Microwave-assisted hydrolysis: efficient synthesis of α-substituted cysteines on multi-gram scale†
Dominic
Fiset
and
André B.
Charette
*
Centre in Green Chemistry and Catalysis, Department of Chemistry, Université de Montréal, P.O. Box 6128, Station Downtown, Montréal, Québec, Canada H3C 3J7. E-mail: andre.charette@umontreal.ca; Fax: (514)-343-7586; Tel: (514)-343-6283
First published on 25th April 2012
Abstract
A microwave-assisted hydrolysis of thiazolidines was developed for the synthesis of enantioenriched α-alkylcysteines. Under optimized conditions, the efficiency of the reaction can be considerably improved and the scope significantly broadened as compared to previously described processes. This approach is shown to be applicable to the multi-gram scale using a high-throughput microwave system.
Quaternary amino acids play a key role in medicinal chemistry as they can be used to modify the conformational properties of a peptide without loss of side-chain functions, while improving its stability toward biological degradation. More specifically, α-alkylcysteines are of particular interest, as they can be used to constrain the structure of a peptide via disulfide bonds. Additionally, the (R)-methylcysteine 5a unit occurs in thiazoline rings of several potent antitumor or anti-HIV natural products such as (–)-thiangazole,1 cyclothiazomycin,2 largazole (vide infra),3 hoiamides,4 bisebromoamide5 and grassypeptolides.6
Although considerable efforts have been devoted to the development of new methodologies for the synthesis of α,α-disubstituted amino acids,7 available methods for the enantioselective synthesis of α-alkylcysteines are still limited. The main reported strategies for their synthesis rely on: (1) hydrolysis of desferrithiocin,8 (2) chiral auxiliaries,9 (3) ring opening of a chiral aziridine or β-lactone with thiolates,10 (4) self reproduction of stereocenters (SRS) from oxazolidinone or thiazolidine derivatives,11 (5) phase transfer alkylation,12 and (6) enzymatic desymmetrization.13 Among these, Pattenden's procedure (Scheme 1),14 which relies on the SRS, remains the most efficient way to synthesize multi-gram quantities of (R)-α-methylcysteine 5a, as showcased in the total syntheses of (–)-didehydromirabazole A,15 (–)-thiangazole,16 micacocidin,17 hoiamide C,18 bisebromoamide,19 and largazole.20
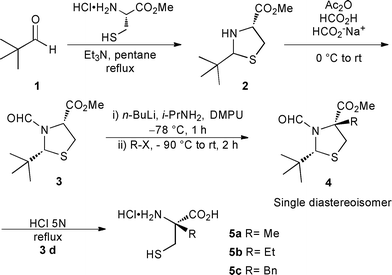 | ||
| Scheme 1 Pattenden's procedure14 | ||
Recently, Àlvarez, Altucci, de Lera and co-workers published a total synthesis of largazole, a potent histone deacetylase (HDAC) inhibitor, in which they investigated the influence of different alkyl groups at the C7 position using Pattenden's procedure (Scheme 2).21
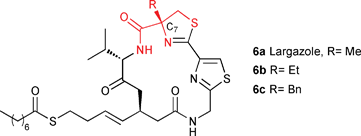 | ||
| Scheme 2 Structure–activity relationship study of largazole | ||
In their report, they mentioned that the major drawback for this structure–activity relationship (SAR) study is the small scope of quaternary amino acids available via this procedure. Even under harsh acidic conditions (HCl 5N under reflux for 3 days), low conversion is obtained in most cases for the hydrolysis of the alkylated thiazolidine 4. Herein, we report a microwave-assisted hydrolysis that can considerably enhance the reaction efficiency and, therefore, broaden the scope of this reaction to access a variety of new α-alkylcysteines. In addition, using MARS, a high-throughput digestion system, we were able to reproduce this reaction on the multi-gram scale with short reaction times.
Results and discussion
Since the 1980s,22 microwave heating has appeared as a valuable alternative to conventional heating processes. In this technique, heat is generated from electromagnetic energy, and its formation strongly depends on the specific polarity of the molecules. Water is an excellent solvent for microwave-mediated reactions because it possesses a polar nature, which accounts for its good potential for microwave absorption.23 Therefore, we were interested in the feasibility of the hydrolysis under microwave irradiation to ultimately enhance the reaction efficiency. To our delight, we found that the reaction can reach completion within an hour at 160 °C while maintaining an excellent yield (Scheme 3).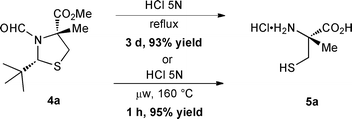 | ||
| Scheme 3 A comparison between thermal and microwave conditions | ||
With these conditions in hand, we were keen to investigate the scope of this reaction (Table 1). In this context, we synthesized thiazolidines 7 starting with the more affordable cysteine ethyl ester.24 Satisfyingly, all of the hydrolysis precursors were obtained as single diastereoisomers.25
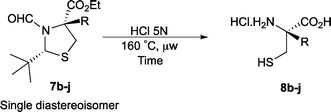
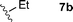
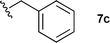
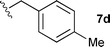
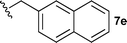
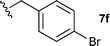
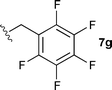
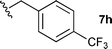
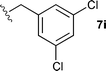

The ethyl 7b and benzyl 7c derivatives (entries 1 and 2) can be respectively hydrolyzed within 1 and 2 h, compared to 3 days under standard thermal conditions, while maintaining high yields. p-Methylbenzyl 7d and naphthylmethyl 7e derivatives proved to be more challenging and thus required longer reaction times (entries 3 and 4). Interestingly, the 4-bromobenzyl 7f derivative could be obtained in 81% yield, which compares favourably to a 33% yield reported in the literature (entry 5).21 In fact, halogenated derivatives are generally not tolerated under standard thermal conditions, as illustrated with a reported 18% yield for the 3,5-difluorobenzyl derivative, while no conversion was observed for the 3,5-bis-trifluoromethylbenzyl.21 Microwave-assisted hydrolysis can be used to circumvent this limitation, as exemplified with the 2,3,4,5-pentafluorobenzyl 7g, 4-trifluoromethylbenzyl 7h and 3,5-dichlorobenzyl 7i derivatives, which were isolated in good to excellent yields (entries 6, 7 and 8). In all the examples mentioned above, the α-substituted cysteines were obtained in a 4-step sequence starting from commercially available cysteine ethyl ester hydrochloride and required a single flash chromatography.24
Unfortunately, the allylated 7j derivative was found to be incompatible under harsh acidic conditions, presumably because of the reactivity of the olefin (entry 9). Nonetheless, we developed a 2-step sequence using this substrate to expand the scope of this methodology (Table 2).
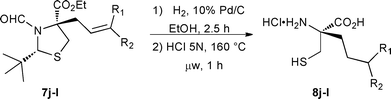
| Entry | R1 | R2 | Yielda (%) | |
|---|---|---|---|---|
| Hydrogenation | Hydrolysis | |||
| a Isolated yield. b Hydrogenation was performed over 16 h. | ||||
| 1 | H | H 7j | 97 | 78 |
| 2 | H | Me 7k | 92 | 83 |
| 3 | Me | Me 7l | 99b | 68 |
Hydrogenation followed by microwave-assisted hydrolysis furnished a propyl 7j analogue in 76% overall yield (entry 1). This strategy could be further extended to the synthesis of butyl 7k and 3-methylbutyl 7l derivatives (entries 2 and 3) in good yields.
Multi-gram synthesis of (R)-α-methylcysteine hydrochloride
Until very recently, a major limitation for the microwave-assisted organic reaction (MAOS) had been the preparation of a substantial amount of material. Considerable efforts have been devoted to address this issue, which have resulted in the development of several large batch reactors, high throughput and flow systems.26 Consequently, we investigated whether our methodology could be reproduced on multi-gram quantities (Table 3).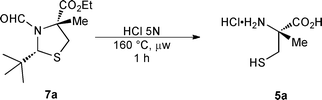
| Entry | Reactor | Scale (g) | Yieldab (%) |
|---|---|---|---|
| a Isolated yield. b Yield based on the recovered starting material in the inlet vial is indicated in brackets. See ESI for details. c See text for details. | |||
| 1 | Biotage Initiator | 1 | 99 |
| 2 | CEM Voyager | 5 | 96 (99) |
| 3c | CEM Voyager | 7.5 | 57 (75) |
| 4 | CEM MARS | 4 | 99 |
| 5 | CEM MARS | 10 | 99 |
To initiate this study, we selected the synthesis of (R)-α-methylcysteine 5a due to its occurrence in natural products and its synthetic utility. First, it should be noted that our conditions are compatible with a standard microwave system for the generation of up to one gram of the amino acid (entry 1). However, we tested our reaction on a Voyager stop-flow system, since it can be used for significantly larger scales.27 Although our first attempt was successful (entry 2), this system was found to be incompatible with the use of corrosive HCl 5N (entry 3).
We then turned our attention to a MARS digestion system, which is typically used for protein hydrolysis. As a proof of concept, we performed our reaction on a four-gram scale (four closed vessels each holding one gram). This method proved to be highly representative of the results obtained on a standard microwave system, as showcased with the excellent reaction efficiency achieved (entry 4). Satisfyingly, this reaction could be further reproduced on a 10 gram scale by running 5 simultaneous reactions (entry 5). Based on these results and on the loading capacity of the MARS system (40 closed vessels, 50 mL each), this methodology is amenable to an 80 gram scale synthesis of 5a on a single run. Given that the sequence for the synthesis of the hydrolysis precursor could be achieved industrially,28 we believe that this process is a significant improvement over existing methods for the preparation of substantial amounts of enantioenriched α-alkylcysteines.
Conclusions
In summary, a microwave-assisted hydrolysis of thiazolidines was developed for the synthesis of α-alkylcysteines. Under microwave irradiation, the reaction efficiency and its scope were significantly enhanced. The effectiveness of these conditions was showcased in the synthesis of halogenated derivatives, paving the way to a new variety of α-substituted cysteines, which can potentially be used in SAR studies of various natural products. Moreover, the feasibility of this method was illustrated on the multi-gram scale using MARS, a high throughput system.Experimental section
General procedure for the microwave-assisted hydrolysis
Substrate 7 (0.20 mmol) was added in a 5 mL vial and purged with argon for 15 min. A solution of aqueous HCl 5N (0.66 mL, 0.30 M), previously degassed by bubbling argon for 30 min, was added. After purging with argon for 15 min, the vial was capped and heated under microwave irradiation (high absorbance) at 160 °C. After 1 h, the reaction was cooled down to room temperature, the pressure was released with a needle and the vial uncapped. The aqueous phase was washed with EtOAc (3 × 15 mL) then the acidic aqueous phase was concentrated under reduced pressure to afford the desired (R)-α-alkylcysteine 8 as a highly hygroscopic solid.General procedure with the MARS digestion system
Thiazolidine 7a (10.0 g, 38.55 mmol) was added to 5 different closed vessels (5 × 2.0 g) and purged with argon for 15 min. A solution of aqueous HCl 5N (5 × 25 mL), previously degassed by bubbling argon for 30 min, was added to each vessel which were rapidly sealed under an atmosphere of argon. The solutions were heated at 160 °C for 1 h using the MARS digestion system. The combined aqueous phases were washed with EtOAc (3 × 75 mL) then the acidic aqueous phases were concentrated under reduced pressure to afford the desired (R)-α-alkylcysteine 5a as a highly hygroscopic beige solid (6.60 g, 99%).Acknowledgements
This work has been supported by NSERC (Canada), the Canada Research Chair Program, the Canada Foundation for Innovation, the Centre in Green Chemistry and Catalysis (CGCC) and the Université de Montréal. The authors would like to thank A. Lemire, F. Galaud and P. Lavallée (Université de Montréal) as well as J. O′Donnell (CEM) for helpful discussions; P. Lapointe (IRIC) is also acknowledged for supplying some of the reagents used in this study.References
- (a) R. Jansen, B. Kunze, H. Reichenbach, E. Jurkiewicz, G. Hunsmann and G. Höfle, Liebigs Ann. Chem., 1992, 1992, 357 CrossRef; (b) R. Jansen, D. Schomburg and G. Höfle, Liebigs Ann. Chem., 1993, 1993, 701 CrossRef.
- Y. Zou, Q. Liu and A. Deiters, Org. Lett., 2011, 13, 4352 CrossRef CAS.
- K. Taori, V. J. Paul and H. Luesch, J. Am. Chem. Soc., 2008, 130, 1806 CrossRef CAS.
- (a) For hoiamide A, see: A. Pereira, Z. Cao, T. F. Murray and W. H. Gerwick, Chem. Biol., 2009, 16, 893 CrossRef CAS; (b) A. Pereira, Z. Cao, T. F. Murray and W. H. Gerwick, Chem. Biol., 2009, 16, 1208 CrossRef CAS; (c) For hoiamide C, see: H. Choi, A. R. Pereira, Z. Cao, C. F. Shuman, N. Engene, T. Byrum, T. Matainaho, T. F. Murray, A. Mangoni and W. H. Gerwick, J. Nat. Prod., 2010, 73, 1411 CrossRef CAS.
- (a) T. Teruya, H. Sasaki, H. Fukazawa and K. Suenaga, Org. Lett., 2009, 11, 5062 CrossRef CAS; (b) X. Gao, Y. Liu, S. Kwong, Z. Xu and T. Ye, Org. Lett., 2010, 12, 3018 CrossRef CAS.
- C. C. Thornburg, M. Thimmaiah, L. A. Shaala, A. M. Hau, J. M. Malmo, J. E. Ishmael, D. T. A. Youssef and K. L. McPhail, J. Nat. Prod., 2011, 74, 1677 CrossRef CAS.
- (a) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 1998, 9, 3517 CrossRef CAS; (b) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2000, 11, 645 CrossRef CAS; (c) Y. Ohfune and T. Shinada, Eur. J. Org. Chem., 2005, 5127 CrossRef CAS; (d) C. Cativiela and M. D. Díaz-de-Villegas, Tetrahedron: Asymmetry, 2007, 18, 569 CrossRef CAS; (e) H. Vogt and S. Bräse, Org. Biomol. Chem., 2007, 5, 406 RSC; (f) C. Cativiela and M. Ordóñez, Tetrahedron: Asymmetry, 2009, 20, 1 CrossRef CAS; (g) V. A. Soloshonok and A. E. Sorochinsky, Synthesis, 2010, 2319 CrossRef CAS; (h) J. Clayden, M. Donnard, J. Lefranc and D. J. Tetlow, Chem. Commun., 2011, 47, 4624 RSC.
- H.-U. Naegeli and H. Zähner, Helv. Chim. Acta, 1980, 63, 1400 CrossRef CAS.
- (a) U. Groth and U. Schöllkopf, Synthesis, 1983, 37 CrossRef CAS; (b) S. Singh, S. J. Rao and M. W. Pennington, J. Org. Chem., 2004, 69, 4551 CrossRef CAS.
- (a) T. Fukuyama and L. Xu, J. Am. Chem. Soc., 1993, 115, 8449 CrossRef CAS; (b) H. Shao, Q. Zhu and M. Goodman, J. Org. Chem., 1995, 60, 790 CrossRef CAS; (c) N. D. Smith and M. Goodman, Org. Lett., 2003, 5, 1035 CrossRef CAS.
- (a) M. A. Walker and C. H. Heathcock, J. Org. Chem., 1992, 57, 5566 CrossRef CAS; (b) G. Pattenden, S. M. Thom and M. F. Jones, Tetrahedron, 1993, 49, 2131 CrossRef CAS; (c) D. Seebach, A. R. Sting and M. Hoffmann, Angew. Chem., Int. Ed. Engl., 1996, 35, 2708 CrossRef CAS.
- T.-S. Kim, Y.-J. Lee, B.-S. Jeong, H-G. Park and S.-S. Jew, J. Org. Chem., 2006, 71, 8276 CrossRef CAS.
- (a) B. L. Kedrowski, J. Org. Chem., 2003, 68, 5403 CrossRef CAS; (b) A. Inoue, H. Komeda and Y. Asano, Adv. Synth. Catal., 2005, 347, 1132 CrossRef CAS; (c) D. S. Masterson, K. Roy, D. A. Rosado and M. Fouche, J. Pept. Sci., 2008, 14, 1151 CrossRef CAS; (d) D. S. Masterson, B. L. Kedrowski and A. Blair, Synlett, 2010, 2941 CrossRef CAS.
- See reference 11b.
- G. Pattenden and S. M. Thom, J. Chem. Soc., Perkin Trans. 1, 1993, 1629 RSC.
- R. J. Boyce, G. C. Mulqueen and G. Pattenden, Tetrahedron, 1995, 51, 7321 CrossRef CAS.
- A. Ino, Y. Hasegawa and A. Murabayashi, Tetrahedron, 1999, 55, 10283 CrossRef CAS.
- L. Wang, Z. Xu and T. Ye, Org. Lett., 2011, 13, 2506 CrossRef CAS.
- (a) X. Gao, Y. Liu, S. Kwong, Z. Xu and T. Ye, Org. Lett., 2010, 12, 3018 CrossRef CAS; (b) W. Li, S. Yu, M. Jin, H. Xia and D. Ma, Tetrahedron Lett., 2011, 52, 2124 CrossRef CAS.
- (a) T. Seiser, F. Kamena and N. Cramer, Angew. Chem., Int. Ed., 2008, 47, 6483 CrossRef CAS; (b) A. K. Ghosh and S. Kulkarni, Org. Lett., 2008, 10, 3907 CrossRef CAS; (c) Y. Ying, K. Taori, H. Kim, J. Hong and H. Luesch, J. Am. Chem. Soc., 2008, 130, 8455 CrossRef CAS; (d) Q. Ren, L. Dai, H. Zhang, W. Tan, Z. Xu and T. Ye, Synlett, 2008, 2379 CAS; (e) Y. Numajiri, T. Takahashi, M. Takagi, K. Shin-ya and T. Doi, Synlett, 2008, 2483 CAS; (f) X. Zeng, B. Yin, Z. Hu, C. Liao, J. Liu, S. Li, Z. Li, M. C. Nicklaus, G. Zhou and S. Jiang, Org. Lett., 2010, 12, 1368 CrossRef CAS; (g) H. Benelkebir, S. Marie, A. L. Hayden, J. Lyle, P. M. Loadman, S. J. Crabb, G. Packham and A. Ganesan, Bioorg. Med. Chem., 2011, 19, 3650 CrossRef CAS; (h) F. Diness, D. S. Nielsen and D. P. Fairlie, J. Org. Chem., 2011, 76, 9845 CrossRef CAS.
- J. A. Souto, E. Vaz, I. Lepore, A.-C. Pöppler, G. Franci, R. Álvarez, L. Altucci and A. R. de Lera, J. Med. Chem., 2010, 53, 4654 CrossRef CAS.
- (a) Bhargava, Naresh, 1980. CA 1127999, 1982; (b) R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett., 1986, 27, 279 CrossRef CAS; (c) R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Tetrahedron Lett., 1986, 27, 4945 CrossRef CAS.
- (a) B. Ondruschka, W. Bonrath and D. Stuerga, in Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH, Weinheim, 2nd edn, 2006, vol. 1, ch. 1, pp. 1-61. Search PubMed; (b) V. Polshettiwar and R. S. Varma, Chem. Soc. Rev., 2008, 37, 1546 RSC.
- See ESI† for details.
- The presence of rotamers was confirmed with 1H NMR by monitoring a sample of 7a heated at 100 °C. See ESI† for details..
- (a) B. Ondruschka, W. Bonrath andD. Stuerga, in Microwaves in Organic Synthesis, ed. A. Loupy, Wiley-VCH, Weinheim, 2nd edn, 2006, vol. 1, ch. 2, pp. 62–107 Search PubMed; (b) M. D. Bowman, J. L. Holcomb, C. M. Kormos, N. E. Leadbeater and V. A. Williams, Org. Process Res. Dev., 2007, 12, 41 CrossRef; (c) C. R. Strauss, Org. Process Res. Dev., 2009, 13, 915 CrossRef CAS.
- J. D. Moseley and E. K. Woodman, Org. Process Res. Dev., 2008, 12, 967 CrossRef CAS.
- M. S. Anson, H. F. Clark, P. Evans, M. E. Fox, J. P. Graham, N. N. Griffiths, G. Meek, J. A. Ramsden, A. J. Roberts, S. Simmonds, M. D. Walker and M. Willets, Org. Process Res. Dev., 2011, 15, 389 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Characterization data and copies of the NMR spectra. See DOI: 10.1039/c2ra20744c |
| This journal is © The Royal Society of Chemistry 2012 |