Formal total synthesis of (−)-exiguolide†
Chada Raji
Reddy
* and
Nagavaram Narsimha
Rao
Division of Natural Products Chemistry, CSIR-Indian Institute of Chemical Technology, Hyderabad 500 007, India. E-mail: rajireddy@iict.res.in; Fax: +91-40-27160512
First published on 22nd June 2012
Abstract
The formal total synthesis of (−)-exiguolide through the chiral-pool approach is described. The key reactions involved for formation of the macrocyclic core from two subunits are Julia–Kocienski olefination and Yamaguchi macrolactonization. The major methylene bis-tetrahydropyran fragment was achieved in a convergent manner from L-glutamic acid and L-aspartic acid involving the oxa-Michael reaction and an aldol-driven reductive etherification as key steps for the formation of a tetrahydropyran ring. The other sulfone subunit was prepared via an Evans aldol reaction.
Introduction
(−)-Exiguolide (1, Fig. 1) was isolated in 2006 by Ohta, Ikegami and co-workers from the marine sponge Geodia exigua Thiele (order Astrophorida, family Geodiidae).1 The structure, along with relative stereochemistry, has been determined by a combination of wide NMR study, conformational analysis based on nuclear Overhauser effect spectroscopy (NOESY) correlations and J-based configuration analysis. Structurally, it is a 20-membered macrolide with a methylene bis-tetrahydropyran (THP) subunit, five carbon–carbon double bonds and seven asymmetric carbons. The presence of the bispyran framework and the exocyclic enoate attached to one of the THP-rings are common motifs that can be found in marine antineoplastic agents, bryostatins.2 (−)-Exiguolide specifically inhibits the fertilization of sea urchin (Hemicentrotus pulcherrimus) gametes but not embryogenesis of the fertilized egg at higher concentrations. Based on these biological and structural features of (−)-1, an assumption has been made that (−)-exiguolide represents a structurally simplified analogue of the bryostatins.2b The limited supply and structural complexity of (−)-1 renders this molecule and its analogues important targets for chemical synthesis towards further evaluation of biological activities.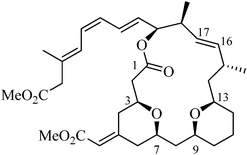 | ||
| Fig. 1 Structure of (−)-exiguolide (1). | ||
In 2008, Lee and co-workers reported the total synthesis of (+)-exiguolide, an enantiomer of the natural product, using ring-closing metathesis, Prins cyclization, radical cyclization and Sonogashira coupling as the key steps.3 From this synthesis, they have determined the absolute stereochemistry of (−)-exiguolide. Later, three total syntheses of (−)-1 were published.4 In the year 2010, Fuwa and Sasaki reported the total synthesis of (−)-1via Julia–Kocienski olefination, reductive etherification, intramolecular oxa-conjugate addition and Suzuki–Miyayura coupling as the key reactions.4a In the same year, Roulland and co-workers accomplished (−)-1 using Yamaguchi macrolactonization, Trost's ruthenium-catalyzed ene–yne cross-coupling, reductive etherification and Sonogashira cross-coupling as steps.4b Recently, Scheidt and co-workers have described the enantioselective synthesis of (−)-exiguolide by dioxinone-directed Prins cyclizations to construct the THP-rings, as well as for macrocyclization and Leibeskind CuI-mediated coupling to stitch the side chain. Further, they have evaluated the ability of (−)-1 to inhibit cell growth on nine cancer cell lines and found significant antiproliferative activity against A549 lung cancer cells compared to others.4c Prior to this work, Fuwa et al. also elucidated the assessment of the growth inhibitory activity of synthetic (−)-exiguolide and its analogues against a panel of human cancer cell lines and observed that (−)-1 is an effective antiproliferative agent against the NCI-H460 human lung large carcinoma (log GI50 = −8.00) as well as the A549 human lung adenocarcinoma cell lines (log GI50 = −6.19).5 Notably (−)-1 has been found to have a 10 to 100-fold greater potency than bryostatin (log GI50 = −5.6 for NCI-H460, −5.4 for A549). They have also investigated the structure–activity relationships, which elucidated that the C5-methoxycarbonylmethylidene group and length of the side chain are important for biological activity.
Our interest in macrolides and THP-containing molecules6 coupled with the distinctive structure of (−)-exiguolide has encouraged us to attempt the synthesis of (−)-1. In this paper, we provide the full details of our work on the formal total synthesis of (−)-exiguolide.7 The retrosynthetic analysis of (−)-exiguolide is illustrated in Scheme 1. We envisioned that the side chain installation on iodo-macrocyclic core 2 would be at the final stage of the synthesis, which may be helpful for preparing new analogs. The macrocycle 2 could be realized from bis-THP subunit 3 and iodo-sulfone 4 through Julia–Kocienski olefination (C16–C17 double bond formation) followed by Yamaguchi macrolactonization. The synthesis of fragment 3 was planned in a convergent fashion from the THP-ketone 5 and aldehyde 6, utilizing an aldol-reaction followed by reductive etherification. Notably, THP-ketone 5 may in turn be obtained from L-glutamic acid using the oxa-Michael reaction, and aldehyde 6 could be derived from L-aspartic acid. The iodo-sulfone precursor 4 was envisaged from ethyl propiolate via iodo acrolein 7 by the Evans aldol approach.
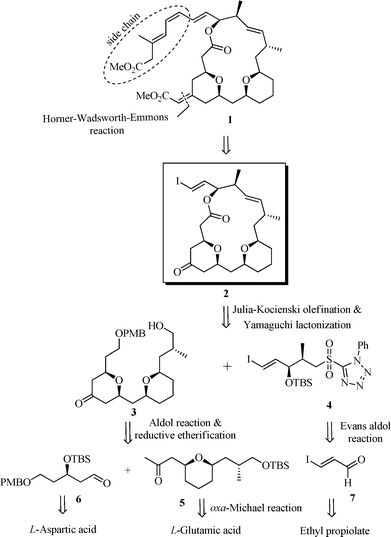 | ||
| Scheme 1 Retrosynthetic analysis of (−)-1. | ||
Results and discussion
Synthesis of THP-ketone, C6–C16 fragment (5)
The synthesis of THP-ketone 5 was initiated by carrying out the preparation of the known tert-butyldiphenyl silyl protected (4S)-hydroxymethyl-4-butanolide 8 from L-glutamic acid using the literature method.8 In the first step, stereoselective methylation of 8 was carried out under LDA/MeI/−78 °C reaction conditions to get 9 in 78% yield. This reaction installed the C-15 methyl stereo center.9 The reductive opening of lactone 9 using lithium borohydride provided the 1,4-diol 10 in 99% yield. Subsequent protection of 1,4-diol 10 under 2,2-DMP/CSA reaction conditions afforded a seven-membered acetonide 11 in 90% yield. Then, the deprotection of the tert-butyldiphenyl silyl group of 11 was accomplished using tetrabutyl ammonium fluoride to give the alcohol 12 in 91% yield. In order to substitute the primary hydroxyl group with a four-carbon chain, a two-step procedure was planned. Accordingly, the alcohol 12 was tosylated (TsCl/Py, 96% yield) to 13, and subsequently tosylate 13 was subjected to a Schlosser copper-catalyzed Grignard coupling reaction with 3-butenyl magnesium bromide in the presence of dilithium tetrachloro cuprate, to give 14 in 88% yield.10 Synthesis of precursor 15 for the oxa-Michael reaction towards the pyran-ring formation was initially accomplished from 14 using a cross-metathesis reaction with methyl vinyl ketone in 56% yield.11 Alternatively, to get a better yield, olefin 14 was subjected to ozonolysis followed by a Wittig olefination (PPh3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOMe), to provide α,β-unsaturated ketone 15 in 84% yield (over two steps). Hydrolysis of acetonide 15 and consequent oxa-Michael addition were accomplished through a p-TSA-catalyzed one-pot reaction to get pyran 16 in 89% yield.12 The NOE effect between H-9 and H-13 (2D NOESY) confirmed the relative syn-relationship of 2,6-disubstituted tetrahydropyran. The protection of the free-hydroxyl group of pyran 16 as tert-butyldimethyl silyl ether using TBSCl/imidazole gave the desired THP-ketone fragment 5 in 95% yield.
CHCOMe), to provide α,β-unsaturated ketone 15 in 84% yield (over two steps). Hydrolysis of acetonide 15 and consequent oxa-Michael addition were accomplished through a p-TSA-catalyzed one-pot reaction to get pyran 16 in 89% yield.12 The NOE effect between H-9 and H-13 (2D NOESY) confirmed the relative syn-relationship of 2,6-disubstituted tetrahydropyran. The protection of the free-hydroxyl group of pyran 16 as tert-butyldimethyl silyl ether using TBSCl/imidazole gave the desired THP-ketone fragment 5 in 95% yield.
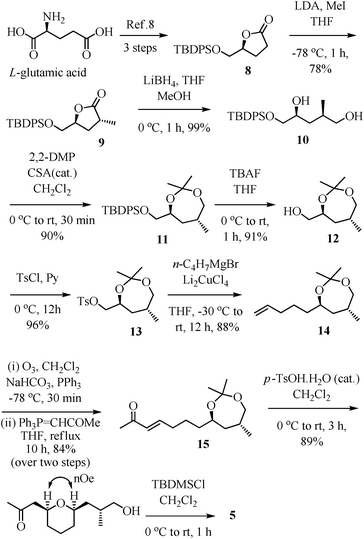 | ||
| Scheme 2 Synthesis of THP-ketone fragment 5. | ||
Synthesis of aldehyde, C1–C5 fragment (6)
Synthesis of the C1–C5 subunit (Scheme 3) commenced with the preparation of epoxide 17 from commercially available L-aspartic acid using a known procedure.13 The conversion of the epoxide 17 to homoallylic alcohol 18 was accomplished through the copper(I)-catalyzed addition of vinyl magnesium bromide in 96% yield.14 Alcohol 18 was protected as tert-butyldimethyl silyl ether 19 in 95% yield (TBSCl/imidazole/DMF). It was then subjected to osmium-catalyzed dihydroxylation of the olefin followed by cleavage of the diol using sodium periodate to furnish aldehyde 6 in 84% yield.15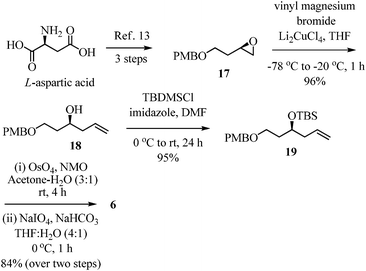 | ||
| Scheme 3 Synthesis of aldehyde fragment 6. | ||
In an alternative approach to 6, a route via the opening of epoxide 17 with 1,3-dithiane was explored (Scheme 4). Thus, the treatment of epoxide 17 with 1,3-dithiane in the presence of n-butyl lithium gave the alcohol 20 in 82% yield. Protection of the hydroxyl group in 20 as tert-butyldimethyl silyl ether gave compound 21 in 86% yield (TBSOTf/2,6-lutidine/CH2Cl2) and subsequent deprotection of thioacetal using MeI/NaHCO3 in CH3CN/H2O afforded the aldehyde 6 in 83% yield.16
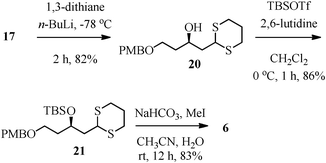 | ||
| Scheme 4 Alternative strategy for aldehyde 6. | ||
Synthesis of sulfone fragment 4
As shown in Scheme 5, synthesis of the sulfone fragment 4 was based on known iodo acrolein 7, which was prepared from ethyl propiolate by a reported three-step sequence.17 For the required syn-propionate homologation, an Evans aldol reaction18 of 7 with the di-n-butylboron enolate of (R)-4-benzyl-3-propionyloxazolidin-2-one (22) was carried out to furnish the syn-product 23 in 74% yield. Exposure of 23 to TBSOTf/2,6-lutidine in CH2Cl2 gave the TBS-ether 24 in 90% yield. Compound 24 was treated with LiBH4 in THF–MeOH for the reductive removal of the auxiliary, to furnish alcohol 25. A Mitsunobu reaction19 of alcohol 25 with 1-phenyl-1H-tetrazole-5-thiol to thio tetrazole 26 (96% yield), followed by ammonium molybdate catalyzed oxidation20 of 26, completed the synthesis of sulfone 4 in 81% yield.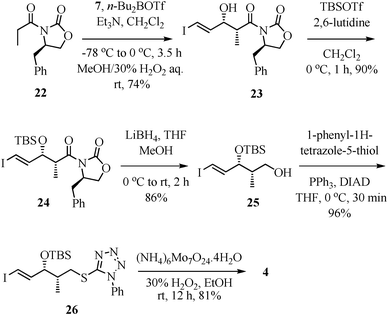 | ||
| Scheme 5 Synthesis of sulfone fragment 4. | ||
Coupling of fragments
Having all the desired fragments 4, 5 and 6 in hand, we next proceeded to the synthesis of methylene bis-tetrahydropyran subunit 3 through an aldol driven reductive etherification strategy (Scheme 6). Hence, an aldol reaction of aldehyde 6 with the lithiated enolate of 5, derived using LDA, formed an aldol product as a inseparable mixture of diastereomers. This mixture was treated with Dess–Martin periodinane to give β-diketone 27 (71% yield over two steps), which entirely existed as the enol form (confirmed by 1H NMR). Deprotection of the TBS groups and cyclization was achieved in a one-pot operation by the reaction of 27 with HF (40% aq. solution) in CH3CN at room temperature, to furnish dihydropyranone 28 in 86% yield.21 Hydrogenation of 28 proceeded with the exclusive formation of cis-tetrahydropyranone 3 in 91% yield.22 The syn-relationship of both the THP rings was confirmed by 2D NOESY experiments.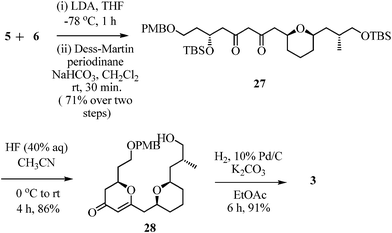 | ||
| Scheme 6 Synthesis of bis-THP subunit 3. | ||
Next, the task was to couple the bis-THP subunit 3 with sulfone 4 by Julia–Kocienski reaction.23 Accordingly, oxidation of alcohol 3 with Dess–Martin periodinane gave the corresponding aldehyde (92% yield), but the following olefination of aldehyde with sulfone 4 using KHMDS as a base failed to give the desired product 29. Varying the reaction conditions (using different bases and solvent systems) also didn't help to get the desired product. These disappointing results prompted us to protect the keto-group present on the THP ring prior to the Julia–Kocienski reaction. Thus, exposure of 3 to trimethyl orthoformate/p-TSA·H2O (cat.) in MeOH afforded the ketal 30 in 82% yield. Now, the primary alcohol in 30 was oxidized to an aldehyde followed by Julia–Kocienski olefination with sulfone 4 using LiHMDS in THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HMPA (4:1), affording 31 in 56% yield (80% brsm) with E-selectivity (>20:1) at the newly formed C16–C17 double bond. Unmasking the p-methoxybenzyl ether of 31 was achieved using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in CH2Cl2:pH 7 buffer (9:1) to provide 32 in 92% yield.24 To advance 32 towards a substrate suitable for the intramolecular Yamaguchi macrolactonization, the primary hydroxyl of 32 was oxidized to carboxylic acid under BAIB–TEMPO reaction25 conditions, followed by deprotection of the TBS-group with tetrabutylammonium fluoride (TBAF), to afford the hydroxy acid 33 in 91% yield (over two steps). Intramolecular macrolactonization of the hydroxy acid 33 under Yamaguchi reaction conditions26 gave the ketal protected macrolactone 34 in 63% yield. Further, hydrolysis of the ketal group of macrolide 34 with p-TSA·H2O (cat.) provided the desired macrocycle 2, whose spectral data matched exactly to that of the same compound prepared by Fuwa et al. for the synthesis of (−)-exiguolide (Scheme 7).4a
:HMPA (4:1), affording 31 in 56% yield (80% brsm) with E-selectivity (>20:1) at the newly formed C16–C17 double bond. Unmasking the p-methoxybenzyl ether of 31 was achieved using 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) in CH2Cl2:pH 7 buffer (9:1) to provide 32 in 92% yield.24 To advance 32 towards a substrate suitable for the intramolecular Yamaguchi macrolactonization, the primary hydroxyl of 32 was oxidized to carboxylic acid under BAIB–TEMPO reaction25 conditions, followed by deprotection of the TBS-group with tetrabutylammonium fluoride (TBAF), to afford the hydroxy acid 33 in 91% yield (over two steps). Intramolecular macrolactonization of the hydroxy acid 33 under Yamaguchi reaction conditions26 gave the ketal protected macrolactone 34 in 63% yield. Further, hydrolysis of the ketal group of macrolide 34 with p-TSA·H2O (cat.) provided the desired macrocycle 2, whose spectral data matched exactly to that of the same compound prepared by Fuwa et al. for the synthesis of (−)-exiguolide (Scheme 7).4a
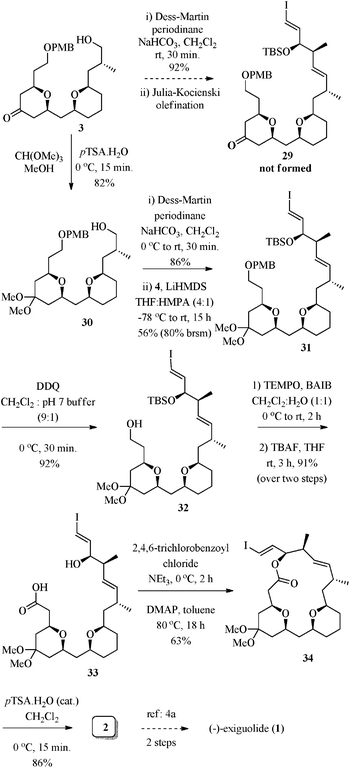 | ||
| Scheme 7 Synthesis of the macrocyclic core of (−)-exiguolide (1). | ||
Conclusions
In summary, a flexible convergent synthesis of a macrocyclic precursor of (−)-exiguolide has been accomplished through a maximum-length linear sequence of 25 steps. The key reactions in the described route are the oxa-Michael reaction and an aldol driven reductive etherification for THP-ring formation, Julia–Kocienski olefination and Yamaguchi macrolactonization for macrocycle synthesis. We believe that the synthetic strategy developed is practical and scalable from inexpensive commercially available natural amino acids.Experimental
General
Reactions were monitored by thin-layer chromatography carried out on silica plates (silica gel 60 F254, Merck) using UV-light and anisaldehyde or β-naphthol for visualization. Column chromatography was performed on silica gel (60–120 mesh) using hexanes, ethyl acetate, methanol and chloroform as the eluent. Evaporation of solvents was conducted under reduced pressure at temperatures less than 45 °C. IR spectra were recorded on a Perkin-Elmer 683, Nicolet Nexus 670 spectrometer. 1H NMR and 13C NMR spectra were recorded in CDCl3 solvent on a 300 MHz, 400 MHz and 500 MHz NMR spectrometer. Chemical shifts δ and coupling constants J are given in ppm (parts per million) and Hz (Hertz) respectively. Chemical shifts are reported relative to the residual solvent as an internal standard for 1H and 13C (CDCl3: δ 7.26 ppm for 1H and 77.0 ppm for 13C). Mass spectra were obtained on a Finnigan MAT1020B, micromass VG 70–70H or LC/MSD trapSL spectrometer operating at 70 eV using a direct inlet system.:EtOAc = 81:15) to give 9 (4.0 g, 78%) as a colorless oil. Rf 0.40 (20% EtOAc in hexanes); IR (KBr): νmax 2931, 2858, 1760, 1191, 1109, 1024, 826, 703 cm−1; [α]20D = +32.0 (c = 1.20, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.70–7.62 (m, 4H), 7.48–7.35 (m, 6H), 4.59–4.50 (m, 1H), 3.85 (dd, J = 11.3, 3.4 Hz, 1H), 3.66 (dd, J = 11.3, 3.2 Hz, 1H), 2.94–2.78 (m, 1H), 2.44 (ddd, J = 12.6, 9.6, 2.8 Hz, 1H), 2.07–1.89 (m, 1H), 1.29 (d, J = 7.3 Hz, 3H), 1.05 (s, 9H); 13C NMR (75 MHz, CDCl3): δ = 180.3, 135.4, 132.8, 132.3, 129.8, 127.8, 77.5, 65.4, 34.2, 32.1, 26.6, 19.1, 16.3; MS (ESI): m/z = 391 [M + Na]+ HRMS: calcd. for C22H32O3SiN (M + NH4)+ 386.2146; found 386.2160.
:EtOAc = 70:30) to give 10 (5.0 g, 99%) as a colorless oil. Rf 0.40 (30% EtOAc in hexanes); IR (KBr): νmax 3415, 2957, 2931, 2861, 1111, 758, 703 cm−1; [α]27D = +3.0 (c = 1.10, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.67–7.60 (m, 4H), 7.46–7.33 (m, 6H), 3.87 (m, 1H), 3.58 (dd, J = 10.0, 3.6 Hz, 1H), 3.55–3.39 (m, 3H), 2.81 (br s, 2H), 1.93–1.77 (m, 1H), 1.52–1.31 (m, 2H), 1.07 (s, 9H), 0.90 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 135.4, 133.0, 129.8, 127.7, 69.2, 67.9, 67.5, 36.4, 32.2, 26.8, 19.1, 16.9; MS (ESI): m/z = 395 [M + Na]+; HRMS: calcd. for C22H32O3SiNa (M + Na)+ 395.2013; found 395.2029.
:EtOAc = 95:5) to give 11 (700 mg, 90%) as a colorless oil. Rf 0.50 (10% EtOAc in hexanes); IR (KBr): νmax 2934, 2861, 1760, 1219, 1110, 1080, 703 cm−1; [α]20D = −14.5 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.70–7.60 (m, 4H), 7.42–7.30 (m, 6H), 3.93–3.81 (m, 2H), 3.59 (dd, J = 10.2, 6.6 Hz, 1H), 3.44 (dd, J = 10.2, 5.8 Hz, 1H), 3.36–3.28 (m, 1H), 1.86–1.74 (m, 1H), 1.54–1.36 (m, 2H), 1.28 (s, 3H), 1.22 (s, 3H), 1.04 (s, 9H), 1.02 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 135.7, 135.6, 129.6, 127.6, 100.7, 68.8, 67.2, 65.9, 37.5, 31.0, 26.8, 25.0, 19.2, 17.1; MS (ESI): m/z = 435.1 [M + Na]+.
:EtOAc = 80:20) to give 12 (460 mg, 91%) as a colorless oil. Rf 0.40 (30% EtOAc in hexane); IR (KBr): νmax 3367, 2927, 2873, 1375, 1219, 1045, 773 cm−1; [α]27D = +11.8 (c = 0.80, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 4.00–3.90 (m, 1H), 3.87 (dd, J = 12.0, 1.5 Hz, 1H), 3.48–3.31 (m, 3H), 1.93–1.79 (m, 1H), 1.55–1.43 (m, 1H), 1.33 (s, 6H), 1.31–1.24 (m, 1H), 1.04 (d, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 108.8, 73.7, 69.6, 67.3, 36.9, 32.8, 26.8, 25.7, 16.7.
:EtOAc = 90:10) to give 13 (3.6 g, 96%) as a colorless oil. Rf 0.50 (20% EtOAc in hexanes). IR (KBr): νmax 2983, 2922, 1362, 1218, 1177, 1063, 983, 812 cm−1; [α]20D = +3.18 (c = 0.88, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.77 (d, J = 8.3 Hz, 2H), 7.32 (d, J = 8.3 Hz, 2H), 4.08–3.97 (m, 1H), 3.95–3.78 (m, 3H), 3.31 (dt, J = 12.0, 2.3 Hz, 1H), 2.46 (s, 3H), 1.88–1.76 (m, 1H), 1.47–1.34 (m, 2H), 1.24 (s, 6H), 1.01 (d, J = 7.5 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 144.6, 133.0, 129.6, 127.7, 100.8, 72.4, 65.6, 65.5, 36.8, 30.5, 24.6, 24.5, 21.4, 16.8; MS (ESI): m/z = 351 [M + Na]+; HRMS: calcd. for C16H24O5SNa (M + Na)+ 351.1237; found 351.1236.
:EtOAc = 95:5) to give olefin 14 (1.8 g, 88%) as a colorless oil. Rf 0.60 (10% EtOAc in hexanes); IR (KBr): νmax 2934, 1378, 1219, 1060 cm−1; [α]20D = −16.8 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 5.88–5.72 (m, 1H), 5.04–4.91(m, 2H), 3.91 (dd, J = 12.0, 1.5 Hz, 1H), 3.87–3.77 (m, 1H), 3.36 (dt, J = 12.0, 3.0 Hz, 1H), 2.06 (dd, J = 14.3, 6.8 Hz, 2H), 1.85–1.73 (m, 1H), 1.59–1.35 (m, 6H), 1.34 (s, 3H), 1.32 (s, 3H), 1.04 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 138.8, 114.3, 100.3, 67.8, 65.7, 41.7, 36.2, 33.8, 31.2, 25.7, 25.1, 17.1.
To a solution of the above aldehyde in THF (30 mL) was added methyl-(triphenylphosphoranylidene)-2-propanone (3.12 g, 9.81 mmol) and the mixture was heated under reflux for 12 h. After cooling, the solvent was removed in vacuo, and the residue was purified by column chromatography (silica gel, hexanes:EtOAc = 90:10) to give enone 15 (1.4 g, 84%) as a colorless oil. Rf 0.50 (20% EtOAc in hexanes); IR (KBr): νmax 2931, 2864, 1712, 1673, 1358, 1218, 1072, 1039 cm−1; [α]26D = +2.6 (c = 1.05, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 6.74 (dt, J = 15.8, 6.8 Hz, 1H), 6.03 (dt, J = 15.8, 1.4 Hz, 1H), 3.85 (dd, J = 12.0, 1.5 Hz, 1H), 3.82–3.72 (m, 1H), 3.31 (dt, J = 12.0, 2.3 Hz, 1H), 2.28–2.17 (m, 2H), 2.22 (s, 3H) 1.83–1.70 (m, 1H), 1.71–1.32 (m, 6H), 1.30 (s, 3H), 1.27 (s, 3H), 1.02 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 198.6, 148.2, 131.3, 100.3, 67.6, 65.7, 41.7, 36.2, 32.4, 31.1, 26.8, 25.1, 25.0, 24.9, 17.1; MS (ESI): m/z = 277 [M + Na]+; HRMS: calcd. for C15H26O3Na (M + Na)+ 277.1774; found 277.1776.
:EtOAc = 70:30) to give 16 (337 mg, 89%) as a colorless oil. Rf 0.40 (50% EtOAc in hexanes); IR (KBr): νmax 3433, 2927, 1709, 1361, 1195, 1037 cm−1; [α]28D = −3.3 (c = 0.90, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 3.85–3.74 (m, 1H), 3.57–3.48 (m, 1H), 3.46 (dd, J = 10.5, 5.3 Hz, 1H), 3.34 (dd, J = 10.5, 6.8 Hz, 1H), 2.67 (dd, J = 15.8, 7.5 Hz, 1H), 2.39 (dd, J = 15.8, 5.3 Hz 1H), 2.15 (s, 3H), 1.93–1.78 (m, 2H), 1.65–1.37 (m, 5H), 1.31–1.12 (m, 3H), 0.9 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 207.3, 75.5, 73.9, 67.6, 49.8, 40.1, 32.0, 31.1, 31.0, 30.9, 23.3, 17.2; MS (ESI): m/z = 237 [M + Na]+; HRMS: calcd. for C12H22O3Na (M + Na)+ 237.1461; found 237.1463.
:EtOAc = 95:5) to give 5 (1.24 g, 95%) as a colorless oil. Rf 0.30 (5% EtOAc in hexanes); IR (KBr): νmax 2930, 2856, 1717, 1360, 1252, 1086, 839, 775 cm−1; [α]27D = −3.2 (c = 1.20, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 3.77–3.65 (m, 1H), 3.44–3.30 (m, 3H), 2.64 (dd, J = 15.1, 7.8 Hz, 1H), 2.35 (dd, J = 15.1, 4.8 Hz, 1H), 2.15 (s, 3H), 1.88–1.65 (m, 2H), 1.64–1.49 (m, 2H), 1.47–1.37 (m, 1H), 1.33–1.06 (m, 4H), 0.89 (s, 9H), 0.87 (d, J = 6.5 Hz, 3H), 0.03 (s, 6H); 13C NMR (75 MHz, CDCl3): δ = 207.7, 76.4, 74.3, 67.7, 50.3, 39.8, 32.3, 31.9, 31.5, 31.4, 31.0, 25.9, 23.5, 17.5, −5.4; MS (ESI): m/z = 351 [M + Na]+; HRMS: calcd. for C18H36O3SiNa (M + Na)+ 351.2326; found 351.2325.
:EtOAc = 80:20) to give 18 (2.61 g, 96%) as a colorless oil. Rf 0.40 (30% EtOAc in hexanes); [α]20D = −4.6 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.22 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 8.7 Hz, 2H), 5.86–5.77 (m, 1H), 5.11–5.05 (m, 2H), 4.44 (s, 2H), 3.86–3.81 (m, 1H), 3.80 (s, 3H), 3.70–3.64 (m, 1H), 3.62–3.56 (m, 1H), 2.76 (br s, 1H), 2.22 (t, J = 6.8 Hz, 2H), 1.76–1.70 (m, 2H); 13C NMR (75 MHz, CDCl3): δ = 159.1, 134.8, 129.9, 129.2, 117.4, 113.7, 72.8, 70.3, 68.5, 55.2, 41.8, 35.7; MS (ESI): m/z = 259.2 [M + Na]+.
:EtOAc = 95:05) to give 19 (1.51 g, 95%) as a colorless oil. Rf 0.70 (20% EtOAc in hexanes); [α]20D = +15.1 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ 7.21 (d, J = 8.5 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 5.86–5.71 (m, 1H), 5.07–4.96 (m, 2H), 4.42 (d, J = 11.5 Hz, 1H), 4.35 (d, J = 11.5 Hz, 1H), 3.94–3.84 (m, 1H), 3.80 (s, 3H), 3.51–3.42 (m, 1H), 2.28–2.13 (m, 2H), 1.81–1.59 (m, 2H), 0.88 (s, 9H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 159.0, 134.8, 130.5, 129.1, 116.8, 113.6, 72.5, 68.8, 66.6, 55.0, 42.2, 36.6, 25.8, 17.9, −4.4, −4.7; MS (ESI): m/z = 373.3 [M + Na]+.
:1) was added OsO4 (4.2 mL, 1 mg/1 mL toluene solution, 0.016 mmol) and NMO (465 mg, 3.98 mmol) at 25 °C and the mixture was stirred for 12 h. The solvent was evaporated and the residue was extracted with ethyl acetate (30 mL). The organic layers were washed with brine (20 mL), dried over Na2SO4 and concentrated in vacuo, affording an oil which was purified by flash column chromatography (silica gel, hexanes:EtOAc = 50:50) to give diol (1.2 g) as a colorless oil.
To a solution of the above diol (1.20 g, 3.12 mmol) in a 30 mL mixture of THF–H2O (4:1) was added NaHCO3 (446 mg, 5.30 mmol) and NaIO4 (2.0 g, 9.4 mmol). After stirring at 0 °C for 30 min, the solid was removed by filtration and the filtrate was extracted with ethyl acetate (2 × 30 mL). The combined organic layers were washed with brine (20 mL), dried with Na2SO4 and concentrated in vacuo. The crude aldehyde was purified by flash chromatography (10% ethyl acetate in hexanes) to give aldehyde 6 (0.98 g, 84%) as a colorless oil. Rf 0.60 (20% EtOAc in hexanes); IR (KBr): νmax 2954, 2931, 2857, 1726, 1613, 1513, 1465, 1250, 1098, 1037, 836, 777 cm−1; [α]20D = +5.2 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 9.79 (s, 1H), 7.24 (d, J = 8.5 Hz, 2H), 6.88 (d, J = 8.5 Hz, 2H), 4.43 (d, J = 11.2 Hz, 1H), 4.38 (d, J = 11.3 Hz, 1H), 4.37–4.32 (m, 1H), 3.80 (s, 3H), 3.51 (t, J = 6.2 Hz, 2H), 2.59–2.51 (m, 2H), 1.9–1.76 (m, 2H), 0.86 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 202.1, 159.1, 130.3, 129.2, 113.7, 72.6, 65.9, 65.6, 55.2, 51.0, 37.5, 25.7, 17.9, −4.6, −4.7; MS (ESI): m/z = 375.2 [M + Na]+.
:EtOAc = 80:20) to give 20 (645 mg, 82%) as a colorless oil. Rf 0.40 (20% EtOAc in hexanes); IR (KBr): νmax 3403, 2921, 2851, 1612, 1511, 1243, 1220, 772 cm−1; [α]20D = −20.7 (c = 0.88, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.24 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 9.0 Hz, 2H), 4.45 (s, 2H), 4.29 (dd, J = 9.0, 5.0 Hz, 1H), 4.17–4.09 (m, 1H), 3.80 (s, 3H), 3.72–3.67 (m, 1H), 3.66–3.60 (m, 1H), 3.08 (br s, 1H), 2.96–2.79 (m, 4H), 2.16–2.09 (m, 1H), 1.96–1.70 (m, 5H); 13C NMR (75 MHz, CDCl3): δ = 159.1, 129.7, 129.2, 113.7, 72.8, 68.4, 67.6, 55.1, 43.8, 42.7, 36.3, 30.3, 29.9, 25.8; MS (ESI): m/z = 329 [M + H]+; HRMS: calcd. for C16H25O3S2 (M + H)+ 329.1240; found 329.1230.
:EtOAc = 70:30) to give 25 (878 mg, 86%) as a colorless oil. Rf 0.40 (40% EtOAc in hexanes); IR (KBr): νmax 3378, 2930, 2858, 1606, 1466, 1362, 1255, 1170, 1117, 1038, 837, 776 cm−1; [α]20D = +38.8 (c = 1.12, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 6.56 (dd, J = 14.3, 5.5 Hz, 1H), 6.25 (d, J = 14.3 Hz, 1H), 4.23 (t, J = 5.5 Hz, 1H), 3.59 (dd, J = 10.0, 8.8 Hz, 1H), 3.50–3.41 (m, 1H), 2.52 (br s, 1H), 1.91–1.82 (m, 1H), 0.88 (s, 9H), 0.81 (d, J = 6.6 Hz, 3H), 0.05 (s, 3H), 0.02 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 146.0, 77.5, 76.8, 65.0, 40.8, 25.7, 18.0, 11.7, −4.6, −5.2; MS (ESI): m/z = 379 [M + Na]+.
5-((2R,3R,E)-3-(tert-Butyldimethylsilyloxy)-5-iodo-2-methylpent-4-enylthio)-1-phenyl-1H-tetrazole (26)
DIAD (0.4 mL, 2.01 mmol) was slowly added at 0 °C to a solution of alcohol 25 (550 mg, 1.55 mmol), PPh3 (488 mg, 1.85 mmol) and 1-phenyl-1H-tetrazole- 5-thiol (330 mg, 1.85 mmol) in THF (20 mL). The mixture was stirred at room temperature for 1 h before the solvent was evaporated. The residue was purified by flash column chromatography (silica gel, hexanes:EtOAc = 90:10) to give 26 (768 mg, 96%) as a colorless oil. Rf 0.40 (10% EtOAc in hexanes); IR (KBr): νmax 2954, 2931, 2857, 1599, 1500, 1465, 1409, 1386, 1252, 1087, 1022, 838, 777 cm−1; [α]20D = +10.7 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.63–7.50 (m, 5H), 6.54 (dd, J = 14.5, 6.2 Hz, 1H), 6.28 (dd, J = 14.5, 1.3 Hz, 1H), 4.21 (dddd, J = 6.0, 4.7, 3.6, 1.3 Hz, 1H), 3.48 (dd, J = 13.0, 6.6 Hz, 1H), 3.23 (dd, J = 13.0, 7.3 Hz, 1H), 2.14–2.05 (m, 1H), 1.02 (d, J = 6.9 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 154.3, 146.2, 133.6, 130.0, 129.7, 123.7, 77.4, 76.9, 39.0, 36.2, 25.7, 18.0, 13.8, −4.4, −5.0; MS (ESI): m/z = 539 [M + Na]+; HRMS: calcd. for C19H30IN4OSSi (M + H)+ 517.0949; found 517.0904.
:10) to give 4 (621 mg, 81%) as a viscous oil. Rf 0.40 (10% EtOAc in hexanes); IR (KBr): νmax 2955, 2930, 2857, 1734, 1607, 1502, 1464, 1341, 1268, 1153, 1100, 761 cm−1; [α]20D = +6.3 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.72–7.66 (m, 2H), 7.64–7.58 (m, 3H), 6.51 (dd, J = 14.0, 6.0 Hz, 1H), 6.36 (dd, J = 14.0, 6.0 Hz, 1H), 4.28 (t, J = 5.0 Hz, 1H), 4.02 (dd, J = 14.0, 4.0 Hz, 1H), 3.48 (dd, J = 14.0, 8.0 Hz, 1H), 2.52–2.41 (m, 1H), 1.15 (d, J = 7.0 Hz, 3H), 0.90 (s, 9H), 0.08 (s, 3H), 0.05 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 153.8, 144.7, 132.9, 131.4, 129.6, 125.0, 78.6, 76.7, 57.8, 34.4, 25.7, 18.1, 14.7, −4.5, −5.0; MS (ESI): m/z = 571 [M + Na]+; HRMS: calcd. for C19H29IN4O3SSiNa (M + Na)+ 571.0667; found 571.0643.
:EtOAc, 80:20) to give the aldol adduct (1.8 g, 74%) as a mixture of diastereomers.
To a stirred solution of the above aldol adduct (1.8 g, 2.64 mmol) in dry CH2Cl2 (20 mL) at room temperature was added NaHCO3 (665 mg, 7.92 mmol) and Dess–Martin periodinane (1.68 g, 3.96 mmol) in one portion. The mixture was stirred for 1 h at room temperature and quenched with saturated aqueous Na2S2O3 (10 mL). The layers were separated and the aqueous phase was extracted with ether (2 × 25 mL). The combined organic layers were washed with brine (10 mL), dried with Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (10% ethyl acetate in hexanes) to give diketone 27 (1.7 g, 96%) as a colorless oil. Rf 0.50 (20% EtOAc in hexanes); IR (KBr): νmax 2928, 2855, 1729, 1612, 1513, 1462, 1248, 1089, 834, 774 cm−1; [α]28D = −7.7 (c = 1.05, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.20 (d, J = 9.0 Hz, 2H), 6.82 (d, J = 9.0 Hz, 2H), 5.55 (s, 1H), 4.41 (d, J = 11.3 Hz, 1H) 4.35 (d, J = 11.3 Hz, 1H), 4.30–4.20 (m, 1H), 3.80 (s, 3H), 3.72–3.62 (m, 1H), 3.47 (t, J = 6.8 Hz, 2H), 3.39 (d, J = 6.0 Hz, 2H), 3.38–3.29 (m, 1H), 2.48 (dd, J = 14.3, 6.7 Hz, 1H), 2.38 (d, J = 6.8 Hz, 2H), 2.28 (dd, J = 14.3, 6.0 Hz, 1H), 1.88–1.67 (m, 4H), 1.64–1.36 (m, 4H), 1.30–1.06 (m, 3H), 0.90 (s, 9H), 0.87 (d, J = 6.8 Hz, 3H), 0.85 (s, 9H), 0.04 (s, 3H), 0.02 (s, 6H), 0.00 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 192.6, 190.6, 159.1, 130.4, 129.2, 113.7, 102.1, 76.6, 74.6, 72.6, 67.8, 67.2, 66.2, 55.2, 46.5, 45.7, 39.7, 37.4, 32.4, 31.5, 31.4, 25.9, 25.8, 23.5, 17.4, −4.7, −5.3; MS (ESI): m/z = 701 [M + Na]+; HRMS: calcd. for C37H66O7Si2Na (M + Na)+ 701.4239; found 701.4234.
:1) to give compound 28 (408 mg, 86%) as a colorless oil. Rf 0.20 (50% EtOAc in hexanes); IR (KBr): νmax 3451, 2927, 2858, 1658, 1606, 1247, 1091, 1034 cm−1; [α]20D = +69.4 (c = 0.50, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.19 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 8.8 Hz, 2H), 5.37 (s, 1H), 4.59–4.51 (m, 1H), 4.41 (s, 2H), 3.80 (s, 3H), 3.61–3.47 (m, 3H), 3.45–3.34 (m, 3H), 2.46–2.35 (m, 2H), 2.26 (dd, J = 14.7, 4.9 Hz, 1H), 1.95–1.80 (m, 3H) 1.60–1.44 (m, 4H), 1.41–1.34 (m, 1H), 1.30–1.17 (m, 3H), 0.90 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 193.0, 174.2, 159.1, 130.0, 129.2, 113.7, 105.5, 75.6, 74.7, 72.7, 67.4, 65.1, 55.2, 41.5, 40.7, 40.0, 34.4, 32.2, 31.2, 31.1, 29.6, 23.4, 17.3; MS (ESI): m/z = 433.2 [M + H]+; HRMS: calcd. for C25H37O6 (M + H)+ 433.2585; found 433.2560.
:EtOAc =70:30) to give dimethyl ketal 30 (126 mg, 82%) as a colorless oil. Rf 0.50 (50% EtOAc in hexanes); IR (KBr): νmax 3452, 2927, 2858, 1610, 1512, 1428, 1358, 1223, 1151, 1033, 751 cm−1; [α]20D = +11.3 (c = 0.70, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 7.25 (d, J = 8.9 Hz, 2H), 6.87 (d, J = 8.9 Hz, 2H), 4.42 (s, 2H), 3.80 (s, 3H), 3.66–3.39 (m, 8H), 3.20 (s, 3H), 3.15 (s, 3H), 2.01–1.89 (m, 2H), 1.88–1.66 (m, 3H), 1.65–1.37 (m, 6H), 1.33–1.12 (m, 6H), 0.92 (d, J = 7.2 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 158.9, 130.4, 129.0, 113.6, 98.9, 75.0, 74.5, 72.5, 70.9, 70.5, 67.5, 66.4, 55.0, 47.5, 47.1, 41.8, 40.4, 38.5, 38.4, 35.8, 32.1, 30.9, 23.5, 17.5; MS (ESI): m/z = 503.3 [M + Na]+.
To a −78 °C solution of sulfone 4 (160 mg, 0.29 mmol) in a mixture of THF (0.8 mL) and HMPA (0.2 mL) was added LiHMDS (0.28 mL of a 1 M solution in THF, 0.28 mmol). After 30 min at that temperature, the above aldehyde (70 mg, 0.15 mmol) was added to a solution of THF (0.3 mL) and HMPA (0.1 mL). The reaction mixture was stirred for 2 h at −78 °C, after which it was slowly warm to room temperature over a period of 3 h and stirred at this temperature for 12 h. The mixture was subsequently diluted with ether (5 mL) and quenched by the addition of a saturated NH4Cl solution (2 mL). The organic phase was washed with water, brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (hexanes:EtOAc, 95:5 to 90:10) to give 31 (67 mg, 56%) and the aldehyde (20 mg, 28%). Rf 0.40 (20% EtOAc in hexanes); IR (KBr): νmax 2932, 2859, 1606, 1512, 1436, 1374, 1253, 1088, 1032, 751 cm−1; [α]20D = +9.17 (c = 0.60, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 7.25 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 6.51 (dd, J = 14.5, 6.2 Hz, 1H), 6.14 (d, J = 14.5 Hz, 1H), 5.26–5.20 (m, 2H), 4.42 (s, 2H), 3.88–3.82 (m, 1H), 3.80 (s, 3H), 3.71–3.39 (m, 5H), 3.32–3.22 (m, 1H), 3.20 (s, 3H), 3.15 (s, 3H), 2.45–2.26 (m, 1H), 2.23–2.11 (m, 1H), 1.99 (t, J = 11.7 Hz, 2H), 1.91–1.67 (m, 2H), 1.64–1.37 (m, 4H), 1.33–1.12 (m, 8H), 0.97 (d, J = 6.6 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.89 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 159.0, 148.0, 137.1, 130.6, 129.9, 129.1, 113.7, 99.0, 79.1, 76.0, 75.5, 74.0, 72.6, 71.0, 70.8, 66.6, 55.2, 47.6, 47.3, 44.0, 43.5, 42.5, 38.8, 38.6, 36.0, 33.0, 32.1, 31.4, 29.6, 25.8, 23.6, 21.7, 18.2, 16.2, −4.4, −4.8; MS (ESI): m/z = 823.4 [M + Na]+; HRMS: calcd. for C39H65IO7SiNa (M + Na)+ 823.3436; found 823.3448.
:EtOAc, 80:20) afforded alcohol 32 (47 mg, 92%) as a colorless oil. Rf 0.30 (20% EtOAc in hexanes); IR (KBr): νmax 3454, 2927, 2858, 1612, 1514, 1452, 1233, 1142, 1033, 772 cm−1; [α]20D = −1.8 (c = 0.50, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 6.50 (dd, J = 14.9, 6.0 Hz, 1H), 6.15 (d, J = 14.9 Hz, 1H), 5.29–5.19 (m, 2H), 3.86 (t, J = 6.0 Hz, 1H), 3.81–3.68 (m, 4H), 3.44–3.35 (m, 1H), 3.29–3.22 (m, 1H), 3.21 (s, 3H), 3.16 (s, 3H), 2.72 (br s, 1H), 2.39–2.25 (m, 1H), 2.17 (dd, J = 13.0, 7.0 Hz, 1H), 2.02 (d, J = 13.0 Hz, 1H), 1.92 (d, J = 13.0 Hz, 1H), 1.86–1.62 (m, 3H), 1.60–1.41 (m, 4H), 1.38–1.11 (m, 7H), 0.97 (d, J = 7.0 Hz, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 3H), 0.01 (s, 3H); 13C NMR (75 MHz, CDCl3): δ = 148.0, 137.1, 130.0, 98.6, 79.0, 76.1, 75.6, 74.7, 74.1, 71.4, 61.6, 47.7, 47.3, 43.9, 43.5, 42.5, 38.6, 38.3, 37.5, 33.0, 31.9, 31.7, 29.6, 25.8, 23.5, 21.7, 18.1, 16.3, −4.4, −4.9; MS (ESI): m/z = 703 [M + Na]+; HRMS: calcd. for C31H57IO6Si (M + Na)+ 703.2861; found 703.2852.
:1, 1 mL) were added TEMPO (4.0 mg, 0.026 mmol) and BAIB (87 mg, 0.27 mmol). After stirring at room temperature for 2 h, the reaction mixture was diluted with CH2Cl2 (5 mL) and then washed with saturated aqueous Na2S2O3 (5 mL). The organic layer was dried over Na2SO4, filtered, and the filtrate was concentrated under reduced pressure to give the crude carboxylic acid, which was used for the next reaction without further purification.
A solution of the above crude carboxylic acid in THF (2 mL) was cooled to 0 °C and TBAF (0.9 mL, 0.9 mmol, 1.0 M solution in THF) was added dropwise. The resulting brown solution was stirred at room temperature for 3 h. The reaction was quenched with saturated aqueous NH4Cl (2 mL) and extracted with EtOAc (2 × 5 mL). The combined organic layer was dried over Na2SO4 and evaporated to dryness under reduced pressure. The residue was purified by flash column chromatography (silica gel, CHCl3:MeOH = 98:2) to give seco acid 33 (47.5 mg, 91%) as a colorless oil. Rf 0.40 (5% MeOH in CHCl3); [α]20D = −4.6 (c = 1.00, CHCl3); 1H NMR (500 MHz, CDCl3): δ = 6.57 (dd, J = 14.0, 5.0 Hz, 1H), 6.33 (d, J = 14.0 Hz, 1H), 5.34–5.26 (m, 2H), 3.98 (t, J = 5.0 Hz, 1H), 3.93–3.85 (m, 1H), 3.73–3.65 (m, 1H), 3.43–3.35 (m, 1H), 3.28–3.22 (m, 1H), 3.21 (s, 3H), 3.17 (s, 3H), 2.56 (dd, J = 15.0, 8.0 Hz, 1H), 2.47 (dd, J = 15.0, 5.0 Hz, 1H), 2.44–2.37 (m, 1H), 2.35–2.23 (m, 1H), 2.07–1.94 (m, 2H), 1.89–1.74 (m, 2H), 1.64–1.40 (m, 6H), 1.38–1.10 (m, 4H), 1.00 (d, J = 7.0 Hz, 3H), 0.96 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 174.9, 146.6, 138.0, 129.7, 98.6, 77.7, 77.5, 75.7, 74.3, 71.5, 70.5, 47.7, 47.4, 43.5, 42.4, 42.2, 40.6, 38.6, 37.8, 33.2, 31.9, 31.2, 29.6, 23.6, 21.6, 15.6; MS (ESI): m/z = 603.3 [M + Na]+.
:EtOAc = 95:05) providing macrolactone 34 (36.5 mg, 63%) as a colorless oil. Rf 0.40 (10% EtOAc in hexanes); IR (KBr): νmax 2926, 2854, 1739, 1660, 1457, 1373, 1282, 1256, 1185, 1098, 1045, 947 cm−1; [α]20D = −11.5 (c = 1.00, CHCl3); 1H NMR (300 MHz, CDCl3): δ = 6.50 (dd, J = 15.1, 6.0 Hz, 1H), 6.33 (d, J = 15.1 Hz, 1H), 5.47 (dd, J = 15.1, 9.0 Hz, 1H), 5.13 (dd, J = 7.5, 2.2 Hz, 1H), 5.07 (dd, J = 15.1, 9.8 Hz, 1H), 3.98–3.86 (m, 1H), 3.36–3.13 (m, 3H), 3.21 (s, 3H), 3.16 (s, 3H), 2.60–2.47 (m, 1H), 2.47–2.42 (m, 2H), 2.40–2.27 (m, 1H), 1.98 (dt, J = 12.8, 2.2 Hz, 1H), 1.86 (dt, J = 13.5, 2.2 Hz, 1H), 1.80–1.61 (m, 2H), 1.53–1.40 (m, 2H), 1.38–1.21 (m, 8H), 1.04 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H); 13C NMR (75 MHz, CDCl3): δ = 170.7, 142.8, 135.9, 131.7, 99.0, 79.4, 79.2, 75.9, 75.3, 71.3, 70.6, 47.6, 47.4, 43.9, 42.9, 41.0, 38.8, 37.6, 32.9, 32.3, 31.6, 29.6, 23.9, 21.7, 14.3. MS (ESI): m/z = 585 [M + Na]+; HRMS: calcd. for C25H39IO6Na (M + Na)+ 585.1684; found 585.1634.
Acknowledgements
NNR thank the Council of Scientific and Industrial Research (CSIR), New Delhi, for the award of fellowship.References
- S. Ohta, M. M. Uy, M. Yanai, E. Ohta, T. Hirata and S. Ikegami, Tetrahedron Lett., 2006, 47, 1957–1960 CrossRef CAS.
- (a) G. R. Pettit, C. L. Herald, D. L. Doubek, D. L. Herald, E. Arnold and J. Clardy, J. Am. Chem. Soc., 1982, 104, 6846–6848 CrossRef CAS; (b) J. Cossy, C. R. Chim., 2008, 11, 1477–1482 CrossRef CAS.
- M. S. Kwon, S. K. Woo, S. W. Na and E. Lee, Angew. Chem., Int. Ed., 2008, 47, 1733–1735 CrossRef CAS.
- (a) H. Fuwa and M. Sasaki, Org. Lett., 2010, 12, 584–587 CrossRef CAS; (b) C. Cook, X. Guinchard, F. Liron and E. Roulland, Org. Lett., 2010, 12, 744–747 CrossRef CAS; (c) E. Crane, T. P. Zabava, R. L. Farmer and K. A. Scheidt, Angew. Chem., Int. Ed., 2011, 50, 9112–9115 CrossRef CAS.
- H. Fuwa, T. Suzuki, H. Kubo, T. Yamori and M. Sasaki, Chem.–Eur. J., 2011, 17, 2678–2688 CrossRef CAS.
- (a) Ch. R. Reddy, D. Suman and N. N. Rao, Synlett, 2012, 23, 272–274 CrossRef CAS; (b) Ch. R. Reddy and N. N. Rao, Eur. J. Org. Chem., 2012, 1819–1824 CrossRef; (c) Ch. R. Reddy, N. N. Rao and B. Srikanth, Eur. J. Org. Chem., 2010, 345–351 CrossRef; (d) Ch. R. Reddy, P. P. Madhavi and S. Chandrashekar, Tetrahedron: Asymmetry, 2010, 21, 103–105 CrossRef CAS; (e) Ch. R. Reddy and B. Srikanth, Synlett, 2010, 1536–1538 CrossRef CAS; (f) Ch. R. Reddy, G. Dharmapuri and N. N. Rao, Org. Lett., 2009, 11, 5730–5733 CrossRef CAS; (g) Ch. R. Reddy and N. N. Rao, Tetrahedron Lett., 2009, 50, 2478–2480 CrossRef.
- For preliminary communication of our work see : Ch. R. Reddy and N. N. Rao, Tetrahedron Lett., 2010, 51, 5840–5842 CrossRef.
- Y. Shiro, K. Kato, M. Fujii, Y. Ida and H. Akita, Tetrahedron, 2006, 62, 8687–8695 CrossRef CAS.
- S. Hanessian, R. J. Roy, M. Petrini, P. J. Hodges, R. Di Fabio and G. J. Carganico, J. Org. Chem., 1990, 55, 5766–5777 CrossRef CAS.
- C. Fouquet and M. Schlosser, Angew. Chem., Int. Ed. Engl., 1974, 13, 82–83 CrossRef.
- Synthesis of 15 via cross-metathesis reaction.
. - J. Liu, J. H. Yang, C. Ko and R. P. Hsung, Tetrahedron Lett., 2006, 47, 6121–6123 CrossRef CAS.
- J. A. Frick, J. B. Klassen, A. Bathe, J. M. Abramson and H. Rapoport, Synthesis, 1992, 7, 621–623 CrossRef.
- Improvement in the yield of the product was observed when Li2CuCl4 was used instead of CuI.
- Y. Wu, X. Liao, R. Wang, X.-S. Xie and J. K. De Brabander, J. Am. Chem. Soc., 2002, 124, 3245–3253 CrossRef CAS.
- The dithiane alternative route was accomplished using simple reagents (Scheme 4) by avoiding cuprate and osmium reagents (Scheme 3).
- (a) I. Marek, C. Meyer and J-F. Normant, Org. Syn., 1997, 74, 194 CAS; (b) B. M. Trost, M. U. Frederiksen, J. P. N. Papillon, P. E. Harrington, S. Shin and B. T. Shireman, J. Am. Chem. Soc., 2005, 127, 3666–3667 CrossRef CAS.
- D. A. Evans, J. Bartoli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127–2129 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1–28 CrossRef CAS.
- H. S. Schultz, H. B. Freyermuth and S. R. Buc, J. Org. Chem., 1963, 28, 1140–1142 CrossRef CAS.
- (a) R. F. Newton and D. P. Reynolds, Tetrahedron Lett., 1979, 20, 3981–3982 CrossRef; (b) D.-R. Le, D.-H. Zhang, C.-Y. Sun, J.-W. Zhang, L. Yang, J. Chen, B. Liu, C. Su, W.-S. Zhou and G.-Q. Lin, Chem.–Eur. J., 2006, 12, 1185–1204 CrossRef.
- P. Wipf and J. T. Reeves, Chem. Commun., 2002, 2066–2067 RSC.
- (a) P. R. Blakemore, W. J. Cole, P. J. Kocienski and A. Morley, Synlett, 1998, 26–28 CrossRef; (b) P. R. Blakemore, J. Chem. Soc., Perkin Trans. 1, 2002, 2563–2585 RSC.
- Y. Oikawa, T. Yoshioka and O. Yonemitsu, Tetrahedron Lett., 1982, 23, 885–888 CrossRef CAS.
- H. Miyaoka, M. Yamanishi, A. Hoshino and A. Kinbara, Tetrahedron, 2006, 62, 4103–4109 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Sacki, T. Hatsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989–1993 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR and 13C NMR spectra of all the new compounds. See DOI: 10.1039/c2ra21161k. |
| This journal is © The Royal Society of Chemistry 2012 |