Effect of pore structure on the selectivity of carbon materials for the separation of CO2/H2 mixtures: new insights from molecular simulation†
K.
Vasanth Kumar
* and
Francisco
Rodríguez-Reinoso
Laboratorio de Materiales Avanzados, Departamento de Química Inorgánica Universidad de Alicante, Spain. E-mail: vasanth@ua.es
First published on 5th September 2012
Abstract
Molecular simulations were performed to study the effect of the nanoporous structure on the selectivity of carbon materials for the adsorption of carbon dioxide from mixtures of carbon dioxide and hydrogen at 298 K and for fluid compositions: xCO2/xH2 = 1/9 and 2/8. Both carbon dioxide and hydrogen were studied using classical Lennard-Jones intermolecular potentials. Typical pore geometries such as slit-shaped pores and nanotubes were considered, along with a hypothetical foam-like structure and a carbon model exhibiting a random porous structure with a wide pore size distribution. Simulation results show that selectivity for carbon dioxide is sensitive to pore structure and composition; the solid/fluid interactions play a decisive role in the selectivity and most of the effects can be explained by the independent analysis of the interactions of carbon dioxide with the pore walls. In the range of pressure and composition studied, nanotubes have the highest selectivity towards carbon dioxide (100–313), followed by slit (9–63), foam-like (29–35) and random porous carbon (8–30). Molecular simulations further indicate that predicting the adsorption behavior for a CO2/H2 mixture from pure component isotherms is inadequate due, to the competing effects of the molecules with the pore walls.
1. Introduction
Recent interest in hydrogen as an alternative fuel has sparked a flurry of research into the technological aspects of its production and distribution. Hydrogen is primarily produced from natural gas via methane reforming (through syngas), but this process is not sustainable and cannot serve long-term demand.1 Coal gasification via a HyPr-RING method2,3 has currently received much attention and seems to be a promising technology for the production of large amounts of hydrogen, especially when considering the large size of the available coal resources.4 This process has several advantages over the conventional coal gasification process as the H2 is produced by co-pyrolysis of CaO and carbon in a single step, integrating the gas production and purification in one reactor.2,3 Despite the apparent advantages of this process, the purification of hydrogen by efficient removal of CO2 (about 10–15%) from the gas stream remains a challenging problem.2,3At this stage more research is needed for the efficient separation of CO2 and H2 mixtures. Among other H2 purification methods,4–6 selective adsorption of CO2 on porous material is considered to be a potential operation for both near term and long-term application during H2 production.4 Several novel carbon-based materials, having different nanoporous structures with excellent adsorption properties, can be potential candidates for H2 purification.7–11 In the context of adsorption of binary mixtures or gas mixtures in general, apart from a few industrial reports, not many experimental studies are available in the literature, due to several technical challenges in performing such experiments in the laboratory. Thus, in most cases12,13 the adsorption behavior of gas mixtures is studied using models based on solution theories.14–17 Despite the usefulness of these solution theories, they have their own limitations, one of which is that they do not consider any mechanism to differentiate solid/fluid interactions from fluid/fluid interactions. The latter are expected to play a crucial role in different carbon pore structures due to the steric and fluid confinement effects, especially at higher pressure. A few of the experimental works reported in the literature claim that, regardless of the morphology of the adsorbent, the storage density of CO2 or H2 of carbonaceous materials correlate fairly well with surface area and micropore volume, depending on a pressure of 298 K.9 In contrast to this, few experimental works18,19 point to the strong influence of the nature of the material, morphology and pore structure (under the same experimental conditions) on the ultimate performance of carbonaceous materials. It is thus not clear what, if any, morphological traits should be designed for any carbonaceous material to be used for the separation of CO2 from syngas.
For the case of the fluid mixture of interest here, a considerable amount of work has been carried out for the single component adsorption of CO2 or H2 on different carbon pore structures,13,20–30 but only a little work was focused on the adsorption of these fluid mixtures on carbon materials.13,30 The evidence from this work implies that the carbon pore geometry should play a significant role in the adsorption of this mixture.
The objective of this work is thus two-fold. Firstly, we explore the effect of the nano-scale morphology on the selectivity of CO2 by studying, under identical conditions, adsorption onto four different nanoporous shapes: (i) a slit pore, (ii) an array of nanotubes that form a unique one-dimensional pore structure, (iii) an open nanoporous foam obtained by fusing nanotubes, and (iv) a random computer-generated model of a disordered carbon. The second objective is to detail the characteristic adsorption behavior of single component and binary mixtures of CO2 and H2 on the different pore structures, in order to explore the possibility of applying theoretical models based on solution theories to predict the selectivity from single-component adsorption data. The carbon structures are carefully selected so that they roughly share some common characteristic properties and thus give a reliable picture at a molecular level of the pore structure on the characteristic adsorption of the fluid mixtures. The simulation studies were performed at 298 K for fluids of composition xCO2 / xH2 = 1/9 and 2/8 (the anticipated composition in the stream gas of a HyPr-RING process). The results could provide some useful information at the molecular level, detailing the effect of pore structure on selectivity, and will also help to select or design an appropriate material for hydrogen production.
2. Simulation method
In this work, GCMC simulations are used to study the adsorption behavior of CO2/H2 gas mixtures on nanoporous carbon materials. Following the work of Cracknell,31 H2 is modeled as a two-site Lennard-Jones fluid with a fixed bond length. Hydrogen interactions are assumed to be classical, as opposed to considering quantum effects. Kumar et al.32 showed that the quantum effects become significant for hydrogen only for temperatures below 100 K. CO2 is modeled as a two centered (dumbbell) Lennard-Jones plus a point quadrupole fluid. The fluid/fluid parameters for CO2 are taken directly from the parameterization of Vrabec et al.33 and give a quantitative representation of both the equilibrium vapor/liquid coexisting bulk densities and the P–V–T behavior of the fluid phases. All the solid structures described in the next section are composed of spherical sp2 hybridized carbon sites fixed in space in the simulation cell with LJ parameters given in Table 1. Monte Carlo simulations were run up to 107 configurations. The first 5 × 106 configurations were used to ensure equilibrium, while the later 5 × 106 configurations were used to obtain the ensemble average. More details are given in the ESI.† Molecular simulations were performed using a program (GCMCv61.f) developed in-house by Prof. Erich Müller, Imperial College London, which was tested and used earlier to study the adsorption behavior of different fluids on carbon structures.22–23,28,34–363. Nanoporous structures
Four nanoporous structures, which included slit-pore, nanotube, carbon foam and model carbon with disordered porous structures, were considered in this work. Hydrogen storage density and methane selectivity from the mixture of methane and hydrogen on these structures were discussed in earlier works.28,36 All these porous media are described in an atomistic way, taking carbon atoms to be represented by Lennard-Jones spheres and their unit cell structures are given in the ESI.† All the carbon models considered here are composed only of spherical sp2 hybridized carbon sites fixed in space in the unit cell with LJ parameters: σCC = 0.34 nm and εCC/k = 28.0 K. No surface heterogeneity is assumed.Briefly, the slit pore model used in this study has a fixed pore width of H = 1.5 nm. In this study, we define the pore width, H, as the distance between the centre of the carbon atom on one wall to the centre of the carbon atom on the opposite pore wall. This slit pore can be approximately modeled using the Steele37 potential, and no significant differences were seen in this study amongst the two (explicit and integrated) potentials. Three layers of graphene are placed on each wall, with an interlayer distance of 0.335 nm, to mimic an essentially isolated micropore. An ideal model for a carbon nanotube bundle is employed consisting of six parallel single-wall carbon nanotubes (SWCNT) with equal diameter D. The simulation cell consists of two rows of three open ended zig-zag (16,0) nanotubes of internal diameter 1.255 nm (measured from the center of the atoms). Only the endohedral adsorption is considered here.
The in silico generated random porous carbon structure, which can be taken as a model of real carbons, was constructed from a collective of flat coronene-shaped graphitic basic units made up of 24 carbon atoms. The random structure was obtained by placing a number of these carbon-building units in a simulation box, avoiding overlapping of the carbon units. There is no connectivity between the structures and they are artificially fixed in space within the cell. The random structure used in this work broadly shares some of the morphological properties as the other, better defined structures (nanotubes and slits).
The hypothetical carbon foam structures described by Yakobson’s group38,39 were originally obtained by conceptually fusing SWCNTs of similar diameters, in order to obtain a three-dimensional porous network. The particular carbon foam used in this work was obtained by welding armchair (10,10) with zigzag (17,0) SWCNTs.
The two structures, carbon foam and random porous carbon (RPC) go beyond the conventional models (slits and tubes) and the four structures reflect the geometry, topology and surface heterogeneity that characterize almost every form of nanoporous carbon. The detailed pore size distribution, surface area and accessible volume of these structures obtained using a geometrical method that involves a Monte Carlo integration procedure40–42 are detailed in the ESI.†
4. Results and discussion
Fig. 1 shows the plot of selectivity at 298 K in different pore structures as a function of pressure at two different compositions. The selectivity plot for nanotubes is shown separately for the sake of clarity. The term selectivity is used here to estimate the effectiveness of the carbon pores for the adsorptive separation of CO2 from the mixture with H2, and is defined as the ratio of molar compositions, x, of the components in the pore with respect to the ratio present in the bulk gas phase: | (1) |
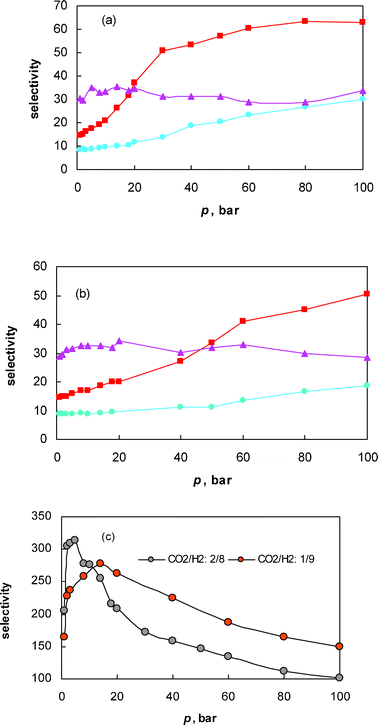 | ||
Fig. 1 Plot of selectivity versus pressure: (a) in different carbon pore structures for xCO2/xH2: 2/8, (b) in different carbon pore structures for xCO2/xH2: 1/9 and (c) in carbon nanotubes at different compositions (plot of the secondary axis corresponds to partial pressure of CO2vs. amount adsorbed, n, from a CO2/H2 mixture of xCO2/xH2: 2/8) ( : slit-pore; : slit-pore;  : foam; : foam;  : random). : random). | ||
Fig. 1 clearly shows that both pore structure and composition play an important role on the selectivity at the range of pressures studied. The carbon with tubular structure exhibits the highest selectivity, ranging from 100 to 313, depending on the molar composition and total bulk gas pressure. In the case of other materials considered here, irrespective of the molar composition, carbon porosity with a slit structure excels at high pressure, whereas carbon foam excels in the lower pressure range and notably, the random structure exhibits a poor selectivity for CO2 at the entire range of pressures considered.
In the case of nanotubes, the selectivity was sensitive to both loading and composition. A peculiar trend between selectivity and pressure is observed in this structure, with two distinct steps: a sharp increase in selectivity with increasing pressure, reaching a maximum, followed by a gradual decrease in selectivity. The peak in the selectivity plot was noticed at a total bulk gas pressure of 5 and 14 bar, for xCO2/xH2 = 2/8 and xCO2/xH2 = 1/9, respectively. In order to get a clear insight into this trend, the binary adsorption isotherms of CO2 and H2 (Fig. 2) are analyzed individually, with some evidential support from snapshots captured during the course of the simulation (Fig. 3). Fig. 2 shows the plot of absolute adsorption of CO2 or H2 (xCO2/xH2 = 2/8 and xCO2/xH2 = 1/9) from their fluid mixtures of different composition in the nanotube as a function of its partial pressure. It can be deduced from the figure that the CO2 isotherm exhibits a sharp increase in adsorption at lower partial pressure, with no significant adsorption thereafter due to pore volume restrictions. In the case of hydrogen, there is not any significant amount of adsorption at lower pressures (Fig. 2); however, although the hydrogen molecules are not preferentially adsorbed on the pore walls at higher pressures, they tend to regularly fill up in between the fluid cavities created by the CO2 molecules, thus reducing the selectivity. Snapshots showing these concepts at 100 bar for a molar concentration of xCO2/xH2 = 2/8 and xCO2/xH2 = 1/9 are given in Fig. 3. The presence of a large number of CO2 molecules within the nanotubes creates some porosity where the hydrogen molecules tend to pack themselves; otherwise surface adsorption of hydrogen molecules would not be observed. A close-up snapshot showing how the hydrogen molecules pack themselves in between the adsorbed CO2 molecules in a nanotube is given in Fig. 3. In other words, the selectivity, especially at higher pressures, was determined by the fluid cavities created by the adsorbed CO2 molecules, rather than by the composition of the bulk fluid. This phenomenon reasonably explains the low selectivity of the nanotubes for CO2 from a binary mixture of CO2 and H2 of composition xCO2/xH2 = 2/8, compared to their selectivity from a mixture of composition xCO2/xH2 = 1/9 (Fig. 1). In any case, in the range of pressure and composition studied, the selectivity always remained higher than unity as the relatively stronger solid/fluid interactions between CO2 and the pore walls dominated the adsorption, significantly depleting the hydrogen from the carbon tubes. The relatively higher selectivity of the nanotubes when compared to the other structures can be explained by considering the enhanced solid/fluid potential between CO2 and the carbon atoms, as all the atoms in the perimeter of the tube add to the interactions. Furthermore, the small effective diameter of the nanotubes enhances the preferential adsorption of CO2, in a one dimensional analogue of the effect reported earlier by Cao and Wu30 for the very narrow slit-shaped pores.
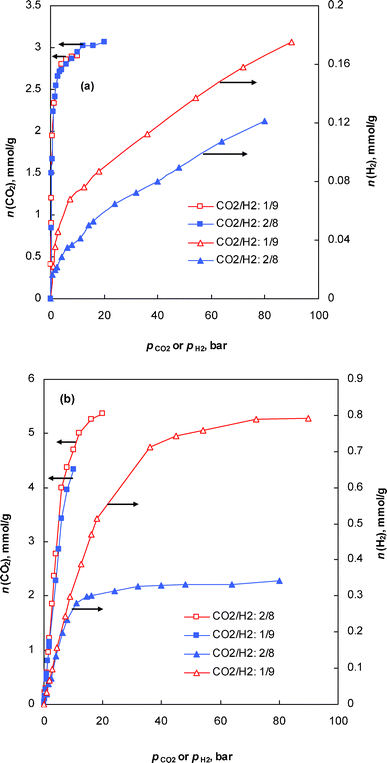 | ||
| Fig. 2 Plot of partial pressure versus absolute adsorption of CO2 or H2 in (a) nanotube and (b) slit-pore structures. | ||
 | ||
| Fig. 3 (a) Adsorption of binary mixtures CO2 (mauve) and H2 (ice-blue) (snapshots are captured at different angles for the sake of clear illustration) in carbon nanotubes at 100 bar of xCO2/xH2: 2/8 (left panel) and at 100 bar of xCO2/xH2: 1/9 (right panel); (b) adsorption of H2 from its pure fluid at 100 bar (left panel) and from a mixture of CO2 and H2 at 100 bar of composition xCO2/xH2: 2/8 (right panel ‡); (c) adsorption of CO2 and H2 from their mixtures of xCO2/xH2: 2/8 in the random structure at 5 bar (left panel), 20 bar (middle panel) and at 100 bar (right panel) and (d) adsorption of CO2 and H2 from their mixtures of xCO2/xH2: 2/8 in carbon foam at 5 bar (left panel), 20 bar (middle panel) and at 100 bar (right panel). | ||
In a slit geometry, which has an open pore structure, unlike in nanotubes, the selectivity globally increases with increasing total gas pressure, irrespective of the gas composition considered here. The increase in selectivity with pressure clearly indicates again that the adsorption is dominated by CO2 throughout the pressure range studied, due to the strong interactions between the CO2 molecules and the carbon atoms, depleting hydrogen from the pores. Fig. 4 shows the extent of the differences between the solid/fluid interactions. Hydrogen molecules experience only a very weak and short-ranged attraction, whereas CO2 molecules are far more strongly adsorbed. In an earlier theoretical study on single component adsorption of hydrogen at 77 K, Wang and Johnson20 showed that a slit-shaped pore that can hold one to two layers of hydrogen has the optimum pore size to store hydrogen at lower pressures due to strong-fluid interactions. Our simulation results do not show this enhanced attraction, either in slit pores or in other geometries, presumably due to the relative high temperature. Thermal motions of hydrogen molecules at 298 K are short ranged and fluid interactions are essentially repulsive, and the fluid/fluid interactions are weak as opposed to the strong interactions (attractive) between CO2 and carbon atoms; thus, the solid/fluid interactions are essentially weaker for any significant amount of adsorption to occur through dispersion forces (Fig. 4). We described earlier36 a similar behavior in a slit pore structure during the adsorption of equimolar binary fluid mixtures of methane and hydrogen at 298 K. The strong energetics of methane dominated the adsorption process, leaving no active sites for hydrogen. In the case of the methane/hydrogen system, the selectivity gradually decreased with pressure, due to the low excess adsorption of methane and the predominance of the hydrogen packing effect at higher pressure. In the present case, the strong energetics of CO2, which is nearly twice that of methane, favors a dense filling of CO2 inside the pores, leaving negligible access to the surface or pore volume for hydrogen. Close-up snapshots showing the difference in the number of hydrogen molecules adsorbed on the surface, or in the available pore volume from its bulk fluid, and from the binary mixture of hydrogen and carbon dioxide, are given in Fig. 3b (left and middle panel). The snapshots also show that for the case of adsorption from the binary fluid mixture of H2 and CO2, the hydrogen molecules are adsorbed in between the adsorbed CO2 molecules and not by surface adsorption (middle and right panel). It is worthwhile to mention that the snapshots captured are for illustrative purposes, depicting the possible equilibrium configuration of the system and they do not characterize the equilibrium state as a whole, which can only be given by statistical analysis of many equilibrium configurations.
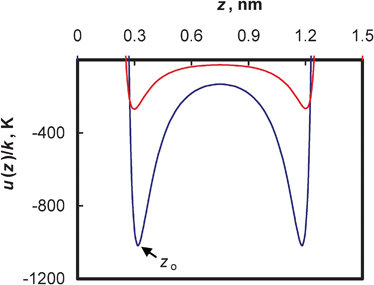 | ||
| Fig. 4 Potential energies, u/k, of a spherical hydrogen site (red) and a CO2 site (blue line) with the walls of the slit-pore structure of width H = 1.5 nm (z0: minimum potential distance). | ||
We further noticed that the composition plays a crucial role in the selectivity in the slit-shaped pore structure. Selectivity remains almost constant at higher pressures during the adsorption from the binary mixture of xCO2/xH2 = 2/8, whereas the selectivity increases with increasing pressure during the adsorption from fluid mixtures of composition xCO2/xH2 = 1/9. This can be easily explained by considering the adsorption pore-filling process of CO2 within this pore structure. In the earlier case, the pore filling process at higher pressure leaves negligible space for hydrogen molecules to adsorb or to pack in between the CO2 molecules, thus maintaining a constant composition of these fluid molecules inside the pores. In the later case, the partial pressure of CO2, pCO2, is not sufficient to fill the pore and we noticed a linear increase in the composition of both CO2 and H2 molecules inside the pores and, in any case, the selectivity tends to increase continuously as the adsorption is dominated by the strong solid/fluid interactions between CO2 and the slit-pore walls throughout the range of pressure studied. A plot of partial pressure of CO2 (pCO2) or H2 (pH2) versus the amount adsorbed from their fluid mixtures at different composition confirming these concepts is given in Fig. 2b.
In the case of a random structure, unlike other geometries, hydrogen molecules tend to adsorb on the active sites of this structure even at low pressure, irrespective of the composition. The pore size distribution plot (see ESI†) suggests that the random structure contains narrow pores and pores of a size that can accommodate one layer of hydrogen molecules, where strong H2 adsorption can be expected to occur, provided the adsorption is not dominated by CO2 due to pore-size restrictions. This explains the reason for the relatively lower selectivity of the random structure for CO2, when compared to the other structures considered here. Some of the captured snapshots in Fig. 2c show the adsorption behavior of CO2 and H2 from their mixtures (xCO2/xH2 = 2/8) at different pressures (total bulk pressure) and support the above arguments. These snapshots also show that, similar to the case of a slit-pore structure, the hydrogen molecules tend to arrange themselves within the fluid cavities created by the linear CO2 molecules. However, the selectivity always remains much higher than unity and it increases with increasing pressure, due to the strong interactions of CO2 with the pore walls. The snapshots further show that, as opposed to the strong confinement of CO2 in a slit pore, the complex porosity of the random structure disturbs the arrangements, especially at lower pressures. In fact, in the entire range of pressure studied, the CO2 density within the random structure is considerably lower when compared to other carbon frameworks.
In the case of carbon foam, with a three-dimensional pore structure that resembles molecular organic frameworks, unlike the other studied structures, the selectivity seems to be independent of pressure and composition. In order to have some clear insight into this behavior, we captured some snapshots depicting the adsorption of CO2/H2 at different pressures (Fig. 2). It can be observed from the snapshots that the hydrogen molecules are energetically favored and adsorbed on the surface of the foam, especially in the neck regions, although there is no adsorption of hydrogen inside the foam structure. An earlier theoretical study based on DFT calculations reported that a curved graphene-like surface binds (atomic) hydrogen more strongly than a flat graphene sheet.43 However, the scenario is completely different inside the pores of this structure, where CO2 molecules with strong energetics are preferentially adsorbed, since all the atoms in the periphery of the carbon framework add to the interactions, thus completely depleting the hydrogen molecules from the pores. In fact, the snapshots clearly show that there is no adsorption of hydrogen at 298 K inside the carbon foam throughout the pressure range studied, due to the strong energetics of CO2 that favor the confinement of CO2 molecules; thus, H2 is completely excluded from the pores. Despite the domination of CO2 within the carbon foam structure, the curved surfaces of the foam energetically favor the progressive adsorption of H2 with increasing pressure, thus maintaining a constant molar concentration of CO2 and H2 within the three-dimensional carbon framework.
To study the possibility of predicting the adsorption of gas mixtures from pure component adsorption isotherms, GCMC simulations were performed at 298 K to obtain pure component isotherms of a particular fluid at a pressure equivalent to the partial pressure of the corresponding component in the binary mixture of xCO2/xH2 = 2/8 (as a case study) and the results are given in Fig. 5. Fig. 5 clearly shows that there is no significant effect of dilution with hydrogen and the adsorption of pure CO2 nearly matches that of the mixture at low pressure; however, there is a very slight decrease in the adsorption of CO2 at higher pressure, indicating a competition of adsorption within the porous structure. In the case of hydrogen adsorption from the binary mixture, there is a significant decrease in hydrogen adsorption in all the carbon pore structures when compared to that of the pure component isotherm. The strong electrostatic forces experienced between the CO2 molecules influence the selectivity, depleting the adsorption of hydrogen by a significant amount, throughout the range of pressure considered. Thus, it would be inappropriate to predict the adsorption behavior of the studied fluid mixtures from single component isotherms. At this stage, it is worth comparing the results obtained from simulations in the recent experimental work of Hong et al.,44 since their experimental conditions match with our simulation conditions (micropore and gas composition of xCO2/xH2 = 1/9). They reported that the CO2 selectivity of activated carbon beads obtained from IAST (ideal adsorbed solution theory) calculations tends to decrease with increasing bulk pressure, whereas our simulation results for a slit-pore structure showed an opposite trend. This could probably be due to the limitations of the IAST theory, which ignores the competing effect of the fluids during adsorption, which seems to play a major role in the studied system as confirmed by our simulation results. It seems to be more probable that at 298 K, the solution theory, at least for the case of microporous carbons and for the fluid compositions studied here, might underestimate the CO2/H2 selectivity values.
 | ||
Fig. 5 Characteristic adsorption of carbon dioxide and hydrogen single components and mixtures xCO2/xH2 = 2/8 at 100 bar and 298 K in different nanoporous carbons (for the case of single components: pCO2 = 20 bar; pH2 = 80 bar). ( : adsorption of CO2 from mixtures; : adsorption of CO2 from mixtures;  : adsorption of pure CO2; : adsorption of pure CO2;  : adsorption of H2 from mixtures; : adsorption of H2 from mixtures;  : adsorption of pure H2.) : adsorption of pure H2.) | ||
Conclusions
In this work, grand canonical Monte Carlo simulations at 298 K were used to discuss the effect of pore structure on the separation of CO2 from CO2/H2 mixtures. The large variability of the parameters that are thought to influence the adsorption on carbons, e.g. pore size distribution, pore geometry and geometrical heterogeneities among others, make it practically difficult to perform such a parametric study from experiments. The GCMC results show that the pore geometry has a significant effect on the selectivity of CO2, which was found to be sensitive to both composition and total bulk pressure. The solid/fluid interactions play a decisive role in the overall selectivity, especially in carbon pores with simpler geometries (slit and tubes). As hydrogen/carbon interactions are relatively weak, most of the effects can be explained by the independent analysis of carbon dioxide interactions. An array of nanotubes with a closed structure shows the highest selectivity towards CO2 due to the enhanced solid/fluid potential, depleting the hydrogen. In the case of the random structure, with a more complex pore size distribution, such effects are partial and the complex porosity also disturbs the arrangement and confinement of the linear CO2 molecules, exhibiting a very low selectivity. The carbon with a three dimensional porous structure provides some specific adsorption sites for hydrogen molecules, exhibiting a selectivity that seems to be independent of pressure. Comparison of single and binary component isotherms indicates that it would be inappropriate to predict the adsorption behavior of a binary mixture from single component information in the carbon structures at the studied operating conditions.Acknowledgements
Support from the Ministerio de Ciencia e Innovación (Joint Spain-Japan Project PLE2009-0052) is acknowledged. KVK would like to thank the Ministerio de Ciencia e Innovación (Spain) for a Juan de la Cierva contract. Authors thank Prof. Erich Muller, Imperial College London for his guidance and the authors acknowledge the contribution of Prof. Boris Yakobson (Rice) for supplying the co-ordinate files used in the studies of the carbon foam structures.References
- J. A. Turner, Science, 1999, 285, 687 CrossRef CAS.
- S. Lin, M. Harada, Y. Suzuki and H. Hatano, Fuel, 2002, 81, 2079 CrossRef CAS.
- S. Y. Lin, M. Harada, Y. Suzuki and H. Hatano, Fuel, 2004, 83, 869 CrossRef CAS.
- Separation Technology R&D Needs for Hydrogen Production in the Chemical and Petrochemical Industries, report submitted for U.S. Department of Energy, available online at: http://www.chemicalvision2020.org/pdfs/h2_report.pdf.
- G. T. Rochelle, Science, 2009, 325, 1652 CrossRef CAS.
- H. Lin, E. V. Wagner, B. D. Freeman, L. G. Toy and R. P. Gupta, Science, 2006, 3, 639 CrossRef.
- A. Wahby, J. M. Ramos-Fernández, M. Martínez-Escandell, A. Sepúlveda-Escribano, J. Silvestre-Albero and F. Rodríguez-Reinoso, ChemSusChem, 2010, 3, 974 CrossRef CAS.
- J. Silvestre-Albero, A. Wahby, A. Sepúlveda-Escribano, M. Martínez-Escandell, K. Kaneko and F. Rodríguez-Reinoso, Chem. Commun., 2011, 47, 6840 RSC.
- A. Wahby, Sintesis de carbones activados especiales para almacenamiento de gases, PhD thesis, University of Alicante, 2011 Search PubMed.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765 CAS.
- J. D. Carruthers, M. A. Petruska, E. A. Sturm and S. M. Wilson, Microporous Mesoporous Mater., 2012, 154, 62–67 CrossRef.
- J. Schell, N. Casas, R. Pini and M. Mazzotti, Adsorption, 2012, 18, 49 CrossRef CAS.
- X. Peng, D. Cao and W. Wang, Chem. Eng. Sci., 2011, 66, 2266 CrossRef CAS.
- W. Sievers and A. Mersmann, Chem. Eng. Technol., 1994, 17, 325 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121 CrossRef CAS.
- M. D. LeVan and T. Vermuelen, J. Phys. Chem., 1981, 85, 3247 CrossRef CAS.
- M. M. Benjamin, Environ. Sci. Technol., 2009, 43, 2530 CrossRef CAS.
- C. Lu, H. Bai, B. Wu, F. Su and J. F. Hwang, Energy Fuels, 2008, 22, 3050 CrossRef CAS.
- S. Beyaz, F. D. Lamari, B. Weinberger and P. Langlois, Int. J. Hydrogen Energy, 2010, 35, 217 CrossRef CAS.
- Q. Wang and J. K. Johnson, J. Phys. Chem. B, 1999, 103, 4809 CrossRef CAS.
- Q. Wang and J. K. Johnson, J. Phys. Chem. B, 1999, 103, 277 CrossRef CAS.
- E. A. Müller, Environ. Sci. Technol., 2005, 39, 8736 CrossRef.
- E. A. Müller, J. Phys. Chem. B, 2008, 112, 8999 CrossRef.
- A. Salih, Molecular simulation of the adsorption and transport properties of carbon dioxide, methane, water and their mixtures in coal-like structures, Ph.D Thesis, Imperial College, London, 2010 Search PubMed.
- M. Tenney and C. M. Lastoski, Environ. Prog., 2006, 25, 343 CrossRef.
- C. M. Lastoskie, Science, 2010, 330, 595 CrossRef CAS.
- M. Rzepka, P. Lamp and M. A. de la Casa-Lillo, J. Phys. Chem. B, 1998, 102, 10894 CrossRef CAS.
- K. V. Kumar, A. Salih, L. Lu, E. A. Müller and F. Rodríguez-Reinoso, Adsorpt. Sci. Technol., 2011, 29, 799 CrossRef CAS.
- D. Levesque, A. Gicquel, F. L. Darkim and S. B. Kayiran, J. Phys.: Condens. Matter, 2002, 14, 9285 CrossRef CAS.
- D. Cao and J. Wu, Carbon, 2005, 43, 1364 CrossRef CAS.
- R. F. Cracknell, Phys. Chem. Chem. Phys., 2001, 3, 2091 RSC.
- A. V. A. Kumar, H. Jobic and S. K. Bhatia, J. Phys. Chem. B, 2006, 110, 16666 CrossRef CAS.
- J. Vrabec, J. Stoll and H. Hasse, J. Phys. Chem. B, 2001, 105, 12126 CrossRef CAS.
- F. J. A. L. Cruz and E. A. Müller, Adsorption, 2009, 15, 1 CrossRef CAS.
- F. J. A. L. Cruz, E. A. Müller and J. P. B. Mota, RSC Adv., 2011, 1, 270 RSC.
- K. V. Kumar, E. A. Müller and F. Rodriguez-Reinoso, J. Phys. Chem. C, 2012, 116, 11820 CAS.
- W. A. Steele, The interaction of gases with solid surfaces, Pergamon Press, New York, 1974 Search PubMed.
- F. Ding, P. O. Krasnov and B. I. Yakobson, J. Chem. Phys., 2007, 127, 164703 CrossRef.
- A. K. Singh, J. Lu, R. S. Aga and B. I. Yakobson, J. Phys. Chem. C, 2011, 115, 2476 CAS.
- T. Düren, F. Millange, G. Férey, K. S. Walton and R. Q. Snurr, J. Phys. Chem. C, 2007, 111(42), 15350 Search PubMed.
- L. D. Gelb and K. E. Gubbins, Langmuir, 1999, 15, 305 CrossRef CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248 CrossRef CAS.
- M. Kayanuma, U. Nagashima, H. Nishihara, T. Kyotani and H. Ogawa, Chem. Phys. Lett., 2010, 495, 251 CrossRef CAS.
- Y. Q. Hong, S. X. Hong and C. D. Peng, Acta Phys. Chim. Sin., 2007, 23(7), 1080 Search PubMed.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20775c |
| ‡ Only hydrogen molecules are shown in the first image for the sake of clear illustration. The close-up shot in the second image shows the packed hydrogen molecules in between the adsorbed CO2 molecules. |
| This journal is © The Royal Society of Chemistry 2012 |