tailed mRNA in such cells. The control of such over expression of poly(A) by the binding of small molecule drugs could be a potential route to RNA targeted therapeutic intervention. Recently, it has been shown that a variety of molecules bind to ss poly(A) and induce unique structural transformation to a self-structure stabilized by adenine–adenine base pairs. In vivo, such molecules that can bind and structurally modulate the polyadenylate tail could stop poly(A) chain elongation, inhibit mRNA function and degrade it, and stop subsequent protein production in the cell.
Polyriboadenylic acid has the unique characteristics to exist in ss helical and parallel stranded double stranded (ds) structure in vitro.2a–c The ds structure of poly(A), formed from two single strands on lowering the pH of the solution to 4.5, is stabilized by base paired protonated adenines. The structure of this ds structure has been established from X-ray crystal analysis.2a However, small molecules that induced self-structure in poly(A) does it at pH 7.0 where only the ss structure can otherwise exist. The mechanism of such self-structure formation at pH 7.0, the nature and mode of the transition, and the structural features of the small molecules that can specifically induce this novel conformational transition is still unclear.2d–h The compounds that were first discovered to induce such structural transitions were the natural alkaloid sanguinarine and the synthetic isoquinoline molecule coralyne.2d,g The parent natural alkaloid berberine was subsequently shown to induce the same structural transformation.3 As berberine is an important natural product with potential anticancer activity, many studies on its derivatives are currently in progress in several laboratories to enhance its efficacy. The significance of 9-position substitutions in berberine's biological activity is well documented in the literature.4 Derivatives of berberine with substitutions at the 9-positions have been shown to possess better anticancer activities in human cancer cells, enhanced DNA and RNA binding, and higher topoisomerase poisoning activities and telomerase inhibition properties.3b,4,5 However, studies on the binding of these 9-substituted analogues to single or double stranded poly(A) have not yet been reported. We have recently studied the DNA binding of a series of novel 9-O-N-aryl/arylalkyl amino carbonyl methyl substituted berberine analogues and showed that they bound to DNA exhibiting remarkably enhanced binding affinities.5b In this investigation, the potential of these 9-O-N-aryl/arylalkyl amino carbonyl methyl analogues of berberine (Fig. 1) to interact with ss poly(A) and induce self-structure has been studied in a search of promising lead compounds for controlling the poly(A) chain elongation and mRNA degradation.
 |
| | Fig. 1 Chemical structures of BER, B1, B2 and B3. | |
Results and discussion
Absorbance and fluorescence spectral studies and evaluation of the binding affinities
The binding of berberine (BER) and its analogues to poly(A) was studied at first using absorption titration in the visible region of 300–600 nm. Typical hypochromic changes and bathochromic effects with three well defined sharp isosbestic points in their spectra were revealed on binding (Fig. S1A†). Analogues B1 and B2 showed larger hypochromic effects. The spectral data were cast into conventional Scatchard plots of r/Cfversus r, where r is the number of moles of alkaloid bound per mole of adenine bases. The binding isotherms of all the three analogues and BER had a positive slope at low r values indicating cooperativity in the binding and were subsequently analyzed by the cooperative binding model of McGhee and von Hippel.6a,b A representative Scatchard plot for B1-poly(A) complexation is presented in Fig. S1B†. Cooperativity for the BER–poly(A) interaction has been reported previously.3b The values of the overall binding affinity (K), which is a product of the cooperative binding affinity (Ki), and the cooperativity factor (ω) are presented in Table 1. For the analogue B1, the affinity was 5.64 × 106 M−1 while for B2 and B3 the values were slightly lower and were at 4.68 × 106 and 3.92 × 106 M−1, respectively. Almost 14 fold higher affinity for the analogue B1 over BER was noteworthy and it can be observed that the magnitude of the binding affinity thereafter slightly reduced and it varied as B1 > B2 > B3 > BER. It is worth mentioning here that a six-fold higher affinity of B1 to ds DNA was reported recently.5b
Table 1 Binding parameters of berberine analogues to poly(A) from spectroscopic studiesa
| Alkaloid/analogue |
Absorbance |
Fluorescence |
|
K
i × 10−4(M−1) |
ω
|
K × 10−5 (M−1) |
K
i × 10−4(M−1) |
ω |
K × 10−5 (M−1) |
|
Average of four determinations. Binding constant (Ki) is the cooperative binding affinity. ω is the cooperativity factor. K is the overall binding affinity obtained as a product of Ki and ω.
|
|
BER
|
1.21 ± 0.04 |
34 |
4.11 ± 0.004 |
0.98 ± 0.02 |
41 |
4.01 ± 0.002 |
|
B1
|
7.05 ± 0.21 |
80 |
56.4 ± 0.021 |
7.01 ± 0.19 |
78 |
54.7 ± 0.019 |
|
B2
|
6.25 ± 0.17 |
75 |
46.8 ± 0.017 |
6.37 ± 0.18 |
72 |
45.8 ± 0.018 |
|
B3
|
5.94 ± 0.12 |
66 |
39.2 ± 0.012 |
5.91 ± 0.11 |
67 |
39.5 ± 0.011 |
Berberine is a weak fluorophore but the analogues are relatively more fluorescent with emission maxima in the range 450–455 nm when excited at 345 nm. Binding to ss poly(A) resulted in many-fold enhancement of the fluorescence intensity of the analogues, revealing that the bound molecules are located in a hydrophobic environment similar to an intercalated state. The fluorescence change of B1 in presence of increasing concentration of poly(A) is shown in Fig. S1C†. The Scatchard plots constructed from fluorescence titration data also revealed cooperative binding for the analogues. A representative Scatchard plot for the binding of B1 is presented in Fig. S1D†. The overall binding constants as per the Scatchard analysis using the cooperative binding model of McGhee–von Hippel yielded binding affinity values of 5.47 × 106 M−1 for B1, 4.58 × 106 M−1 for B2 and 3.95 × 106 M−1 for B3 against a value of 4.01 × 105 M−1 for BER (Table 1). These values are in excellent agreement with the results obtained from the spectrophotometric titration analysis (Table 1).
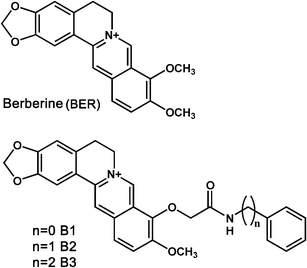
Conformational changes associated with the strong binding of berberine analogues to ss poly(A) were probed through circular dichroism (CD) studies. Single stranded poly(A) has a characteristic CD spectrum with a large positive band around 266 nm and a less intense negative band around 246 nm (curve 1 in Fig. 2). This is in agreement with the spectrum reported previously.2f,3b Nucleic acids in their A-conformation show a large positive band around 260 nm (due to base stacking) and a negative band at around 240 nm (due to polynucleotide helicity). CD spectral changes accompanying the interaction of the berberine analogues with ss poly(A) was characterized by a remarkable decrease in the positive 266 nm peak with a comparatively less pronounced decrease in the negative CD band ellipticity. The changes in the CD spectrum of the ss poly(A) with increasing concentration of BER and analogues B1, B2 and B3 are depicted in Fig. 2. Concomitant with the intrinsic CD changes, there occurred the formation of extrinsic CD bands in the 330–370 nm region with negative and positive ellipticity. The positive band (∼365 nm) had a weaker intensity compared to the negative band (∼335 nm) for all the analogues in comparison to berberine indicating significant differences in the conformational changes on complexation with the analogues. The ellipticity decrease of the 266 nm band of poly(A) on complexation with the analogues was more pronounced at a lower D/P (alkaloid/nucleotide molar ratio) compared to BER, indicating greater binding efficacy of the analogues. Thus, the CD changes revealed that the analogues bind strongly to poly(A) depending on their functional groups. This also indicated that the CD of the poly(A)-alkaloid complexes has similar CD spectral characteristics although there were some differences in the absolute ellipticity values. The final spectra in each case resembled the CD spectrum of the poly(A)-self-structure induced by sanguinarine, coralyne and europium–amino acid complex.2d,g,7a The presence of the induced CD band in the visible absorption region on complexation with poly(A) further established the strong environment of the bound molecules inside the poly(A) helix. It may be noted that the decrease of the long wavelength band ellipticity has been correlated to both helix winding angle and base pair twist.7b,c More often, structural change from the A-form to the B-form and from the B-form to the C-form in double stranded DNA results in such a large decrease of the long wavelength band ellipticity. It is pertinent to observe that the A-form CD spectrum of poly(A) undergoes structural change to a more organized self-structure formation as the spectral changes observed here are similar to that has been reported previously with sanguinarine.2d
 |
| | Fig. 2 Representative circular dichroism spectra of poly(A) (50 μM) treated with increasing concentrations of BER (A), B1 (B), B2 (C) and B3 (D). Inset: Induced circular dichroism spectra for the complexation of BER (A), B1 (B), B2 (C) and B3 (D) with poly(A). | |
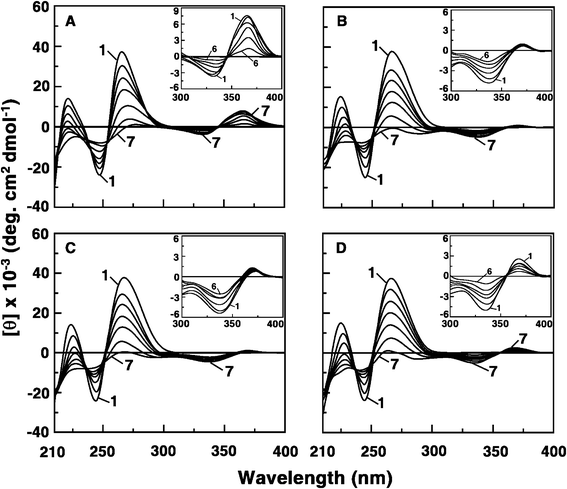
FTIR spectroscopy
The interaction of the analogue B1 that showed highest binding with ss poly(A) was also monitored by FTIR spectroscopy. Comparative IR spectra in the range 1800–800 cm−1 for poly(A) in presence of B1 at three D/P values are presented in Fig. 3. The characteristic adenine band at 1607 cm−1 in poly(A) remained unaltered at 1607 cm−1 at D/P = 0.2 but shifted to 1617 cm−1 at D/P = 0.6 and to 1605 cm−1 at D/P = 1. However, the characteristic phosphate (PO2) stretching band at 1213 cm−1 of poly(A) shifted at first to 1221 cm−1 at D/P = 0.2 and then to 1243 cm−1 at D/P = 0.6 and then finally to 1226 cm−1 at D/P = 1. The ribose-phosphate band that was centred at 875 cm−1 in ss poly(A) shifted to 861 cm−1 at D/P = 0.2, to 854 cm−1 at D/P = 0.6, and ultimately to 835 cm−1 at D/P = 1. The ribose phosphate band shift was also accompanied by an increase in the intensity. These results provide unambiguous evidence for the major interaction of B1 with the bases and phosphates of ss poly(A).8
 |
| | Fig. 3 FTIR spectra of poly(A) and poly(A) complexed with analogue B1 at varying molar ratios. | |
Site size and mode of binding: fluorescence quenching studies
For determining the stoichiometry of the binding, the alkaloid analogue![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :poly(A) molar ratio was varied, keeping the total molar concentration constant. The stoichiometry of the binding was determined from the molar ratio where maximal binding was observed. The plot of difference fluorescence intensity (ΔF) at the wavelength maxima versus the mole fraction of the alkaloids (Fig. S2†) revealed a single binding mode in each case. From the inflection point, χalkaloid was found to be 0.22, 0.40, 0.34 and 0.33, respectively, for BER, B1, B2 and B3. Thus, the number of adenine bases bound per berberine analogue was estimated to be around 3.54, 1.50, 1.94 and 2.03, respectively, for BER, B1, B2 and B3.
:poly(A) molar ratio was varied, keeping the total molar concentration constant. The stoichiometry of the binding was determined from the molar ratio where maximal binding was observed. The plot of difference fluorescence intensity (ΔF) at the wavelength maxima versus the mole fraction of the alkaloids (Fig. S2†) revealed a single binding mode in each case. From the inflection point, χalkaloid was found to be 0.22, 0.40, 0.34 and 0.33, respectively, for BER, B1, B2 and B3. Thus, the number of adenine bases bound per berberine analogue was estimated to be around 3.54, 1.50, 1.94 and 2.03, respectively, for BER, B1, B2 and B3.
Fluorescence quenching experiments provide an effective method for investigating the mode of binding of small molecules to nucleic acids.9 In a drug–nucleic acid complex, the molecules that are free or bound weakly on the surface may be readily available to an anionic quencher, while those bound between bases or inside the polynucleotide may be shielded from quenching. The electrostatic barrier of the negatively charged phosphate groups limits the penetration of an anionic quencher into the interior of the helix. Hence, very little or no quenching may be observed in the presence of an anionic quencher, if the binding involves intercalation/deep stacking. Consequently, the magnitude of the Stern–Volmer quenching constant (Ksv) of the ligands that are bound inside will be lower than that of the free molecules. It has been observed that binding to ss poly(A) resulted in the decreased quenching of the fluorescence intensity of analogues (Fig. S3†). The quenching constants (Ksv) calculated were in the range 200–230 M−1 for the unbound BER and analogues and were 71, 41, 44 and 54 M−1, respectively, for BER, B1, B2 and B3. The trend in the values varied as B1 < B2 < B3 < BER. This data suggests that the bound analogue molecules are located in a protected environment compared to BER confirming stronger intercalative binding on poly(A).
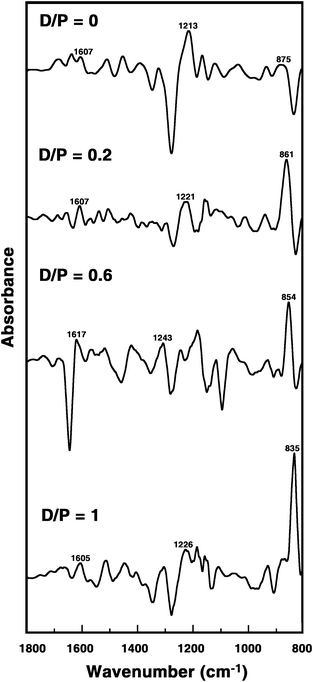
Optical melting experiments
Single stranded poly(A) does not show a cooperative melting pattern, but the self-structure reveals cooperative melting profile in absorbance and CD melting and this is a critical test for self structure formation.2e,3b In order to ascertain the ability of these berberine analogues to induce self-structure formation in poly(A) UV and CD melting experiments were performed. Sharp and cooperative UV melting profiles were observed for poly(A) complexes in presence of these analogues clearly revealing the formation of self-structure in poly(A). In Fig. 4A and B the representative UV melting profiles of poly(A) in absence and presence of analogue B1 are presented. The melting temperatures of the poly(A) self-structure were about 58, 58 and 60 °C, respectively, for B1, B2 and B3 and this is in excellent agreement with the melting temperatures reported in the literature for poly(A) self-structure induced by other small molecules.2d,g,3b
 |
| | Fig. 4 UV (A and B) and CD (C and D) melting profiles of poly(A) and poly(A) complexed with analogue B1. | |
Circular dichroic melting experiments of poly(A) in the presence of these analogues further confirmed the induction of self-structure formation in poly(A). Cooperative CD melting profiles were observed for poly(A) complexes in presence of these analogues confirming, unequivocally, the formation of self-structure in poly(A). In Fig. 4C and D the CD melting profiles of poly(A) and poly(A) in presence of analogue B1 are presented. The melting temperature of the poly(A) self-structure was about 57–60 °C and this is in agreement with the values reported in the literature.2d,g,3b Thus the absorbance and CD melting results confirm the induction of self-structure in poly(A) by all the berberine analogues.
Dilution experiments
To provide additional support for the induction of self-structure in poly(A) we probed the binding by dilution method of Hud and colleagues.3a From the data analysis of the curves presented in Fig. 5, the binding constant for BER, B1, B2 and B3 were deduced to be 1.0 × 104, 3.95 × 105, 3.03 × 105 and 2.62 × 105 M−1 and the cooperativity factors were obtained as 1.20, 1.32, 1.38 and 1.42, respectively The binding constant of BER obtained is in agreement with the value reported in the literature.3b In each case, the cooperativity factor was greater than 1, which indicated that the binding process was cooperative. Furthermore, it is noteworthy that the magnitude of both the binding constant and the cooperativity factor is significantly greater for the analogues compared to berberine , as it most likely reflects the greater ease of formation of the self-structure of poly(A) with the analogues in comparison to BER.
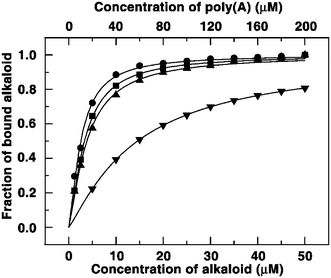 |
| | Fig. 5 Plot of fraction of bound BER (▼), B1 (•), B2 (■) and B3 (▲) against their concentration obtained from dilution experiment. | |
Thermodynamics of the interaction
Thermodynamic characterization of the binding of these berberine analogues to poly(A) was performed by isothermal titration calorimetry (ITC). This is an effective and sensitive tool to characterize the binding of small molecules to biomacromolecules,10a–c providing key insights about the molecular forces that drive the complex formation.10 The advantage of this technique is that from a single titration, a complete thermodynamic profile of the interaction can be built up together with information on the affinity constant and stoichiometry of the binding. A representative calorimetric titration of poly(A) into solutions of the analogues B1, B2 and B3 at 20 °C is presented in Fig. 6. Each heat burst curve in the upper panel of Fig. 6 corresponds to the heat generated in a single injection. These injection heats were corrected by subtracting the corresponding dilution heats derived from the injection of identical amounts of poly(A) into the buffer alone (shown at the top portion of the upper panel, curves off set for clarity). In the lower panels of Fig. 6, the resulting corrected heats are plotted against the molar ratio. The data points denote the experimental points and the continuous lines represent the calculated best fit of the data to the binding model. It was observed that the binding was characterized by exothermic heats in each case. The corrected isotherms showed single site binding in each case indicating that one type of complexation is formed exclusively and justifying the fitting of the data to a single site protocol. Fitting to one site model was also supported by the results from the Job plot analysis that revealed only one binding mode. The results of the ITC experiments are presented in Table 2. A large entropy contribution was seen for the binding of the analogues and this may be interpreted in terms of binding-induced release of bound water and condensed sodium ions. The binding affinity values (Ka) obtained are of the order of 106 M−1 for all the analogues which is remarkably higher in comparison to BER that had affinity of the order of 105 M−1. The binding affinity of B1 was enhanced more than eight times in comparison to BER and the affinity varied in the order B1 > B2 > B3 > BER which follows the same trend as obtained from spectroscopic studies. The site size (n) values, which are the reciprocal of stoichiometry (N) values, in the range 1.50–2.03 also enhanced going from B1 to B3 and are in excellent agreement with the stoichiometry values obtained from Job plot analysis. The binding of BER was reported to be driven largely by negative enthalpy (ΔHo) and a small favorable entropy change (ΔSo).3b The binding of all the analogues was favoured by strong entropy contribution and a comparatively small negative enthalpic contribution. The strongest binding was observed for analogue B1, which was predominantly entropy driven (TΔSo =7.09 kcal mole−1) with an enthalpy contribution of only −1.92 kcal mole−1. This was in sharp contrast to the binding of BER to ss poly(A) where the enthalpy contribution was −7.04 kcal mole−1 and the entropic contribution was only 0.80 kcal mole−1.3b The binding affinities of analogues B2 and B3 were weaker compared to B1 but was also favoured by relatively large entropy and smaller enthalpy contributions. The binding free energy (ΔGo) was −7.84 kcal mole−1 for BER that enhanced for all the analogues indicating higher binding preference. Comparison of the thermodynamic parameters helps to understand the molecular forces that govern the complexation. First of all, with the introduction of the substituent at 9-position a remarkable enhancement in binding affinity was observed. Secondly, the addition of the side chain at the 9-position enhanced the interaction free energy (in absolute values) by an additional 1.17 kcal mole−1 for B1 compared to BER, indicating more favorable contacts by the side chain on the assembly of adenine bases. The trend observed here in the thermodynamic parameters is generally typical for intercalative interaction of small molecules to nucleic acids.10f–h The strong and dominating positive entropy term for each of the analogues compared to BER is suggestive of the disruption and release of bound water molecules on intercalation within the assembly of adenine bases. The binding affinity values observed form ITC may be equated with K values obtained from spectroscopy.
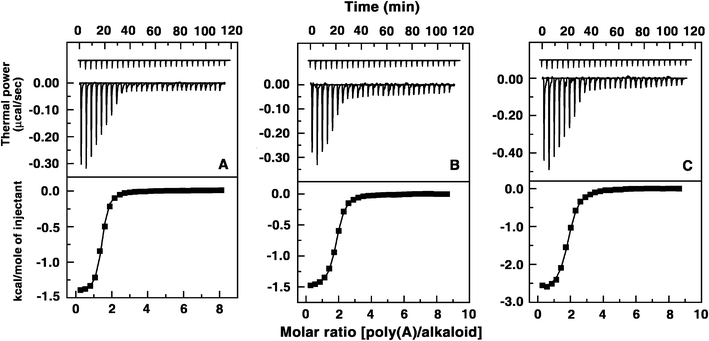 |
| | Fig. 6 Top panel: Representative ITC profile for the binding of alkaloids to poly(A) indicating the raw data for sequential injection of 10 μL of poly(A) into (A) B1, (B) B2, (C) B3 solution (curves on the bottom) and poly(A) dilution control (curves at the top offset for clarity). Bottom panel: Plot of integrated heat data after correction of heat of dilution of poly(A) against the molar ratio of the alkaloid to poly(A). The data were fitted to a one-site model and the solid curves represent the best fit. | |
Table 2 Thermodynamic parameters for the association of alkaloid analogues with poly(A) from ITCa
| Alkaloids |
K
a × 10−6 (M−1) |
N
|
ΔHo (kcal mole−1) |
TΔSo (kcal mole−1) |
ΔGo (kcal mole−1) |
|
All the data in this table are derived from ITC experiments and are average of four determinations. Ka (binding affinity), N (stoichiometry) and ΔHo (enthalpy change) values were determined from ITC profiles fitting to Origin 7.0 software as described in the text. The values of ΔGo (free energy change) were determined using the equations and ΔGo = ΔHo − TΔSo where TΔSo is the entropy contribution. All the ITC profiles were fit to a model of single binding site.
|
|
BER
|
0.65 ± 0.015 |
0.30 ± 0.002 |
−7.04 ± 0.035 |
0.80 |
−7.84 ± 0.017 |
|
B1
|
5.43 ± 0.033 |
0.74 ± 0.007 |
−1.92 ± 0.021 |
7.09 |
−9.01 ± 0.040 |
|
B2
|
4.39 ± 0.028 |
0.56 ± 0.004 |
−2.68 ± 0.025 |
6.21 |
−8.89 ± 0.031 |
|
B3
|
3.08 ± 0.021 |
0.56 ± 0.005 |
−4.82 ± 0.031 |
3.90 |
−8.72 ± 0.027 |
Effect of ionic strength and temperature on the binding
Like BER, the analogues have the quaternary nitrogen atom at the 7-position of the isoquinoline chromophore that is positively charged (Fig. 1). To provide an insight into the nature of the binding, the effect of salt concentration on the binding was studied by ITC experiments and the data was analyzed by van't Hoff plot for analogues B1, B2 and B3. The data at three [Na+] concentrations are presented in Table 3. The binding constants decreased as the salt concentration was increased. The ΔGo, ΔHo and TΔSo values slightly decreased. A relation between the binding constant and the [Na+] ion concentration was described previously11a as δlog(Ka)/δlog([Na+]) = −Zψ where Z is the apparent charge of the bound ligand and ψ is the fraction of [Na+] bound per phosphate group. A van't Hoff plot of log Kaversus log[Na+] is presented in Fig. S4†. The observed Gibbs energies of the interaction are in the range −8.10 to −9.10 kcal mole−1 (Table 3). In order to elucidate the dependence of Ka on [Na+], the observed free energy values were partitioned between the polyelectrolytic (ΔGope) and non polyelectrolytic (ΔGot) contributions. The contribution to the free energy from the electrostatic interaction (polyelectrolytic) can be quantitatively determined from the relationship ΔGope = ZψRTln([Na+]) where Zψ is the slope of the van't Hoff plot. At 50 mM [Na+] contributions from the ΔGope to the Gibbs energy have been determined to be around −1.30, −1.30 and −1.25 kcal mole−1 for B1, B2 and B3 (Table 3) which are about 15% of the total Gibbs energy change. This is similar to that observed for berberine, palmatine and coralyne binding to ds DNA and RNAs where majority of the free energy has been suggested to arise from non polyelectrolytic forces.11b,c At all salt concentrations, the ΔGot that had a large magnitude in each case, remained almost invariant. Thus, although positively charged like BER the binding of these analogues is mainly dominated by hydrophobic forces and forces other than electrostatic.
Table 3 Thermodynamic parameters for the association of alkaloid with poly(A) from ITC at different salt concentrationsa
| |
[Na+] (mM) |
K
a × 10−6 (M−1) |
ΔHo (kcal mole−1) |
TΔSo(kcal mole−1) |
ΔGo (kcal mole−1) |
ΔGot (kcal mole−1) |
ΔGope (kcal mole−1) |
|
All the data in this table are derived from ITC experiments conducted in CP buffer of different [Na+], pH 7.0 and are average of four determinations. Ka (binding affinity) and ΔHo (enthalpy change) values were determined from ITC profiles fitting to Origin 7.0 software as described in the text. The values of ΔGo (free energy change) and TΔSo (entropy contribution) were determined using the equations ΔGo = −RTlnKa, and TΔSo = ΔHo − ΔGo. All the ITC profiles were fit to a model of single binding site.
|
|
B1
|
10 |
5.43 ± 0.033 |
−1.92 ± 0.021 |
7.17 |
−9.09 ± 0.040 |
−7.09 |
−2.00 |
| 20 |
3.46 ± 0.024 |
−1.92 ± 0.020 |
6.90 |
−8.82 ± 0.032 |
−7.12 |
−1.70 |
| 50 |
1.67 ± 0.017 |
−1.85 ± 0.017 |
6.55 |
−8.40 ± 0.025 |
−7.10 |
−1.30 |
| 10 |
4.39 ± 0.028 |
−2.68 ± 0.025 |
6.28 |
−8.96 ± 0.031 |
−6.96 |
−2.00 |
|
B2
|
20 |
2.80 ± 0.021 |
−2.62 ± 0.023 |
6.08 |
−8.70 ± 0.029 |
−7.00 |
−1.70 |
| 50 |
1.35 ± 0.015 |
−2.57 ± 0.022 |
5.70 |
−8.27 ± 0.024 |
−6.97 |
−1.30 |
| 10 |
3.08 ± 0.021 |
−4.82 ± 0.031 |
3.93 |
−8.75 ± 0.027 |
−6.83 |
−1.92 |
|
B3
|
20 |
2.04 ± 0.017 |
−4.76 ± 0.030 |
3.75 |
−8.51 ± 0.025 |
−6.88 |
−1.63 |
| 50 |
0.99 ± 0.011 |
−4.65 ± 0.027 |
3.44 |
−8.09 ± 0.022 |
−6.84 |
−1.25 |
The constant pressure heat capacity changes (ΔCpo) of BER and analogues-ss poly(A) interaction were determined from the temperature dependent enthalpy results employing the standard relationship, ΔCpo = δΔHo/δT. Heat capacity change data can provide valuable insights into the type and magnitude of forces involved in the complexation. For this, ITC experiments were performed at three temperatures, viz. 10, 20 and 30 °C, and the thermodynamic parameters were evaluated. These data are depicted in Table 4. The BER analogues exhibited unusual temperature dependence upon complexation with ss poly(A). As the temperature increased, the association constant (Ka) of the complexation also increased significantly in each case thereby indicating that the complexation was favored with increasing temperature. A similar observation was reported for BER–poly(A) interaction and has been suggested to be due to the higher affinity of BER to poly(A) at 30 °C.11d It is likely that the self-structure formation may be favoured at higher temperatures that is enhancing the affinity of these analogues at higher temperature. For the analogue B1, the Ka value increased from 3.8 × 106 M−1 to 6.9 × 106 M−1 on increasing the temperature from 10 to 30 °C. Furthermore, there were remarkable changes in the enthalpy and entropy contributions with increasing temperature; while the binding ΔH° values increased, the favorable TΔSo values decreased. Such trends were observed for many biomolecular interactions11b,12a,b and were suggested to be due to the involvement of significant hydrophobic interactions in the binding. The variation of ΔHo with temperature (Fig. S5†) afforded the ΔCp° values. The slopes of the straight lines gave values of −78.8, −65.0 and −56.5 cal mole−1 K−1, respectively, for the binding of B1, B2 and B3. Negative heat capacity values have been observed for a large variety of small molecules binding to DNA and RNA structures.5b,10e,11b,12c–e Furthermore, a negative ΔCpo value is usually associated with change in hydrophobic or polar group hydration and is considered as an indicator of dominant hydrophobic interactions in the binding process. The heat capacity change values decreased gradually on going from B1 to B3 probably indicating that the complexation is disfavoured with increasing chain length. The differences in the ΔCpo values may also indicate differences in the release of structured water consequent to the transfer of nonpolar groups into the interior of the helix. First of all, the values of ΔCpo are non-zero and negative, indicating temperature dependence of the enthalpy change and significant hydrophobic contribution in the binding process. Secondly, in almost all the cases the values are very close to the value of 100–500 cal mole−1 K−1 that is frequently observed for ligand–nucleic acid interactions.12c,d Slightly higher ΔCpo value for B1 compared to the other analogues may suggest conformational differences in its structure and also differences in the disruption of the water structure around adenine bases on complexation. DNA recognition by small molecules involve sequence specific, non-specific, minimal sequence specific or structure specific binding.12f Small negative ΔCpo values, as observed here, are considered to be the hallmark of a minimal sequence specific binding and may denote structure specific binding. For intercalators, a significantly large hydrophobic contribution to the binding Gibbs energy is thought to be generated from their aromatic ring system and the binding should be energetically favourable.12g From the Record's relationship,12g ΔGohyd = 80(± 10) × ΔCpo, the Gibbs energy contribution from the hydrophobic transfer step of binding of these molecules were calculated. The values of ΔGohyd for B1, B2 and B3 binding to ss poly(A) were deduced to be −6.30, −5.20, and −4.52 kcal mole−1, respectively. These are well within the range that is observed for DNA and RNA intercalating small molecules.11b,12c,d,h
Table 4 Thermodynamic parameters for the association of alkaloids with poly(A) from ITC at different temperaturesa
| |
T (°C) |
K
a × 10−6 (M−1) |
ΔHo (kcal mole−1) |
TΔSo (kcal mole−1) |
ΔGo (kcal mole−1) |
ΔCpo (cal mole−1 K−1) |
ΔGohyd (kcal mole−1) |
|
All the data in this table are derived from ITC experiments conducted in citrate-phosphate buffer of 10 mM [Na+], pH 7.0 and are average of four determinations. Ka (binding affinity) and ΔHo (enthalpy change) values were determined from ITC profiles fitting to Origin 7.0 software as described in the text. The values of ΔGo (free energy change) were determined using the equation ΔGo = ΔHo − TΔSo where TΔSo is the entropy contribution. All the ITC profiles were fit to a model of single binding site.
|
|
B1
|
10 |
3.83 ± 0.027 |
−1.07 ± 0.011 |
7.44 |
−8.51 ± 0.038 |
|
|
| 20 |
5.43 ± 0.033 |
−1.92 ± 0.021 |
7.09 |
−9.01 ± 0.040 |
−78.8 |
−6.30 |
| 30 |
6.89 ± 0.039 |
−2.65 ± 0.025 |
6.85 |
−9.50 ± 0.044 |
|
|
| 10 |
3.17 ± 0.024 |
−2.17 ± 0.022 |
6.25 |
−8.42 ± 0.030 |
|
|
|
B2
|
20 |
4.39 ± 0.028 |
−2.68 ± 0.025 |
6.21 |
−8.89 ± 0.031 |
−65.0 |
−5.20 |
| 30 |
5.54 ± 0.034 |
−3.47 ± 0.027 |
5.88 |
−9.35 ± 0.042 |
|
|
| 10 |
2.08 ± 0.021 |
−4.31 ± 0.030 |
3.88 |
−8.19 ± 0.029 |
|
|
|
B3
|
20 |
3.08 ± 0.021 |
−4.82 ± 0.031 |
3.90 |
−8.72 ± 0.027 |
−56.5 |
−4.52 |
| 30 |
3.81 ± 0.027 |
−5.44 ± 0.035 |
3.70 |
−9.14 ± 0.041 |
|
|
Conclusions
The binding and associated thermodynamics of self-structure formation in poly(A) by three 9-O-N-aryl/arylalkyl amino carbonyl methyl substituted BER analogues were studied. The binding affinities of the analogues were higher by more than eight times compared to BER. Thus, with the introduction of 9-O-N-aryl/arylalkyl amino carbonyl methyl substituent the affinity enhanced remarkably. The highest binding affinity for B1 was 5.43 × 106 M−1. The cooperative binding of BER was propagated in the analogues also. After the threshold chain length of the substituent the binding affinity decreased gradually. This trend is identical to that observed for the binding of these analogues to DNA. Ferrocyanide quenching conclusively proved stronger intercalative geometry of the analogues compared to BER, the best being B1. FTIR studies provided further testimony for the strong interaction of the analogues with significant perturbations of the phosphates and adenine bases of poly(A). Stronger binding of the analogues was also inferred from circular dichroism studies as the secondary structural changes observed were more pronounced at a lower D/P. The presence of an induced CD band in the visible absorption region on complexation with poly(A) further established the strong environment of the bound molecules inside the poly(A) helix. Absorbance and CD melting studies along with the unique dilution experiment provided unambiguous evidence for the more rapid induction of self-structure in ss poly(A) by these analogues compared to BER. This data suggest that cooperative binding of these molecules and not planarity may be the criteria for the induction of self-structure. It is pertinent to mention here that the DNA binding affinities of these analogues were found to be more than six fold higher than that of BER. In all the cases the binding was dominated by non electrostatic forces that contributed to at least 85% of the binding Gibbs energy. Temperature dependent ITC studies revealed that the heat capacity values are negative which in turn indicated the involvement of significant hydrophobic and other multiple weak noncovalent forces in the binding reaction in agreement with the salt dependent studies. This study proves that the substitution at the 9-position of BER with aryl/arylalkyl amino carbonyl methyl substituent remarkably enhanced the binding affinity of BER to poly(A) and also proved that a threshold length of the side chain is critical for the strong binding to occur. Furthermore, results reiterate the importance of 9-O-substitution in BER for stronger nucleic acid binding. These results provide new insights into the 9-position derivatization of the natural alkaloid berberine in enhancing the binding with ss poly(A) and self structure induction that may be useful for the design and development of novel berberine based therapeutic agents with higher efficacy.
Experimental
General procedures
Polyriboadenylic acid [poly(A)], as potassium salt was purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA) and used as received. Concentration of poly(A) in terms of nucleotide phosphate (hereafter nucleotide) was determined by absorbance measurements at 257 nm using molar extinction coefficient (ε) values of 10000 M−1 cm−1.13 Berberine chloride was purchased from Sigma-Aldrich. B1, B2 and B3 were synthesized, purified and characterized as reported.5b The concentrations of B1, B2 and B3 were determined by applying the molar extinction coefficient (ε) values of 22500, 25000, 26000 and 26500 M−1 cm−1, respectively, at 345 nm. All buffer salts and other reagents were of analytical grade or better. Solutions were freshly prepared in the buffer and kept protected in the dark. All experiments were conducted in filtered 10 mM citrate-phosphate (CP) buffer, pH 7.0, containing 5 mM of Na2HPO4 prepared in deionised and triple distilled water. pH was adjusted with citric acid.
Spectroscopic titration experiments and binding data evaluation
The absorption spectral titrations were performed on a Jasco V660 unit (Jasco International Co, Hachioji, Japan) at 20 ± 0.5 °C following generally the methods standardized in our laboratory and described in details earlier.6b,14 Steady state fluorescence measurements were performed either on a Hitchi-F4010 fluorescence spectrometer (Hitachi Ltd., Tokyo, Japan) or Shimadzu RF 5301 PC unit (Shimadzu Corporation, Kyoto, Japan) in fluorescence free quartz cuvettes of 1 cm path length as described previously.6b,14a The excitation wavelength for BER and analogues was 345 nm. All measurements were performed keeping an excitation and emission band pass of 5 nm. The sample temperature was maintained at 20 ± 1.0 °C using an Eyela Uni Cool U55 water bath (Tokyo Rikakikai Co. Ltd., Tokyo, Japan). Binding data obtained from spectrophotometric and spectrofluorimetric titrations were converted into Scatchard plot of r/Cfversus r and were analyzed using the cooperative McGhee–von Hippel equation6a as described earlier.6b
Continuous variation analysis (Job plot)
Job's continuous variation method15a,b was employed to determine the binding stoichiometry in each case in fluorescence. The difference in fluorescence intensity of the alkaloids in the absence and presence of poly(A) (ΔF) was plotted as a function of the input mole fraction of each alkaloid as reported previously.14b Break point in the resulting plot corresponds to the mole fraction of the bound alkaloid in the complex. The stoichiometry was obtained in terms of poly(A)-alkaloid[(1 − χalkloid)/χalkaloid] where χalkaloid denotes the mole fraction of the respective analogue. The results presented are average of at least three experiments.
Thermal melting studies
Absorbance versus temperature profiles (melting curves) of the complexes were measured on a Shimadzu Pharmaspec 1700 spectrophotometer equipped with a peltier controlled TMSPC-8 model microcell accessory as reported previously.14 In a typical melting experiment, poly(A) samples were mixed with the respective alkaloid analogue and diluted into the degassed buffer into the eight chambered micro optical cuvette. The temperature of the microcell accessory was raised at a heating rate of 0.5 °C min−1. continuously monitoring the absorbance change at 258 nm. Melting curves revealed hyperchromic change at 258 nm that enabled the estimation of melting temperature, Tm as the midpoint of the hyperchromic transition obtained from the first derivative.
Circular dichroism spectra and CD melting profiles were recorded on a Jasco J815 unit (Jasco International Co.) equipped with a temperature controller and thermal programmer model PFD 425L/15 in strain free quartz cuvette of 1 cm path length as described earlier.4a,14
Dilution experiments
Dilution experiment were performed at 20 ± 0.5 °C following generally the methods reported by Hud.3a Briefly, a fixed concentration of poly(A)–alkaloid complex (200 μM poly(A) + 50 μM alkaloid analogue) was taken in the cell and the spectra was recorded. Then the solution was half diluted. If the pattern of spectrum remained same then the alkaloids are fully bound. The solution was diluted gradually and the absorbance at λmax was measured each time. The absorbance of free alkaloids and free poly(A) at λmax of the alkaloids (345 nm) was measured and their values were added. This value was taken as Af. The absorbance of the fully bound complex was taken as Ab and the value of absorbance at each dilution was taken as A. The molar extinction coefficient was determined for each case by dividing with the concentration. The molar extinction coefficient for free, bound, and intermediate alkaloid was taken as εf, εb, ε, respectively. So the fraction of bound alkaloid (θ) is obtained as (εf − ε)/(εf − εb). For BER, spectra of fully bound alkaloid could not be obtained even at the highest sample concentration investigated. Therefore, the amount of bound ligand could not be determined by the same two-spectra fitting procedure as described above. As an alternative suggested by Hud,3a the ratio of average absorbance between 458 nm and 478 nm (representing bound berberine) to average absorbance between 398 nm and 418 nm (representing unbound berberine) were taken as fraction of bound alkaloid. The spectra were corrected for background using the average absorbance between 510 nm and 530 nm. To determine the binding constant, θ was fitted into the Hill equation according to the procedure described in details earlier.3b
Infra red spectroscopic studies
Infrared spectra were recorded on a Jasco FTIR 4200 spectrophotometer using ATR cell and the analysis was done using the software supplied by the manufacturer. For each spectrum 512 scans were performed at a resolution of 4 cm−1. The spectra were measured in triplicate (three individual samples of the same poly(A) and alkaloid concentrations).
Isothermal titration calorimetry
All isothermal titration calorimetry experiments were performed using a MicroCal VP-ITC unit (MicroCal, Inc., Northampton, MA, USA) at 20 °C as reported previously.10e,12d Aliquots of degassed poly(A) solution were injected from a rotating syringe (610 rpm) into the isothermal sample chamber containing the alkaloid solutions (1.4235 mL). Corresponding control experiments to determine the heat of dilution of poly(A) were performed by injecting identical volumes of same concentration of poly(A) into the buffer. The instrument control, injection and analysis were all controlled by the dedicated Origin software. The area under each heat burst curve was determined by integration to give the measure of the heat associated with the injection. The heat associated with each RNA-buffer mixing was subtracted from the corresponding heat associated with the RNA injection to the alkaloid to give the heat of alkaloid binding to poly(A). The resulting corrected injection heats were plotted as a function of the molar ratio and fit with a model for one set of binding sites and analyzed using Origin 7.0 software to estimate the binding affinity (Ka), the binding stoichiometry (N) and the enthalpy of binding (ΔHo) as reported earlier.14b
Acknowledgements
Financial support from the CSIR network project on “Comparative genomics and biology of noncoding RNA in the human genome” (NWP0036) is gratefully acknowledged. A. Basu thanks the University Grants Commission, Government of India for the Senior Research Fellowship. The authors thank all the colleagues of the Chemistry and Biophysical Chemistry Laboratories for cooperation and help during the course of this work. The critical comments of the anonymous reviewers that enabled us to improve the manuscript are also appreciated.
References
-
(a) S. L. Topalian, S. Kaneko, M. I. Gonzales, G. L. Bond, Y. Ward and J. L. Manley, Mol. Cell. Biol., 2001, 21, 5614–5623 CrossRef CAS;
(b) S. L. Topalian, M. I. Gonzales, Y. Ward, X. Wang and R. F. Wang, Cancer Res., 2002, 62, 5505–5509 CAS.
-
(a) A. Rich, D. R. Davies, F. H. C. Crick and J. D. Watson, J. Mol. Biol., 1961, 3, 71–86 CrossRef CAS;
(b) W. M. Scovell, Biopolymers, 1978, 17, 969–984 CrossRef CAS;
(c) A. G. Petrovic and P. L. Polavarapu, J. Phys. Chem. B, 2005, 109, 23698–23705 CrossRef CAS;
(d) P. Giri and G. Suresh Kumar, Biochim. Biophys. Acta, 2007, 1770, 1419–1426 CrossRef CAS;
(e) P. Giri and G. Suresh Kumar, Arch. Biochem. Biophys., 2008, 474, 183–192 CrossRef CAS;
(f) A. Das, K. Bhadra, B. Achari, P. Chakraborty and G. Suresh Kumar, Biophys. Chem., 2011, 155, 10–19 CrossRef CAS;
(g) F. Xing, G. Song, J. Ren, J. B. Chaires and X. Qu, FEBS Lett., 2005, 579, 5035–5039 CrossRef CAS;
(h) M. Hossain, A. Kabir and G. Suresh Kumar, Dyes Pigm., 2012, 92, 1376–1383 CrossRef CAS.
-
(a) P. Centikol and N. V. Hud, Nucleic Acids Res., 2009, 37, 611–621 CrossRef;
(b) M. M. Islam, A. Basu and G. Suresh Kumar, Med. Chem. Commun., 2011, 2, 631–637 RSC.
-
(a) P. Krishnan and K. F. Bastow, Anti Cancer Drug Des., 2000, 15, 255–264 CAS;
(b) W. J. Zhang, T. M. Ou, Y. J. Lu, Y. Y. Huang, W. B. Wu, Z. S. Huang, J. L. Zhou, K. Y. Wong and L. Q. Gu, Bioorg. Med. Chem., 2007, 15, 5493–5501 CrossRef CAS;
(c) Y. Ma, T. M. Ou, J. Q. Hou, I. J. Lu, J. H. Tan, L. Gu and Q. Z. S. Huang, Bioorg. Med. Chem., 2008, 16, 7582–7591 CrossRef CAS.
-
(a) M. M. Islam, A. Basu, M. Hossain, G. Sureshkumar, S. Hotha and G. Suresh Kumar, DNA Cell Biol., 2011, 30, 123–133 CrossRef CAS;
(b) A. Basu, P. Jaisankar and G. Suresh Kumar, Bioorg. Med. Chem., 2012, 20, 2498–2505 CrossRef CAS;
(c) J. Y. Pang, Y. Qin, W. H. Chen, G. A. Luo and Z. H. Jiang, Bioorg. Med. Chem., 2005, 13, 5835–5840 CrossRef CAS;
(d) J. Y. Pang, Y. H. Long, W. H. Chen and Z. H. Jiang, Bioorg. Med. Chem. Lett., 2007, 17, 1018–1021 CrossRef CAS.
-
(a) J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS;
(b) K. Bhadra, M. Maiti and G. Suresh Kumar, Chem. Biodiversity, 2009, 6, 1323–1342 CrossRef CAS.
-
(a) H. Zhang, H. Yu, J. Ren and X. Qu, FEBS Lett., 2006, 580, 3726–3730 CrossRef CAS;
(b) B. B. Johnson, K. S. Dahl, I. Tinoco Jr., V. I. Ivanov and V. B. Zhurkin, Biochemistry, 1981, 20, 73–78 CrossRef CAS;
(c) M. R. Dufff Jr., V. K. Mudhivarthi and C. V. Kumar, J. Phys. Chem. B, 2009, 113, 1710–1721 CrossRef.
-
(a) S. Nafisi, A. Shadaloi, A. Feizbaksh and H. A. Tajmir-Riahi, J. Photochem. Photobiol., B, 2009, 94, 1–7 CrossRef CAS;
(b) S. Nafisi, M. Hashemi, M. Rajabi and H. A. Tajmir-Riahi, DNA Cell Biol., 2008, 27, 433–442 CrossRef CAS.
-
(a)
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 1983 Search PubMed;
(b) R. Sinha and G. Suresh Kumar, J. Phys. Chem. B, 2009, 113, 13410–13420 CrossRef CAS;
(c) P. Giri and G. S. Kumar, Mol. BioSyst., 2008, 4, 341–384 RSC.
-
(a) A. Rajendran, C. Zhao, B. Rajendar, V. Thiagarajan, Y. Sato, S. Nishizawa and N. Teramae, Biochim. Biophys. Acta, 2010, 1800, 599–610 CrossRef CAS;
(b) B. Rajendar, A. Rajendran, Z. Ye, E. Kanai, Y. Sato, S. Nishizawa, M. Sikorski and N. Teramae, Org. Biomol. Chem., 2010, 8, 4949–4959 RSC;
(c) B. Rajendar, A. Rajendran, Y. Sato, S. Nishizawa and N. Teramae, Bioorg. Med. Chem., 2009, 17, 351–359 CrossRef CAS;
(d)
R. O'Brien and I. Haq, Applications of Biocalorimetry: Binding, Stability and Enzyme Kinetics, in Biocalorimetry 2, ed. J. E. Ladbury and M. Doyle, John Wiley and Sons Ltd., West Sussex, UK, 2004, pp. 1–34. Search PubMed;
(e) M. Hossain and G. Suresh Kumar, J. Chem. Thermodyn., 2009, 41, 764–774 CrossRef CAS;
(f) I. Haq, Arch. Biochem. Biophys., 2002, 403, 1–15 CrossRef CAS;
(g) M. Hossain and G. Suresh Kumar, Mol. BioSyst., 2009, 5, 1311–1322 RSC;
(h) I. Saha, M. Hossain and G. Suresh Kumar, Phys. Chem. Chem. Phys., 2010, 12, 12771–12779 RSC.
-
(a) M. T. Jr. Record, C. F. Anderson and T. M. Lohman, Q. Rev. Biophys., 1978, 11, 103–178 CrossRef;
(b) M. M. Islam, S. Roy Chowdhury and G. Suresh Kumar, J. Phys. Chem. B, 2009, 113, 1210–1224 CrossRef CAS;
(c) K. Bhadra, M. Maiti and G. Suresh Kumar, DNA Cell Biol., 2008, 27, 675–685 CrossRef CAS;
(d) R. C. Yadav, G. Suresh Kumar, K. Bhadra, P. Giri, R. Sinha, S. Pal and M. Maiti, Bioorg. Med. Chem., 2005, 13, 165–174 CrossRef CAS.
-
(a) J. B. Chaires, Arch. Biochem. Biophys., 2006, 453, 26–31 CrossRef CAS;
(b) L. Jen-Jacobson, L. E. Engler and L. A. Jacobson, Structure, 2000, 8, 1015–1023 CrossRef CAS;
(c) K. M. Guthrie, A. D. Parenty, L. V. Smith, L. Cronin and A. Cooper, Biophys. Chem., 2007, 126, 117–123 CrossRef CAS;
(d) J. Ren, T. C. Jenkins and J. B. Chaires, Biochemistry, 2000, 39, 8439–8447 CrossRef CAS;
(e) A. Das and G. Suresh Kumar, Mol. BioSyst., 2012, 8, 1958–1969 RSC;
(f) F. V. Murphy and M. E. Churchill, Structure, 2000, 15, R83–R89 CrossRef;
(g) J. H. Ha, R. S. Spolar and M. T. Record Jr., J. Mol. Biol., 1989, 209, 801–816 CrossRef CAS;
(h) S. Roy Chowdhury, M. M. Islam and G. Suresh Kumar, Mol. BioSyst., 2010, 6, 1265–1276 RSC.
- C. Ciatto, M. L. D'Amico, G. Natile, F. Secco and M. Venturini, Biophys. J., 1999, 77, 2717–2724 CrossRef CAS.
-
(a) K. Bhadra, M. Maiti and G. Suresh Kumar, Biochim. Biophys. Acta, 2007, 1770, 1071–1080 CrossRef CAS;
(b) M. M. Islam, R. Sinha and G. Suresh Kumar, Biophys. Chem., 2007, 125, 508–520 CrossRef CAS;
(c) S. Das and G. Suresh Kumar, J. Mol. Struct., 2008, 872, 56–63 CrossRef CAS.
-
(a) P. Job, Ann. Chim., 1928, 9, 113–203 CAS;
(b) C. Y. Huang, Methods Enzymol., 1982, 27, 509–525 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, absorbance and fluorescence spectra of berberine (BER) and analogues binding to poly(A) and Scatchard plots; Fig. S2, Job’s plot for the binding; Fig. S3, Stern–Volmer plots for fluorescence quenching; Fig. S4, the plot of log Kaversus log [Na+]; Fig. S5, plot of variation of enthalpy of binding (ΔHo) with temperature. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.