Formation of unexpected α-amino amidine through three-component ‘UGI condensation reaction’†
Abu T.
Khan
*a,
Sidick Basha
R
a,
Mohan
Lal
a and
Mohammad Hedayetullah
Mir
b
aDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati 781 039, India. E-mail: atk@iitg.ernet.in; Fax: +91 361 2582349; Tel: +91 361 2582305
bDepartment of Chemistry, Aliah University, DN41 Salt Lake, Kolkata 700 091, India
First published on 20th April 2012
Abstract
The synthesis of a wide variety of α-amino amidine derivatives was accomplished through a one-pot three-component reaction from aromatic aldehydes, aromatic amines and isocyanides, catalyzed by bromodimethylsulfonium bromide (BDMS), in good yields. Some of the salient features of the present protocol are: mild reaction conditions, shorter reaction times, non-aqueous work-up procedure and no need for chromatographic separation.
Introduction
In recent years, multi-component reactions (MCRs) have gained considerable attention due to the easy accessibility of complex structures in a single step from readily available starting materials.1 The most important aspects of these reactions are high convergence, superior atom-economy, simpler reaction procedures, lower costs, high variability, high bond forming efficiency (BFE)2 and being environmentally benign.Amidines3 are naturally occurring compounds which display a wide range of biological activities, as shown in Fig. 1.
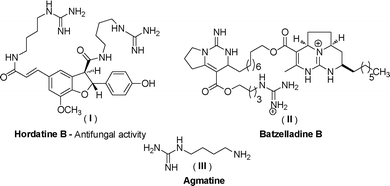 | ||
| Fig. 1 Biologically active compounds containing the amidine skeleton. | ||
Among them, α-amino amidines are important building blocks for the synthesis of antibiotics,4 iminopeptides,5 novacaine derivatives6 and intermediates for the study of the bioluminescence of Cypridina hilgendorfii.7 In 1956, Mengelberg first reported the synthesis of an α-amino amidine, starting from N-protected α-amino iminoethers as an intermediate.8 McFarland reported that the reaction of isobutyraldehyde, dimethyl amine and cyclohexyl isocyanide in methanol can provide a mixture of products such as α-amino amides along with α-hydroxy amides. However, by tuning the reaction conditions, the synthesis of the α-amino amidine was achieved using the same combination.9 Later on, Barluenga et al. reported10 the synthesis of α-amino amidines by oxidative addition of primary aromatic amines to prop-2-ynol in the presence of mercury(II) acetate. Afterward, various other research groups have also demonstrated the synthesis of amidines from nitriles.11
A few years ago, Keung et al. reported the synthesis of α-amino amidines on the micromolar scale from a primary or secondary amine, an aliphatic or aromatic aldehyde and cyclohexyl isocyanide, using the one-pot three-component ‘Ugi condensation reaction’, by employing scandium(III) triflate as a catalyst.12 Furthermore, surveying the literature reveals the existence of only a few methods for the synthesis of α-amino amidines. Earlier reported methods had some demerits, such as a longer reaction time, tedious work-up procedure and requirement of an expensive catalyst, as well as it being difficult to isolate the product for characterization. Keung et al. also mentioned that it is difficult to interpret the 1H-NMR spectra of the α-amino amidines due to broad peaks. Consequently, there is further scope to devise a new methodology for the synthesis of these compounds in the context of the paucity of the method.
As part of our ongoing efforts in developing multi-component syntheses to access scaffolds with potential for biological activities,13 we report here a one-pot three-component reaction for the synthesis of α-amino amidines by the reaction of aromatic aldehydes, aromatic amines and isocyanides, using bromodimethylsulfonium bromide as a catalyst (Scheme 1).
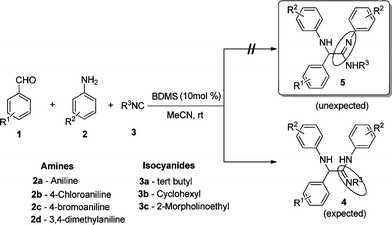 | ||
| Scheme 1 Synthesis of α-amino amidine using a three-component reaction. | ||
Results and discussion
Bromodimethylsulfonium bromide (BDMS) is a readily available, cheap and highly efficient brominating reagent as well as a catalyst for various organic transformations.14 For the present study, the catalyst, BDMS, was prepared by the following reported method.14a A combination of benzaldehyde (1a), aniline (2a) and tert-butyl isocyanide (3a) were chosen as the model substrates. The reaction conditions were optimized for the synthesis of the α-amino amidines in terms of the yield and reaction time. Several reactions were scrutinized using various mol% of catalysts in different solvents such as CH2Cl2, CH3CN, and MeOH. In the initial experiments, 2 mol%, 4 mol%, and 5 mol% of BDMS were used, and the yields of compound 5a were obtained as 57%, 61% and 68%, respectively (Table 1, entries 4–6). However, in the presence of 10 mol% of the catalyst, the reaction proceeded smoothly and the yield of the product 5a increased significantly to 92% (Table 1, entry 7).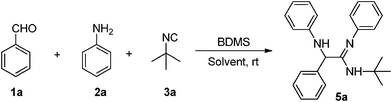
| Entry | BDMS used (mol%) | Solvent | Time/h | Yield (%)b |
|---|---|---|---|---|
| a The reactions were performed in 1 mmol benzaldehyde (1a), tert-butyl isocyanide (3a) and 2 mmol aniline (2a), in 5 mL solvent at room temp., respectively. b Isolated yield. | ||||
| 1 | — | CH2Cl2 | 22 | 0 |
| 2 | 2 | CH2Cl2 | 7 | 52 |
| 3 | 2 | CH3OH | 6 | 45 |
| 4 | 2 | CH3CN | 5 | 57 |
| 5 | 4 | CH3CN | 4 | 61 |
| 6 | 5 | CH3CN | 3 | 68 |
| 7 | 10 | CH3CN | 1 | 92 |
| 8 | 15 | CH3CN | 2 | 91 |
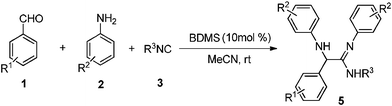
It is to be noted that no product was formed in the absence of the catalyst. From this observation, it is clear that the catalyst plays an important role in the formation of the product, and 10 mol% of BDMS was sufficient to carry out the reaction, in terms of time and yield effectiveness. Similarly, the role of the solvent in this reaction was studied by performing the same reaction in various solvents such as dichloromethane, acetonitrile and methanol. It is worth mentioning that acetonitrile is the most suitable solvent for the synthesis. Moreover, the isolated product of the representative compound 5a was characterized based on its 1H NMR spectrum which shows a peak at 1.46 as a singlet for the tertiary butyl proton, 4.92 as a singlet for the –CH proton, two broad peaks at 3.71 and 5.90 for the –NH proton and the aromatic proton which appears in the region of 6.47–7.30 ppm. Subsequently, the carbon signal of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the 13C NMR spectrum appears at 154 ppm. In addition, the structure of α-amino amidine 5a was determined by single-crystal X-ray crystallographic data which revealed that 5a crystallized in P21/c. The crystal structure shows that the N attached to the alkyl group is protonated (N3–H3A = 0.86 Å and C7–N3 = 1.35 Å) and thus the position of the CN (C7–N1 = 1.27 Å) was also determined by single-crystal X-ray data as shown in Fig. 2.
N in the 13C NMR spectrum appears at 154 ppm. In addition, the structure of α-amino amidine 5a was determined by single-crystal X-ray crystallographic data which revealed that 5a crystallized in P21/c. The crystal structure shows that the N attached to the alkyl group is protonated (N3–H3A = 0.86 Å and C7–N3 = 1.35 Å) and thus the position of the CN (C7–N1 = 1.27 Å) was also determined by single-crystal X-ray data as shown in Fig. 2.
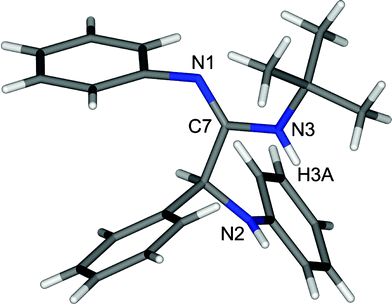 | ||
| Fig. 2 Crystal structure of 5a (CCDC no. 855248). | ||
After optimization of the reaction conditions, a variety of aromatic aldehydes containing electron donating and electron withdrawing groups in the aromatic ring were reacted with various substituted aromatic amines and isocyanides. In all cases, the corresponding α-amino amidine derivatives were obtained in good yields under similar reaction conditions, which are summarized in Table 2. From these successful results, we may conclude that aniline has a shorter reaction time as compared to chloroaniline and provides a better yield (Table 2, entries 13 and 15). Similarly, chloroaniline requires a shorter reaction time than bromoaniline and gives a better yield (Table 2, entries 2 & 3, 9 & 11). It is also noted that 2-morphilinoethyl isocyanide provides a smaller yield (Table 2, entry 17), due to lower reactivity of the isocyanide as compared to other isocyanides. All the products were characterized through 1H-NMR, 13C-NMR and elemental analysis. To compare the effectiveness of BDMS over HBr, we have examined the reaction of benzaldehyde (1a), aniline (2a) and tert-butyl isocyanide (3a) with an equivalent amount of 48% aqueous HBr at room temperature. It was noted that the experimental conditions not only provided a low yield (40%) of product 5a, but also required a longer reaction time (4 h).
| Entry | Aldehyde | Amine | Isocyanide | Product | Time/h | Yield (%)b |
|---|---|---|---|---|---|---|
| a All the reactions were performed using aldehyde (1 mmol) in MeCN (5 mL) at room temperature with amine (2 mmol), isocyanide (1 mmol) and BDMS (10 mol %). b Isolated yield. | ||||||
| 1 | C6H5 | 2a | 3a | 5a | 1 | 92 |
| 2 | 4-BrC6H4 | 2b | 3a | 5b | 1 | 85 |
| 3 | 4-BrC6H4 | 2c | 3a | 5c | 2.5 | 77 |
| 4 | 4-ClC6H4 | 2b | 3a | 5d | 0.5 | 93 |
| 5 | 4-FC6H4 | 2b | 3a | 5e | 1.5 | 89 |
| 6 | 4-NO2C6H4 | 2c | 3a | 5f | 2.5 | 76 |
| 7 | 3,4,5-OMeC6H2 | 2c | 3a | 5g | 3 | 82 |
| 8 | 2-NO2C6H4 | 2c | 3a | 5h | 2 | 75 |
| 9 | 2-ClC6H4 | 2c | 3a | 5i | 3 | 65 |
| 10 | 2,4-OMeC6H3 | 2b | 3a | 5j | 3.5 | 78 |
| 11 | 2-ClC6H4 | 2b | 3a | 5k | 1 | 87 |
| 12 | 3-OHC6H4 | 2b | 3a | 5l | 2 | 82 |
| 13 | C6H5 | 2a | 3b | 5m | 1.5 | 85 |
| 14 | 4-ClC6H4 | 2b | 3b | 5n | 0.5 | 90 |
| 15 | C6H5 | 2b | 3b | 5o | 3.5 | 80 |
| 16 | 4-FC6H4 | 2d | 3b | 5p | 4 | 76 |
| 17 | 4-FC6H4 | 2b | 3c | 5q | 6 | 35 |
The structures of the compounds 5b and 5n were also determined by single-crystal X-ray crystallography for further confirmation. Both the compounds are isostructural to 5a and the N attached to the alkyl group is protonated (C23–N1 = 1.35 Å in 5b and C14–N3 = 1.34 Å in 5n), as shown in Fig. 3a and 3b. Interestingly, 5n forms a 1D zigzag chain, propagated along the b-axis via inter-molecular hydrogen bonding interactions through N–H···N bonds (H···N = 2.24 Å, N···N = 3.074 Å, ∠N–H···N = 164°) as shown in Fig. 3c.
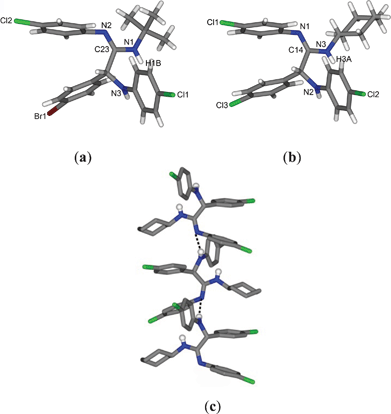 | ||
| Fig. 3 Molecular structures of (a) 5b and (b) 5n; (c) zigzag hydrogen bonded chain of 5n propagated along the b-axis. | ||
The formation of the α-amino amidine can be explained as follows: initially the aromatic aldehyde reacts with the aromatic amine to form an intermediate imine 1a and water. The released water molecule reacts with BDMS to generate HBr, dimethyl sulfide and HOBr in the reaction medium. The in situ generated HBr activates the imine 1a to give the reactive intermediate 1b, which reacts instantaneously with isocyanide to form species 1c. Subsequently, another molecule of amine attacks the intermediate 1c to give α-amino amidine 4 which undergoes further [1,3] hydrogen shift to provide the α-amino amidines 5 as shown in Scheme 2. Mechanistically, the structure of the reaction products may also be able to exist as isomeric α-amino amidine derivative 4 if we use a secondary amine. In addition, HBr also reacts with aniline to form anilinium hydrobromide, which also assists the formation of the product observed by McFarland.9
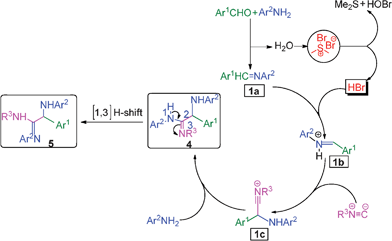 | ||
| Scheme 2 Plausible mechanism for the formation of the α-amino amidine. | ||
Conclusion
In summary, we have demonstrated a new synthetic protocol for the synthesis of α-amino amidines by the one-pot three-component reaction of an aromatic aldehyde, aromatic amine and isocyanide, by employing an inexpensive and eco-friendly catalyst, BDMS, which has not previously been reported. The significant features of this protocol are: easy reaction procedure, simple separation and purification process, followed by compatibility with a wide variety of substrates under mild reaction conditions. In addition, the present protocol is superior in terms of yield, reaction time and conditions, and environmentally benign as compared to the earlier methods. Nevertheless, we have established that the structure of the α-amino amidine obtained from the reaction of aromatic aldehyde, isocyanide and aromatic amine exists as a different structure from the reaction of the secondary amine, which has not been reported previously.General procedure for the synthesis of α-amino amidine
Into a 25 mL round bottomed flask was placed a mixture of aromatic amine (2 mmol), aromatic aldehyde (1 mmol) and isocyanide (1 mmol) in 5 mL acetonitrile at room temperature. Then, the catalyst bromodimethylsulfonium bromide (0.022 g, 0.1 mmol) was added into the above reaction mixture and stirring was continued. After completion of the reaction, as checked by TLC, the solvent was removed in a rotary evaporator. Then the crude residue was treated with 5 mL methanol and the crystalline product was separated out and filtered through a Büchner funnel. The solid product was washed with cold methanol followed by diethyl ether and finally it was dried under reduced pressure.Acknowledgements
Sidick Basha R and Mohan Lal are thankful to CSIR, New Delhi, India for their research fellowships. We also acknowledge Department of Science & Technology, New Delhi for financial support for creating a single crystal XRD facility in the Department of Chemistry under the FIST programme. The authors are grateful to the Director, IIT Guwahati for providing general laboratory facilities. We are grateful to the referees for their valuable comments and constructive suggestions on our manuscript. This paper is dedicated to my mentor, Professor Dr. R. R. Schmidt, Universitaet Konstanz, Germany on the occasion of his 77th birthday.References
- (a) J. Zhu, H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, Germany, 1st edn, 2005 Search PubMed; (b) B. U. W. Maes, R. V. A. Orru, E. Ruijter, Synthesis of Heterocycles via Multicomponent Reaction I & II, Springer, New York, 2010 Search PubMed.
- (a) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS; (b) P. A. Wender, S. T. Handy and D. L. Wright, Chem. Ind., 1997, 765 CAS.
- (a) J. B. Harborne, H. Baxter and G. P. Moss, Phytochemical Dictionary, Taylor & Francis, London, 1999, pp. 91–94 Search PubMed; (b) A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. D. Brosse, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Pottst, J. Org. Chem., 1995, 60, 1182 CrossRef CAS; (c) L. Heys, C. G. Moore and P. J. Murphy, Chem. Soc. Rev., 2000, 29, 57 RSC; (d) S. Patai, The Chemistry of Amidines and Imidates, John Wiley and Sons, New York, 1991, 2 Search PubMed.
- H. Umezuma, T. Takita, Y. sugiura, M. Otsuka, S. Kobayashi and M. Ohno, Tetrahedron, 1984, 40, 501 CrossRef.
- (a) T. Yamada, K. Suegane, S. Kuwata and H. Watanabe, Bull. Chem. Soc. Jpn., 1977, 50, 1088 CrossRef CAS; (b) T. Yamada, K. Suegane, S. Miyazawa, S. Kuwata and H. Watanabe, Bull. Chem. Soc. Jpn., 1978, 51, 878 CrossRef CAS.
- H. P. Kaufmann, J. Budwig and K. Mohnke, Chem. Ber., 1942, 75, 1585 CrossRef.
- T. Goto, M. Isobe, D.A. Coviello and Y. Kishi, Tetrahedron, 1973, 29, 2035 CrossRef CAS.
- M. Mengelberg, Chem. Ber., 1956, 89, 1185 CrossRef CAS.
- J. W. McFarland, J. Org. Chem., 1963, 28, 2179 CrossRef CAS.
- J. Barluenga, F. Aznar and R. Liz, J. Chem. Soc., Chem. Commun., 1981, 1181 RSC.
- (a) T. Ueda, Y. Okamoto, T. Tsuji and M. Muraoka, Chem. Pharm. Bull., 1968, 16, 2355 CrossRef CAS; (b) A. Pinner, Die Iminoather und ihre Derivate, R. Oppen-heim, Berlin, 1892 Search PubMed; (c) R. S. Garigipati, Tetrahedron Lett., 1990, 31, 1969 CrossRef CAS; (d) R. A. Moss, W. Ma, D. C. Merrer and S. Xue, Tetrahedron Lett., 1995, 36, 8761 CrossRef CAS.
- W. Keung, F. Bakir, A. P. Patron, D. Rogers, C. D. Priest and V. Darmohusodo, Tetrahedron Lett., 2004, 45, 733 CrossRef CAS.
- (a) A. T. Khan, M. Lal and M. M. Khan, Tetrahedron Lett., 2010, 51, 4419 CrossRef CAS; (b) A. T. Khan, D. K. Das and M. M. Khan, Tetrahedron Lett., 2011, 52, 4539 CrossRef CAS; (c) A. T. Khan, M. Lal, S. Ali and M. M. Khan, Tetrahedron Lett., 2011, 52, 5327 CrossRef CAS.
- (a) L. H. Choudhury, T. Parvin, A. T. Khan, Tetrahedron, 2009, 65, 9513 and references cited therein Search PubMed; (b) A. T. Khan, S. Ali, A. A. Dar and M. Lal, Tetrahedron Lett., 2011, 52, 5157 CrossRef CAS; (c) A. T. Khan, R. S. Basha and M. Lal, Tetrahedron Lett., 2012, 53, 2211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H,13C-NMR spectra and crystallographic data in CIF. Complete crystallographic data of 5a, 5b and 5n for the structural analysis with CCDC reference numbers 855248, 838468 and 837583, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20539d |
| This journal is © The Royal Society of Chemistry 2012 |