Amination of benzylic and cinnamic alcohols via a biocatalytic, aerobic, oxidation–transamination cascade†
Michael
Fuchs
a,
Katharina
Tauber
a,
Johann
Sattler
a,
Horst
Lechner
a,
Jan
Pfeffer
b,
Wolfgang
Kroutil
a and
Kurt
Faber
*a
aDepartment of Chemistry, Organic & Bioorganic Chemistry, University of Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: Kurt.Faber@Uni-Graz.at; Fax: +43-316-380-9840; Tel: +43-316-380-5332
bEvonik Degussa GmbH, Paul-Baumann-Straße 1, 45772 Marl, Germany
First published on 12th June 2012
Abstract
The amination of benzylic and cinnamic alcohols was achieved via a biocatalytic, one-pot oxidation–transamination cascade in aqueous medium at physiological conditions. Alcohol oxidation by galactose oxidase at the expense of O2 furnished the corresponding aldehydes, which were aminated using ω-transaminases in situ. The applicability of this method was demonstrated by a short synthesis of the antifungal agent naftifine.
Among the most prominent functional groups present in chemical products, the hydroxy moiety is most abundant (∼40%), followed by the –CO2H (∼22%) and the amine moiety (∼16%).1 Whereas renewable resources are rich in OH-groups (e.g. in carbohydrates), amines are considerably more scarce. Thus, the functional group interconversion2 of alcohols into the corresponding amines is an important issue for the generation of high-value building blocks. Recent research in this field revealed that Ru-based catalysis in the presence of ammonia can convert primary and sec-alcohols into the corresponding amines at elevated temperature and pressure.3 This transformation proceeds in situ via oxidation and reductive amination mediated by the same catalytic species. Inspired by the biocatalytic deracemisation of rac-sec-alcohols through a simultaneous reduction/oxidation sequence,4 we envisaged to devise a one-pot sequence for the synthesis of amines from alcohols by separating the reductive from the oxidative reaction via the enzyme exclusion volume.5
The groups of Arnold and Turner have demonstrated the potential of galactose oxidase from Fusarium NRRL 2903 for chemoselective, O2-dependent oxidation reactions.6,7 In nature, galactose oxidase converts the primary C6-hydroxy functional group of D-galactose into the corresponding aldehyde at the expense of O2 as a ‘green’ oxidant, but it is unable to accept sec-alcohols.8 These findings together with our own experience with the M1 variant11 made this enzyme a suitable starting point for the oxidation step.
For the transformation of the aldehyde to the amine, an ω-transaminase (ω-TA) was chosen together with a suitable amine-donor.9 In order to shift the unfavourable equilibrium towards amine formation, alanine proved to be an ideal amine-donor, which can be efficiently recycled from pyruvate via (formal) reductive amination using alanine dehydrogenase and NADH-recycling, which efficiently drives the reaction towards completion (Scheme 1).10 For the direct interconversion of a hydroxy group into the corresponding amine analog, both reactions have to run simultaneously including cosubstrate and cofactor recycling.
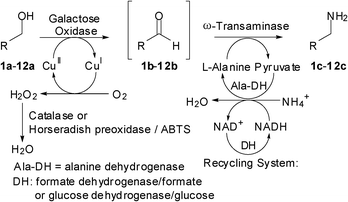 | ||
| Scheme 1 Biocatalytic oxidation–transamination cascade. | ||
As starting point for the oxidation reaction, we chose the optimized conditions recently developed.11 Good to excellent conversions to the corresponding aldehydes were obtained for benzylic and cinnamic alcohols 1a–12a. In the presence of horseradish peroxidase (HRP) and 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS), no over-oxidation was observed. In order to improve the atom economy of the process, catalase from Micrococcus lysodeikticus was successfully employed as an alternative to the HRP/ABTS system. On the other hand, the transamination reaction required some optimisation. Although ω-transaminases have been extensively employed for the amination of ketones, aldehydes have been investigated to a much lesser extent.12,13 Hence, we investigated the transamination of benzaldehyde (1b) with several ω-transaminases (Table 1).
| Entry | Source of ω-TA | Conv. [%]b |
|---|---|---|
| a Reaction conditions: Substrate (50 mM), ω-TA [20 mg, lyophilized whole cells of E. coli BL21(DE3) host containing over-expressed ω-TA], Pi buffer (100 mM, pH 7.0, 1 mM pyridoxal-5-phosphate), ammonium formate (175 mM, 3.5 equiv.), L-alanine (250 mM, 5 equiv.), alanine dehydrogenase (Ala-DH) from Bacillus subtilis (7.5 μg, 0.013 U), formate dehydrogenase (FDH, 4.4 U), shaking at 30 °C for 16 h. b Amine formation was determined by GC-FID analysis. c Only trace amounts of amine were observed. | ||
| 1 | Bacillus megaterium SC6394 | Tracec |
| (BM-ωTA)14,15 | ||
| 2 |
Chromobacterium violaceum DSM 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 191 (CV-ωTA)12,14 191 (CV-ωTA)12,14 |
30 |
| 3 | Alcaligenes denitrificans Y2k-2 | 83 |
| (AD-ωTA)14,16 | ||
| 4 | Pseudomonas putida KT2440 | 83 |
| (Pp1-ωTA, Gen PP5182)17 | ||
| 5 | Pseudomonas putida KT2440 | 82 |
| (Pp2-ωTA, Gen PP2180)17 | ||
| 6 | Paracoccus denitrificans | 91 |
| (Pd-ωTA)17,18 | ||
| 7 | Vibrio fluvialis | 96 |
| (Vf-ωTA) | ||
Surprisingly, there were significant differences in the conversions of ω-TAs in the amination of benzaldehyde: whereas Bacillus megaterium BM-ω-TA and Chromobacterium violaceum CV-ω-TA showed very low conversions, all other ω-TAs formed benzylamine (1c) in >80% yield. Best results were obtained with Paracoccus denitrificans Pd-ω-TA and Vibrio fluvialis Vf-ω-TA. Consequently, the oxidation–transamination cascade was optimised using galactose oxidase from Fusarium NRRL 2903 and Vf-ω-TA. The relevant parameters are listed in Table 2.
| Entry | L-Alanine [mM] | NH4HCO2 [mM] | Conv. [%]b | ||
|---|---|---|---|---|---|
| 1a | 1b | 1c | |||
| a Reaction conditions: substrate (50 mM), galactose oxidase [20 mg, lyophilized whole cells of E. coli BL21(DE3) host containing overexpressed galactose oxidase], horseradish peroxidase (HRP, 0.075 mg mL−1), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, 0.075 mg mL−1), Vf-ω-TA [20 mg, lyophilized whole E. coli BL21(DE3) host cells containing overexpressed Vf-ω-TA], Pi buffer (100 mM, pH 7.0, 1 mM pyridoxal phosphate, 10 mM CuSO4), alanine dehydrogenase (Ala-DH) from Bacillus subtilis (7.5 μg, 0.013 U), formate dehydrogenase (FDH, 4.4 U), ammonium formate, shaking at room temperature for 20 h. b Amine formation was determined by GC-FID analysis. c GDH (20 U)/glucose was used for NADH-recycling, NH4Cl (100 mM, 2 equiv.) was used as the amine donor. | |||||
| 1 | 250 | 175 | 66 | 8 | 31 |
| 2 | 150 | 175 | 58 | 4 | 38 |
| 3 | 100 | 175 | 54 | 14 | 32 |
| 4 | 250 | 105 | 51 | 7 | 42 |
| 5 | 250 | 70 | 44 | <1 | 56 |
| 6 | 150 | 70 | 19 | <1 | 81 |
| 7c | 150 | — | <1 | <1 | >99 |
High alanine loading (5 equiv., entry 1), required for the reductive amination of ketones, could be significantly reduced and the optimum turned out to be 3 equiv. (entry 2); below this value (2 equiv., entry 3) the conversion decreased. There was an even more pronounced effect with ammonium formate: reducing the amount from 3.5 to 1.4 equiv. almost doubled amine formation (entries 1 and 5) and it became clear that the oxidation was rate limiting under these conditions (81% benzylamine, but 19% remaining alcohol, entry 6). Since ammonium formate was shown in additional experiments to cause the strongest inhibition of galactose oxidase (cf. entries 1, 4 and 5), it was replaced by ammonium chloride (as NH3-donor) and glucose dehydrogenase (GDH) with glucose (3 equiv.) for NADH-recycling. This latter system proved to be ideal for the cascade process, yielding solely benzylamine 1c in >99% yield. The inhibition was not caused by a change in pH, as the pH shift for ammonium formate and L-alanine at their highest levels was ± 0.1.
With optimized cascade conditions in hand, a series of benzylic alcohols were tested to explore the scope and limitations of the method (Table 3).
| Entry | R = | ω-TAc | Conv. [%]b | ||
|---|---|---|---|---|---|
| a | b | c | |||
| a Reaction conditions: Substrate (50 mM), galactose oxidase [20 mg, lyophilized whole cells of E. coli BL21(DE3) host containing overexpressed galactose oxidase], horseradish peroxidase (HRP, 0.075 mg mL−1), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS, 0.075 mg mL−1), ω-TA [20 mg, lyophilized whole E. coli BL21(DE3) host cells containing ω-TA], Pi buffer (100 mM, pH 7.0, 1 mM PLP, 10 mM CuSO4), alanine dehydrogenase (Ala-DH) from Bacillus subtilis (7.5 μg, 0.013 U), glucose dehydrogenase (GDH, 20 U), NH4Cl (100 mM, 2 equiv.), glucose (120 mM, 2.4 equiv.), shaking at room temperature for 20 h. b Amine formation was determined by GC-MS analysis. c Vf-ω-TA = Vibrio fluvialis ω-transaminase, Pd-ω-TA = Paracoccus denitrificans ω-transaminase. d Trace amounts of product. e Same results were obtained using catalase from Micrococcus lysodeikticus (10 μL, 1500 units). | |||||
| 1e | H5C6-(1a–c) | Vf-ωTA | <1 | <1 | >99 |
| 2 | o-Cl-C6H4-(2a–c) | Vf-ωTA | 99 | <1 | traced |
| 3 | o-MeO-C6H4-(3a–c) | Vf-ωTA | 33 | 49 | 18 |
| 4 | m-Cl-C6H4-(3a–c) | Vf-ωTA | <1 | 4 | 96 |
| 5 | m-MeO-C6H4-(3a–c) | Vf-ωTA | <1 | 19 | 81 |
| 6 | m-Me-C6H4-(3a–c) | Pd-ωTA | <1 | <1 | >99 |
| 7 | p-Cl-C6H4-(3a–c) | Vf-ωTA | 18 | 38 | 44 |
| 8 | p-MeO-C6H4-(3a–c) | Pd-ωTA | <1 | 32 | 68 |
| 9 | p-Me-C6H4-(3a–c) | Pd-ωTA | 16 | 2 | 82 |
| 10 | p-F-C6H4-(3a–c) | Pd-ωTA | 21 | 4 | 75 |
| 11 | Piperonyl (3a–c) | Vf-ωTA | <1 | 65 | 35 |
| Pd-ωTA | <1 | 64 | 36 | ||
| 12e | 3-Phenylallyl (3a–c) | Vf-ωTA | 12 | 85 | 3 |
| Pd-ωTA | 8 | <1 | 92 | ||
In contrast to benzyl alcohol (1a), o-chlorobenzylalcohol (2a) and the corresponding o-methyl derivative (data not shown) were converted at very low levels by galactose oxidase11 and therefore only trace amounts of the amine were detected (entry 2). In contrast, the o-methoxy derivative 3a was oxidized at higher levels and higher amounts of the amine were observed. m-Substituents seemed to exert less steric hindrance and the corresponding amines 4c–6c were formed with good to excellent conversions. The nature of p-substituents clearly had some impact on the reactivities: whereas the p-chloro derivative 7a gave only 44% conversion, the p-F-(10a), p-MeO-(8a), and p-Me-analogs (9a) gave significantly better results. The bulky piperonyl alcohol (11a) gave amine 11c in modest yield with both transaminases. Cinnamyl alcohol (12a) was smoothly oxidised, but sluggishly transaminated by Vf-ω-TA. However, when Paracoccus denitrificans ω-TA was used, the sensitive amine 12c was formed in 92% yield. The mild reaction conditions ensured that the (E)-configuration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-bond was not affected.
C-bond was not affected.
In comparison to the enzymatic cascade-process, standard protocols for the preparation of amine 12c are either more complex (e.g. Mitsunobu-conditions) or are not applicable due to the sensitivity of the CC double bond towards over-reduction (e.g. amide reduction using LiAlH4).19 Reductive amination (NaBH3CN/MeOH/NH4HCO2) showed insufficient selectivity for the primary amine.
In order to demonstrate the applicability of the enzymatic oxidation–amination cascade, the amine 12c was used as the starting material for the preparation of the potent antifungal agent naftifine (14)20 as outlined in Scheme 2. Thus, reductive amination of 12c with 1-naphthaldehyde gave sec-amine 13 in 73% yield. N-Methylation by formaldehyde/NaBH(OAc)3 proceeded quantitatively to yield naftifine (14) in 51% overall yield via four steps using the chemo-enzymatic protocol (69 mg isolated yield).
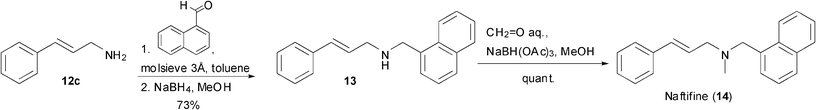 | ||
| Scheme 2 Synthesis of the antifungal agent naftifine (14). | ||
In conclusion, we have accomplished the direct conversion of benzylic and cinnamic primary alcohols to the corresponding amines based on a one-pot, multi-enzyme cascade comprising five enzymatic reactions through concurrent alcohol-oxidation and reductive amination. The benefits of this process were highlighted by the sensitive product 3-phenylallylamine (12c), which was used as the precursor for the antifungal agent naftifine (14).
Experimental
Screening of ω-transaminases
Lyophilized whole cells of the corresponding ω-TA (20 mg) were rehydrated in sodium phosphate buffer (500 μL, 100 mM, pH 7.0, 2 mM PLP, 2 mM NAD+) for 30 min at 30 °C and 120 rpm (horizontal position). A solution of L-alanine (22 mg, 0.25 mmol) and ammonium formate (11 mg, 0.17 mmol) in sodium phosphate buffer (500 μL, 100 mM, pH 7.0), formate dehydrogenase (20 μL), alanine dehydrogenase from Bacillus subtillis (10 μL, 7.5 mg protein/mL stock solution) and the substrate alcohol (0.05 mmol) were added in the above order and the reaction mixture was shaken for 16 h at 30 °C and 120 rpm (horizontal position). The obtained mixture was basified with NaOH (100 μL, 10 M in H2O) and extracted with EtOAc (2 × 500 μL). The combined organic phase was dried over Na2SO4 and subjected to GC-MS analysis.General cascade procedure
Whole cell preparations of the corresponding ω-transaminase (20 mg, lyophilised dry weight) and galactose oxidase from Fusarium NRRL 2903 (20 mg, lyophilised dry weight) were re-suspended in sodium phosphate buffer (500 μL, 100 mM, pH = 7.0, 2 mM PLP, 2 mM NAD+, 6 mg mL−1 CuSO4·5H2O) and were shaken at 30 °C and 120 rpm for 30 min in a horizontal position. A solution of L-alanine (13 mg, 0.15 mmol) and ammonium formate (5 mg, 0.07 mmol) in sodium phosphate buffer (500 μL, 100 mM, pH 7.0), formate dehydrogenase (20 μL), alanine dehydrogenase from Bacillus subtilis (10 μL, 7.5 mg protein mL−1 stock solution), horse radish peroxidase (15 μL, 10 mg mL−1 stock solution), ABTS (15 μL, 10 mg mL−1 stock solution) and the substrate alcohol (0.05 mmol) were added in the above order and the reaction mixture was placed into the oxygen apparatus. The apparatus was primed with oxygen (technical grade) for about 1 min and pressurized to 4 bar. The whole apparatus was shaken at rt and 170 rpm for 20 h (MG5 vials in vertical position). The obtained mixture was basified with NaOH (100 μL, 10 M in H2O) and extracted with EtOAc (2 × 500 μL). The combined organic phase was dried over Na2SO4 and subjected to GC-MS analysis. When the GDH/glucose recycling system was used, formate dehydrogenase was replaced by glucose dehydrogenase (20 μL, 20 mg mL−1 stock solution), and ammonium chloride (5.3 mg, 0.1 mmol) was used instead of ammonium formate, and glucose (21.6 mg, 0.12 mmol) was added to reaction mixture.Preparation of (E)-N-methyl-N-(1-naphthylmethyl)-3-phenylallylamine (naftifine, 14) from N-[(1E)-1-naphthalenylmethylene]-3-phenyl-2-propen-1-amine via one-pot procedure
N-[(1E)-1-naphthalenylmethylene]-3-phenyl-2-propen-1-amine (66 mg, 0.24 mmol) was dissolved in MeOH (15 mL). Sodium borohydride (48 mg, 1.26 mmol) was added and the reaction mixture was stirred at room temperature for 4 h. Formaldehyde (31 μL of a 37% aqueous solution, 11 mg, 0.38 mmol), sodium sulfate (18 mg, 0.13 mmol) and sodium triacetoxyborohydride (176 mg, 0.83 mmol) were added and the reaction mixture was stirred for additional 2 h. The solvent was removed under reduced pressure, the remaining white solid was treated with HClaq. (2 M) to remove the excess reagent, basified to pH > 10 by addition of NaOHaq. (10 M) and extracted with EtOAc (3 × 20 mL). The combined organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure to give compound 14 (69 mg, 0.24 mmol, > 99%). Physical properties are described in the electronic supporting information†.Acknowledgements
Financial support by the BMBF (Germany, project ‘Bioxamine’) is gratefully acknowledged. Plasmids of ω-transaminases from Pseudomonas putida, Paracoccus denitrificans and Vibrio fluvialis, and of alanine dehydrogenase from Bacillus subtilis were kind gifts of Alexandra Lerchner and Arne Skerra (Munich). Thomas Haas (Marl) is thanked for fruitful discussions.References
- S. M. Glueck, S. Gümüs, W. M. F. Fabian and K. Faber, Chem. Soc. Rev., 2010, 39, 313–328 RSC.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 CAS.
- (a) For a selection see: S. Imm, S. Baehn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126–8129 CrossRef CAS; (b) D. Pingen, C. Mueller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130–8133 CrossRef CAS; (c) B. Blank, S. Michlik and R. Kempe, Chem.–Eur. J., 2009, 15, 3790–3799 CrossRef CAS; (d) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661–8664 CrossRef CAS.
- C. V. Voss, C. C. Gruber, K. Faber, T. Knaus, P. Macheroux and W. Kroutil, J. Am. Chem. Soc., 2008, 130, 13969–13972 CrossRef CAS.
- For a recent review on biocatalytic cascade processes see: E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS.
- F. Escalettes and N. J. Turner, ChemBioChem, 2008, 9, 857–860 CrossRef CAS.
- (a) L. Sun, T. Bulter, M. Alcalde, I. P. Petrounia and F. H. Arnold, ChemBioChem, 2002, 3, 781–783 CrossRef CAS; (b) L. Sun, I. P. Petrounia, M. Yagasaki, G. Bandara and F. H. Arnold, Protein Eng., Des. Sel., 2001, 14, 699–704 CrossRef CAS; (c) B. Yuan, A. Page, C. P. Worrall, F. Escalettes, S. C. Willies, J. J. W. McDouall, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 7010–7013 CrossRef CAS.
- (a) N. Ito, S. E. V. Phillips, C. Stevens, Z. B. Ogel, M. J. McPherson, J. N. Keen, K. D. S. Yadav and P. F. Knowles, Nature, 1991, 350, 87–90 CrossRef CAS; (b) J. W. Whittaker and M. M. Whittaker, J. Biol. Chem., 1988, 263, 6074–6080 Search PubMed.
- (a) M. Höhne and U. T. Bornscheuer, ChemCatChem, 2009, 1, 42–51 CrossRef; (b) D. Koszelewski, K. Tauber, K. Faber and W. Kroutil, Trends Biotechnol., 2010, 28, 324–332 CrossRef CAS; (c) P. Tufvesson, J. Lima-Ramos, J. S. Jensen, N. Al-Haque, W. Neto and J. M. Woodley, Biotechnol. Bioeng., 2011, 108, 1479–1493 CrossRef CAS; (d) J. H. Seo, D. Kyung, K. Joo, J. Lee and B.-G. Kim, Biotechnol. Bioeng., 2011, 108, 253–263 CrossRef CAS; (e) N. J. Turner and M. D. Truppo, in Chiral Amine Synthesis: Methods, Developments and Applications, ed. T. C. Nugent, Wiley-VCH, Weinheim, 2010, pp. 431–459 Search PubMed; (f) H. C. Hailes, P. A. Dalby, G. J. Lye, F. Baganz, M. Micheletti, N. Szita and J. M. Ward, Curr. Org. Chem., 2010, 14, 1883–1893 CrossRef CAS; (g) J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914–1927 CrossRef CAS.
- (a) M. Höhne, S. Kühl, D. Robins and U. T. Bornscheuer, ChemBioChem, 2008, 9, 363–365 CrossRef; (b) D. Koszelewski, I. Lavandera, D. Clay, D. Rozell and W. Kroutil, Adv. Synth. Catal., 2008, 350, 2761–2766 CrossRef CAS; (c) D. Koszelewski, I. Lavandera, D. Clay, G. M. Guebitz, D. Rozell and W. Kroutil, Angew. Chem., Int. Ed., 2008, 47, 9337–9340 CrossRef CAS; (d) M. D. Truppo, J. D. Rozzell, J. C. Moore and N. J. Turner, Org. Biomol. Chem., 2009, 7, 395–398 RSC.
- M. Fuchs, M. Schober, J. Pfeffer, W. Kroutil, R. Birner-Gruenberger and K. Faber, Adv. Synth. Catal., 2011, 353, 2354–2358 CrossRef CAS.
- U. Kaulmann, K. Smithies, M. E. B. Smith, H. C. Hailes and J. M. Ward, Enzyme Microb. Technol., 2007, 41, 628–637 CrossRef CAS.
- (a) D. Koszelewski, D. Clay, K. Faber and W. Kroutil, J. Mol. Catal. B: Enzym., 2009, 60, 191–194 CrossRef CAS; (b) J.-S. Shin and B.-G. Kim, Biotechnol. Bioeng., 1999, 65, 206–211 CrossRef CAS.
- D. Koszelewski, M. Göritzer, D. Clay, B. Seisser and W. Kroutil, ChemCatChem, 2010, 2, 73–77 CrossRef CAS.
- R. L. Hanson, B. L. Davis, Y. Chen, S. L. Goldberg, W. L. Parker, T. P. Tully, M. A. Montana and R. N. Patel, Adv. Synth. Catal., 2008, 350, 1367–1375 CrossRef CAS.
- H. Yun, S. Lim, B.-K. Cho and B.-G. Kim, Appl. Environ. Microbiol., 2004, 70, 2529–2534 CrossRef CAS.
- V. Sieber, K. Grammann, B. Rühmann, T. Haas, J. Pfeffer, K. Doderer, C. Rollmann, A. Skerra, C. Rausch and A. Lerchner, World Intellectual Property Organization, 2010 ( WO 2010/089171 A2; Chem. Abstr. , 153 , 284693 ) Search PubMed.
- E. Park, M. Kim and J.-S. Shin, Adv. Synth. Catal., 2010, 352, 3391–3398 CrossRef CAS.
- C. R. Walter, J. Am. Chem. Soc., 1952, 74, 5185–5187 CrossRef CAS.
- (a) D. Berney and K. Schuh, Helv. Chim. Acta, 1978, 61, 1262–1273 CrossRef CAS; (b) G. Petranyi, N. S. Ryder and A. Stuetz, Science, 1984, 224, 1239–1241 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional optimisation data, source of chemicals and enzymes, additional experimental data, analytical methods and NMR spectra. See DOI: 10.1039/c2ra20800h |
| This journal is © The Royal Society of Chemistry 2012 |