Atomic layer deposition of lithium nitride and carbonate using lithium silylamide
Erik
Østreng
*,
Ponniah
Vajeeston
,
Ola
Nilsen
and
Helmer
Fjellvåg
Centre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, P.O. Box 1033 Blindern, N-0315 Oslo, Norway. E-mail: erik.ostreng@smn.uio.no; Fax: +47 2285 5565; Tel: +47 2285 5558
First published on 14th June 2012
Abstract
Lithium silylamide, LiN(SiMe3)2, has been explored as precursor for the successful deposition of thin films of lithium nitride, Li3N, and of lithium carbonate, Li2CO3, by atomic layer deposition. Deposition of Li2CO3 has been used as a tool in the method development as the compound is stable in air, contrary to Li3N. Self limiting growth was demonstrated for both Li3N and Li2CO3. The crystalline state of Li3N depends on the deposition conditions, and varies from amorphous to a phase mixture of α-Li3N and β-Li3N. The growth rate of Li3N is 0.95 Å cycle−1. The Li2CO3 is well crystalline and highly oriented with (002) parallel to the substrate as deposited, and has, according to XPS, a low content of silicon at deposition temperatures between 89 and 332 °C. The growth rate of Li2CO3 is 0.35 Å cycle−1. A geometrical model has been applied to rationalise the observed growth rates. This is the first example of deposition of nitrides using silylamides, and the first route towards lithium nitride by ALD.
Introduction
Lithium-containing functional materials have a wide range of applications. Lithium enters into ferroelectric materials such as in LiNbO3 and as a dopant in ZnO. However, the most intense field of current developments relates to lithium-ion batteries. Electrode and electrolyte materials containing lithium span several material classes; phosphates like LiFePO4 and “LiPON”, carbonates Li2CO3, oxides LiCoO2 and (Li1 − xLax)TiO3, and nitrides like Li3N and Li1 − xFexN.1–6 The present work on thin films focuses on two such classes; deposition of carbonates, which typically occur in SEI (solid electrolyte interface) layers, and depositions of nitrides as electrolyte or electrode materials. Lithium nitride (Li3N) has further been investigated in relation to imide-/amide-based hydrogen storage materials7 and is a very good lithium ion conductor,2 however, its breakdown voltage is too low for practical use in batteries in its pure state.8The growth of lithium containing thin films by ALD was demonstrated by Putkonen et al.,9 followed by growth of ion conducting lithium lanthanum titanate and lithium aluminium oxide by Aaltonen.6,10 Recently, deposition of lithium silicate has been reported by Hämäläinen et al.11 Lithium forms monovalent cations, and as pointed out in Ref. 9 this gives rise to challenges with respect to achieving self-limiting growth as required for ALD-processes.
The currently explored precursor, LiN(SiMe3)2, adopts a trimeric structure12 in the solid state, however, dimers exist in the gas phase according to gas electron diffraction.13 The hygroscopic nature of Li2O and its tendency to react with CO2, represents a major challenge and makes deposition of pure Li2O virtually impossible from a lithium precursor and water or ozone. The range of precursors investigated for lithium ALD is at this moment still limited. The need to develop novel material chemistries for use in improved lithium-ion batteries, calls for efforts to investigate new precursor systems.
Several nitrides of different metallic elements have previously been deposited by ALD using different classes of precursors. Examples are TiN, TaN, MoN and NbN which are deposited from halides,14 whereas organometallic and metal–organic precursors like alkyls, amines and amides are reported for deposition of AlN,15 TiN,16 TaN,17 Hf3N4,18 Zr3N418 and Mo2N.19
The current precursor, lithium silylamide, is an amide where the functional groups are silyl groups and the carbon is replaced by a metal, in this case lithium. Silylamide complexes are well known as reagents in organic and metal–organic chemistry. Such molecules have already been used in ALD, for example homoleptic silylamides of bismuth,20 lanthanum21 and praseodymium22 have been used for growth of BiOx, Sr–Bi–Ta–O and Bi4Ti3O12, La2O3 and LaAlO3, and PrOx respectively. Bi[N(SiMe)3]3 was used at 190 °C, but the deposition of binary BiOx was not sufficiently reproducible. La[N(SiMe3)2]3 and H2O has been used to deposit La2O3 in the range 150 to 250 °C, with the incorporation of 3.5 to 8 at% of silicon. Also the Pr[N(SiMe3)2]3 precursor results in 4–12 at% silicon impurities in the temperature range 200–300 °C. These studies also report high contents of hydrogen, which decrease with increasing deposition temperature.
Heteroleptic silylamides like dichlorobis[(bis(trimethylsilyl)] amido hafnium and zirconium have been studied for the deposition of hafnium silicate23,24 and zirconium oxide25 in the temperature ranges 150–400 °C and 150–350 °C, respectively. Also these studies report silicon impurities that increase in amount with increasing deposition temperature. Nam et al. reports silicon contents as high as 30 at% when reacting HfCl2[N(SiMe3)2]2 with water.23 Common for all these prior studies is a high growth rate for the silylamides that decreases with temperature. To our knowledge, no prior studies have focused on the deposition of nitrides using silylamides.
In the present contribution we demonstrate the deposition of Li3N and Li2CO3 from the silylamide, LiN(SiMe3)2. This represents furthermore the first deposition of lithium nitride by ALD and is the first example of the application of silylamide precursors for nitride growth with ALD. In this respect, the work may open up new possibilities within ALD precursor chemistry, both generally for the deposition of nitrides and for the nitrogen doping of lithium compounds such as LiPON.
As Li3N is air and moisture sensitive we use Li2CO3 as a model system to study the chemistry of this precursor and determine the temperature window and decomposition temperature of the precursor. We will therefore first describe the growth of Li2CO3 before elaborating on the growth of Li3N.
Experimental
Thin films were deposited in a F-120 Sat ALD-reactor (ASM Microchemistry Ltd.) using LiN(SiMe3)2 (Aldrich 98%) Mo(CO)6 (Aldrich 98%), deionised H2O, CO2 (95%, AGA) and NH3 (Linde, anhydrous 99.999%) as precursors. LiN(SiMe3)2 was sublimated inside the reactor at 75 °C.Nitrogen was used as a carrier gas in all experiments and supplied at 500 cm3 min−1. Nitrogen was generated with a Schmidlin UHPN3001 N2 purifier that provides better than 99.999% N2 + Ar in the carrier gas. The carrier gas was further dried by P2O5 and purified for remains of O2 by a Mykrolis gas purifier.
Thin films were deposited on soda-lime glass, polished titanium plates and 2.5 × 2.5 cm2 single crystal Si(100) wafers polished for epitaxial growth, with native oxide. The substrate is considered to have insignificant roughness compared to the samples deposited. The soda-lime glass substrates were cleaned with ethanol. The single crystals were blown dry with pressurized air, and otherwise used as supplied.
Spectroscopic ellipsometry data were collected with a Woollam Alpha SE ellipsometer between 380 and 900 nm and analyzed by fitting a Cauchy model to the whole dataset. Ellipsometry was used to determine the refractive index and thickness of all Li2CO3 samples with an estimated uncertainty of below 1%.
Characterization by X-ray diffraction (XRD) in θ-2θ-mode was performed with a Bruker AXS D8 powder diffractometer equipped with a Ge(111) monochromator providing Cu-Kα1 radiation and using a LynxEye detector. X-ray reflectometry (XRR) and ω-scans was performed using a Bruker AXS D8 diffractometer with a thin film stage and an asymmetric double bounce Ge(220) monochromator and 0.2 mm slits to provide Cu-Kα1 radiation. The XRR-data were fitted using the GENX software package. The total uncertainty of XRR is estimated to be less than 1%.
Surface topography was studied for selected samples with atomic force microscopy (AFM) using a Park Instruments XE-70 and analyzed using the XEI software.
XPS spectra were collected with a Kratos Axis UltraDLD instrument using monochromatic Al Kα X-ray radiation. The resolution was 0.54 eV as determined by the full width at half maximum of the Ag 3d5/2 peak. Low energy electrons were used to compensate for surface charging on the Li2CO3 reference sample. Energy referencing is based on the C 1s peak of adventitious carbon set to 285.0 eV binding energy (BE). Peak fitting was performed using Voight functions after subtraction of a Shirley type background in CASA XPS. Instrument manufacturer's sensitivity factors were employed for quantification.
TGA experiments were performed using a Perkin Elmer TGA 7 at a heating rate of 2 °C min−1 in N2. Thermal decomposition studies of the precursor were done using the equipment described by Nilsen et al.26 The precursor was heated inside 8 mm sealed quartz tubes at 115 °C for five days.
Raman spectroscopy was preformed with a Spectra-Physics Millennia Pro 12sJS Nd:YVO4 solid state laser using 532 nm wavelength operating at 200 mW.
Computational details
The first-principles calculation was performed based on the density functional theory and the pseudopotential methods, which were implemented in the CASTEP code.27 Ultrasoft pseudopotentials were employed to describe the electron-ion interactions, and the plane-wave cut-off energy was 600 eV. The exchange and correlation terms were described with generalized gradient approximations in the scheme of Perdew–Burke–Ernzerhof.28 The geometric optimization of the unit cell was carried out with the BFGS minimization algorithm provided in this code. For each phase, the lattice parameters and atomic positions were fully optimized. The k-points were generated using the Monkhorst–Pack method with a grid size of 10 × 10 × 4 and 10 × 10 × 8 for the α- and β-phase respectively, structural optimization. Iterative relaxation of atomic positions was stopped when the change in total energy between successive steps was less than 1 meV cell−1. With this criterion, the forces generally acting on the atoms were found to be less than 10−3 eV Å−1.Density functional perturbation theory (DFPT)29 was used for the Raman calculations. For the Raman calculation we have used norm-conserving pseudopotentials with 850 eV energy cut-off for all atoms together with a 15 × 15 × 12, mesh of k points, with the energy conversion threshold of 0.01 meV atom−1, maximum displacement of 0.001 Å and maximum force of 0.03 eV Å−1, yielding a high accuracy for the energy and atomic displacements. For Li and N atoms the valence states were modelled using the 2s1 and 2s2, 2p3 electrons, respectively. In general, in most of the cases the calculated and observed Raman data vary within 5%.30,31
Results
The LiN(SiMe3)2 compound, being orange to yellow-brown and sticky, reacts slowly with air and moisture. The compound must nevertheless be stored and handled under inert conditions to assure sufficient reproducibility of experiments. Fresh precursor was therefore transferred directly from a glovebox prior to each experiment.The precursor was studied by TGA in order to determine a suitable sublimation temperature of 75 °C, at which the residue was about 1 wt%. The precursor decomposition test resulted in a white ring of decomposed precursor in the quartz tube at 375 °C suggesting a potential maximum in the ALD-window around 375 °C, which is consistent with the prior art.21,22
Reactions of LiN(SiMe3)2 with H2O
The initial experiments using alternating pulses of LiN(SiMe3)2 and H2O resulted in films with rather uncontrolled growth and large gradients. These films were in addition highly reactive towards air which made characterization extremely difficult. The uncontrolled growth is likely caused by a reservoir effect where the bulk of the film absorbs water providing a large supply of hydroxide groups in addition to the self limiting surface reaction. Any LiOH thereby formed in the bulk of the film may react with the subsequent pulses of LiN(SiMe3)2 leading to a growth rather limited by the thickness of the deposited film. This finding is also supported by earlier results of Aaltonen.10 In order to circumvent the problem, a pulse of CO2 was introduced after each water pulse in order to form the stable and non-hygroscopic Li2CO3. This allowed studies of the temperature window and pulse parameters for the selected precursor in a simpler way than directly studying the Li3N.The pulse and purge parameters were optimized at a deposition temperature of 186 °C, as shown in Fig. 1, resulting in an LiN(SiMe3)2 pulse of 4 s and an H2O pulse of 0.25 s in order to obtain surface limited growth.
 | ||
| Fig. 1 Pulse parameters for LiN(SiMe3)2, H2O and CO2versus growth per cycle (GPC) as measured by ellipsometry for films deposited using 2000 cycles at 186 °C. The other pulse and purge parameters were kept constant during the screening are: 4 s Li—1 s purge—1 s H2O—1 s purge—7.5 s CO2—1 s purge. Non-uniformities were measured to be below 8%. | ||
It was found necessary to use 7.5 s CO2 pulses to provide samples that were stable in air. Samples made using 2.5 and 5 s CO2 pulses turned milky white over the course of a few days, probably due to the reaction of unreacted Li2O or LiOH in the film with ambient CO2.
The suggested wide ALD window from the thermal decomposition experiments, see above, was indeed observed experimentally in Fig. 2. Samples were deposited using 2000 cycles of a 4 s Li pulse, a 0.5 s water pulse and a 7.5 s CO2 pulse at temperatures between 89 and 429 °C. Visually uniform films with low silicon contents were achieved up to 380 °C, however non-uniformity was measured as high as 31% at 89 °C, but was below 8% in most cases. Fig. 1 and 2 show the average thickness over a 10 cm length and the error bar is the non-uniformity. At 429 °C the resulting film was black, showed gradients and the extraction of growth rate data was cumbersome. XPS analyses were undertaken in order to clarify the impurity level of silicon as a function of deposition temperature. 70 nm thick films of Li2CO3 were obtained on polished titanium substrates after 2000 deposition cycles, Fig. 3. Previous works indicate that thermal decomposition of the precursor at higher temperatures will result in increased silicon content.21,22 The present analysis shows that the films deposited at temperatures up to 380 °C contain between 0.04 and 0.65 at% silicon impurities with no clear pattern in the variation with deposition temperature. However, the sample made at 428 °C contains as much as 5.8 at% silicon. The carbon and oxygen levels in samples deposited between 89 and 380 °C are consistent with the lithium carbonate powder used as reference. The good correspondence of the lithium and oxygen contents between the reference and the deposited films indicates a low hydrogen content in the film. As the main possibility of hydrogen incorporation will be as hydroxide or bicarbonate the incorporation of hydrogen in the film will then be accompanied by an increase in oxygen relative to lithium. The formation of Li2CO3 films is further supported by the density as measured by XRR (not shown) of 2.01 g cm−3 compared to a theoretical density32 of 2.10 g cm−3. It was not possible to extract the thickness of the deposited films from the XRR due to the high roughness of the samples.
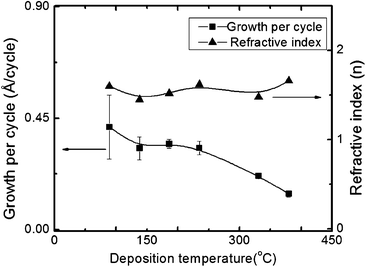 | ||
| Fig. 2 Ellipsometric measurements of the growth rate (GPC) and index of refraction as a function of deposition temperature using pulse parameters of a 4 s LiN(SiMe3)2 pulse, a 0.5 s H2O pulse and a 7.5 s CO2 pulse, and 1 s purge. The error bars represent the variation over the length of the chamber, ca. 10 cm. | ||
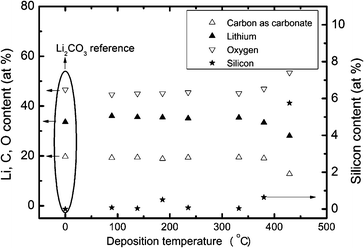 | ||
| Fig. 3 Analyzed composition by XPS versus deposition temperature for films deposited on titanium substrates after 2000 cycles using pulse parameters of a 4 s LiN(SiMe3)2 pulse, a 0.5 s H2O pulse and a 7.5 s CO2 pulse, and 1 s purge. | ||
The crystallinity of the films deposited during 2000 cycles on Si(100) at different temperatures was characterized with XRD, Fig. 4. As expected, the crystallinity increases with increasing deposition temperature. However, surprisingly the samples deposited at 380 °C turned out be X-ray amorphous, but no apparent explanation could be found. All crystalline samples show only the (002) of the Li2CO3 phase (zabuyelite) proving that this process yields an oriented film on silicon. Rocking curve measurements (ω-scan) of (002) for the sample deposited at 332 °C using 2000 cycles show a FWHM (full width at half maximum) of 2.05°, supporting the claim of a highly oriented growth.
 | ||
| Fig. 4 XRD data versus deposition temperatures for films deposited using 2000 cycles and pulse parameters of a 4 s LiN(SiMe3)2 pulse, a 0.5 s H2O pulse and a 7.5 s CO2 pulse, and 1 s purge. The sharp peak at 33° is from the silicon substrate whereas the peak at 31.65° is identified as (002) from Li2CO3, zabuyelite; the bars in the lower panel indicate the expected diffraction from a (randomized) powder sample. | ||
Analysis of the topography by AFM shows that the morphology varies notably with deposition temperature, Fig. 5. The sample deposited at 138 °C is X-ray amorphous even though AFM analysis shows structures that resemble crystallites with dimensions of some 100 nm. At 331 °C larger grains are formed with sizes up to 1 μm.
 | ||
| Fig. 5 AFM images of Li2CO3 samples deposited on silicon(100) at (left) 138 °C and (right) 331 °C using 2000 cycles. The RMS roughness is measured to 12 and 48 nm respectively. The pulse and purge parameters are: 4 s Li—1 s purge—1 s H2O—1s purge—7.5 s CO2—1 s purge. | ||
Depositions of Li–N
Deposition of Li3N was achieved on soda lime glass, silicon and titanium substrates by alternating pulses of LiN(SiMe3)2 and NH3. The deposition was facilitated by first depositing a 5 nm adhesion layer of MoNx prior to the Li–N growth. The final Li3N product was capped with a 20 nm layer of MoNx in order to prevent reactions with the ambient air and to allow ex-situ characterization. Initial studies proved that the Li3N films were by far more reactive to ambient air than lithium oxide films. This sandwiching of the Li3N film between two films of a denser material also amplifies its signal in the XRR analysis, as the reflectance comes from the difference in electronic densities between the layers, thus making the analysis easier. Depositions without the adhesion layer often resulted in powder-like depositions rather than a homogeneous film, regardless of the pulse and purge parameters; however successful depositions were carried out on several different substrates without an adhesion layer. With an adhesion layer, a continuous layer was always formed.The pulse parameters were optimized at 167 °C using 200 cycles of a 5 s LiN(SiMe3)2 pulse and a 5 s NH3 pulse, modifying one parameter at a time (see Fig. 6 and 7). The ammonia pulse could be as low as 1 s, however, then yielding products with reduced uniformity. For a 2.5 s ammonia pulse, the uniformity was optimal throughout the whole reactor chamber. For longer ammonia pulses, a decrease in density was observed. A satisfactory explanation is still lacking. The shorter LiN(SiMe3)2 pulse required for growth of Li3N suggests that the reaction between LiN(SiMe3)2 and the carbonate surface is quite slow compared to the reaction between LiN(SiMe3)2 and an –NHx-terminated surface, as the pulse required for self limited growth of Li3N can be as short as 1 s.
 | ||
| Fig. 6 Growth rate and density of Li3N measured by XRR as function of LiN(SiMe3)2 pulse length. Samples are deposited at 167 °C using 200 cycles and a 5 s NH3 pulse and 1 s purge. | ||
 | ||
| Fig. 7 Growth rate and density of Li3N measured by XRR as function of NH3 pulse length. Samples are deposited at 167 °C using 200 cycles and a 5 s LiN(SiMe3)2 pulse and 1 s purge. | ||
When depositing LiN(SiMe3)2 and NH3 using 100, 200 and 500 cycles, a linear relationship between growth rate and thickness of the Li–N layers were found, see Fig. 8. Linear regression gives a growth rate of 0.95 Å cycle−1.
 | ||
| Fig. 8 Thickness as function of number of cycles for Li3N, deposited using 5 s pulses of LiN(SiMe3)2 and of NH3 at 167 °C. Solid line corresponds to a growth rate of 0.95 Å cycle−1 according to linear regression. | ||
Samples deposited at 167 °C were amorphous as deposited. In order to obtain crystalline films, 95 nm thick films were annealed at 300 or 600 °C for 1 min. However, these samples turned milky already after a few minutes exposure to air after the annealing, probably due to deterioration of the capping layer during annealing. On the other hand, crystalline samples were obtained by deposition of 1000 cycles of LiN(SiMe3)2 and NH3 at 332 °C. X-ray diffraction shows the (110), (111) and (102) reflections of α-Li3N, in addition to a few weak reflections attributed to β-Li3N, see Fig. 9. The XRD-analysis also shows Li2CO3 and LiOH, however, these are believed to result from post deposition reactions with ambient air. The refined unit cell dimensions of hexagonal α-Li3N are a = 3.696 Å and c = 3.895 Å, in good agreement with literature.33
 | ||
Fig. 9 XRD pattern of a sample deposited with 1000 cycles at 332 °C, using a 5 s LiN(SiMe3)2 pulse, a 5 s NH3 pulse and 1 s purges. The pattern contains reflections identified as α-Li3N ( ), β-Li3N ( ), β-Li3N ( ), LiOH (•), and Li2CO3 (+). The inset shows the (222) and (204) reflections from α-Li3N. ), LiOH (•), and Li2CO3 (+). The inset shows the (222) and (204) reflections from α-Li3N. | ||
Raman spectroscopy was used as an additional tool for proving the chemical state of the amorphous samples of lithium nitride, Fig. 10. For amorphous samples, the Raman spectra confirmed the presence of both α- and β-Li3N, and the peak at ca. 760 cm−1 is attributed to a LiSi2N3 phase, however this cannot be confirmed by XRD. The theoretically calculated modes are all within a Raman shift error of ±5%, in line with expectations as described in the experimental section. The relative intensities of the calculated modes concur well with observations.
 | ||
| Fig. 10 Raman spectra recorded for 100 nm thick, X-ray amorphous sample deposited on SiO2 at 167 °C using 1000 cycles of a 5 s LiN(SiMe3)2 pulse, a 5 s NH3 pulse and 1 s purges. Black line is measured data, red, blue and pink lines are theoretically modelled spectra of α-Li3N and β-Li3N, and LiN3Si2 respectively. | ||
The silicon content of a sample of Li3N deposited at 168 °C on MoNx-buffered titanium was analyzed with XPS. The sample was deposited without a capping layer and allowed to oxidize before the analysis to avoid effects from sputtering through the capping layer. The silicon content was found to be ca. 6 atomic percent when assuming a film composition of Li3N.
Discussion
The differences in growth rate of Li2CO3 as a function of temperature, as well the difference in growth rate between Li2CO3 and Li3N, are considered in view of models by Ylilammi.34 Obviously, the surface chemistry is rather different between these cases. The carbonate surface is quite unreactive while the |–NHx surface is more reactive towards LiN(SiMe3)2 due to its ability to react with protons at the surface. To explore the surface chemistry we calculate the theoretical growth rate as: | (1) |
We here assume that the precursor ligand (–N(SiMe3)2) can be approximated as a sphere with 6 Å diameter in correspondence with the report by Fjeldberg,13 and that the reactive site is at the centre of the anion. In the case of Li2CO3 growth, we assume that the precursor is physisorbed on the surface with both ligands intact and that the Li2CO3 film grows along the 001 direction leaving a rough surface. This indicates that the growth is limited by surfaces at some angle to this growth direction, having growth rates notably lower than the 001 direction. We are unable to identify the specific terminating surfaces, and limit our theoretical considerations to the 001 surface which should represent the upper limit of what is observed along this direction.
We identify two possibilities regarding the arrangement of precursor molecules on the surface, being either free to rotate and spanning a circular area (case I), or being constrained to an area that is better represented by an ellipse (II). In the case of Li3N growth we assume that the densely packed (111)-surface is exposed and furthermore that the precursor loses one ligand and can be represented by a 6 Å diameter sphere, and bonds to either every second (III) or every third nitrogen atom on the surface (IV), see Fig. 11. The calculated surface densities and growth rates resulting from these considerations are given in Table 1
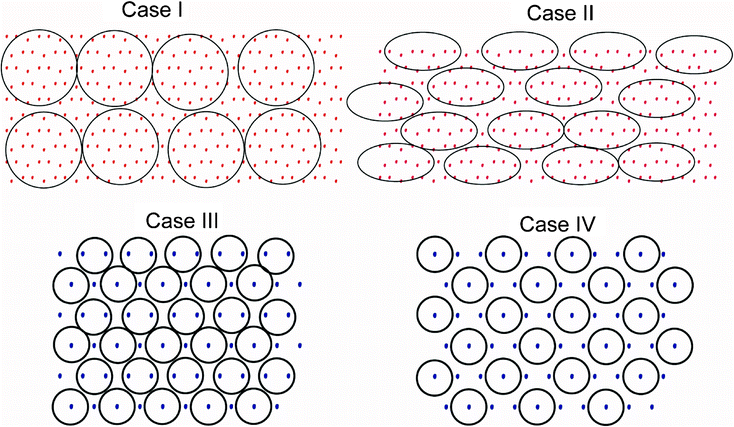 | ||
| Fig. 11 Schematic illustrations of cases I–IV, only the film anions are drawn, see text. Red dots symbolize oxygen atoms, blue dots nitrogen and the black circle outlines the adsorbed precursor on the surface. | ||
| Case | Material | Temp. (°C) | n p | n UC | GPC (Å cycle−1) |
|---|---|---|---|---|---|
| I | Li2CO3 | — | 0.16 | 0.04 | 0.23 |
| II | Li2CO3 | — | 0.29 | 0.073 | 0.42 |
| III | Li3N | — | 0.5 | 0.33 | 1.1 |
| IV | Li3N | — | 0.33 | 0.22 | 0.73 |
| Exp | Li2CO3 | 331 °C | 0.23 | ||
| Exp | Li2CO3 | 89 °C | 0.41 | ||
| Exp | Li3N | 167 °C | 1.1 |
By comparing calculated and experimental growth rates in Table 1, Case I corresponds well with the Li2CO3 growth rate at 331 °C, Case II corresponds quite well with the Li2CO3 growth rate at 89 °C. The difference in growth rates between these temperatures may indicate that elevated temperature provides enough thermal energy to allow the precursor molecules to rotate freely, while they are locked in at lower temperatures. Case III corresponds well with the experimental growth rate of Li3N at 167 °C. This may indicate that the reason for the high growth rate of Li3N is caused by the loss of one ligand of the precursor when reacting with the surface. The reduction in growth rate of Li2CO3 at high temperatures is probably due to rotation of ligands, see Fig. 2.
The importance of a buffer layer underneath Li3N was unexpected. In our case, layers of Mo-nitride turned out to be successful. Previously, it is known that noble metals benefit from similar adhesion layers, both metals35 and Al2O336 can act as adhesion or nucleation layers. Aaltonen et al.35 reports that platinum films do not grow at low temperature unless there is a nucleation layer. It may be that the very reactivity of the Li3N requires a barrier layer to prevent reactions with the substrate. On the other hand, it may be equally likely that the chemistry for depositing Li3N requires a nitride surface in order to form a metal-nitrogen-metal bond. A full explanation requires experiments beyond the current data and will be highly relevant for the deposition of nitrides from silylamide complexes in general.
There are few examples of deposition of orientated carbonates using ALD in the literature. One such examples is given by Nilsen et al.37 who deposited CaCO3 by using Ca(thd)2, ozone and CO2. The calcite deposited was oriented in either with the (104) plane normal to the growth direction at 350 °C or with the (006) plane normal to the growth direction at 250 °C. For the latter orientation, the CO32−-groups are arranged parallel to the growth direction. In our experiments, which show a strong (001) orientation, the CO32− groups are also arranged in a plane parallel to growth direction.
Silylamides are well known precursors in atomic layer deposition of oxides and silicates. They have, however, not previously been used as precursors in the growth of nitrides by ALD. We have here proven deposition of Li3N and anticipate that the process can be extended to nitrides more generally. It must be a necessary, but not sufficient criterion, for thermal ALD of any compound that the ligand of the cation precursor is more basic than the anion precursor and thus able to strip the anion precursor of hydrogen. For deposition of nitrides by ALD using ammonia as nitrogen source, the ligand should therefore be more basic than ammonia and thus enable the ligand to strip ammonia of hydrogen. Considering that LiN(SiMe3)2 is a very strong non-nucleophilic base,38,39 silylamides should be well suited as precursors for deposition of other nitrides. Since complexes with sufficient thermal stability are known for both transition metals40 and rare earth metals,41 this may open up a new field within low temperature growth of nitrides by ALD.
Conclusion
The precursor Li-silylamine has been explored and used to deposit Li2CO3 and Li3N by ALD. The potential for deposition of Li2O and LiOH is also considered. Self limiting growth and deposition of amorphous and crystalline samples are proved for Li2CO3 and Li3N. The obtained Li2CO3 is almost free of silicon, crystalline above 235 °C and oriented and grows with the (001) plane normal to the growth direction at temperatures between 235 and 332 °C. The growth rate of Li2CO3 is about 0.35 Å cycle−1. In the case of deposition of Li3N, it proved beneficial to deposit an adhesion layer and a capping layer of MoNx in order to obtain uniform film growth and adhesion to the surface, and to prevent sample oxidation in ambient air. Crystalline samples of Li3N were deposited at 332 °C and contained both α-Li3N and β-Li3N, and some impurities considered as mainly Li2CO3 and LiOH. The presence of the two Li3N-phases is supported by Raman-spectroscopy. The growth rate of Li3N is 0.95 Å cycle−1. The difference in growth rates between Li2CO3 and Li3N, as well temperature dependence of the growth rate, has been discussed in connection to packing and chemistry of the precursor molecules at the surface.Acknowledgements
The authors thank Martin F. Sunding for performing XPS measurement and discussions. Dr Niels Højmark Andersen is thanked for experimental Raman data.The research leading to these results has received funding from the European Union Seventh Framework Programme ([FP7/2007-2013]) under grant agreement n° 227541.
References
- J. B. Goodenough and Y. Kim, Chemistry of Materials, 2009 Search PubMed.
- R. A. Huggins, Electrochim. Acta, 1977, 22, 773–781 CrossRef CAS.
- M. Nishijima, T. Kagohashi, M. Imanishi, Y. Takeda, O. Yamamoto and S. Kondo, Solid State Ionics, 1996, 83, 107–111 CrossRef CAS.
- S. D. Culligan, H. W. Langmi, V. B. Reddy and G. Sean McGrady, Inorg. Chem. Commun., 2010, 13, 540–542 CrossRef CAS.
- A. Yamada, S. Matsumoto and Y. Nakamura, J. Mater. Chem., 2011, 21, 10021–10025 RSC.
- T. Aaltonen, M. Alnes, O. Nilsen, L. Costelle and H. Fjellvag, J. Mater. Chem., 2010, 20, 2877–2881 RSC.
- T. Hao, M. Matsuo, Y. Nakamori and S.-i. Orimo, J. Alloys Compd., 2008, 458, L1–L5 CrossRef CAS.
- V. Thangadurai and W. Weppner, Ionics, 2006, 12, 81–92 CrossRef CAS.
- M. Putkonen, T. Aaltonen, M. Alnes, T. Sajavaara, O. Nilsen and H. Fjellvag, J. Mater. Chem., 2009, 19, 8767–8771 RSC.
- T. Aaltonen, O. Nilsen, A. Magraso and H. Fjellvag, Chem. Mater., 2011, 23, 4669–4675 CrossRef CAS.
- J. Hämäläinen, F. Munnik, T. Hatanpää, J. Holopainen, M. Ritala and M. Leskela, J. Vac. Sci. Technol., A, 2012, 30, 01A106–101-105 Search PubMed.
- D. Mootz, A. Zinnius and B. Boettcher, Angew. Chem., Int. Ed. Engl., 1969, 8, 378–379 CrossRef CAS.
- T. Fjeldberg, M. F. Lappert and A. J. Thorne, J. Mol. Struct., 1984, 125, 265–275 CrossRef CAS.
- L. Hiltunen, M. Leskela, M. Makela, L. Niinisto, E. Nykanen and P. Soininen, Thin Solid Films, 1988, 166, 149–154 CrossRef CAS.
- D. Riihelä, M. Ritala, R. Matero, M. Leskelä, J. Jokinen and P. Haussalo, Chem. Vap. Deposition, 1996, 2, 277–283 CrossRef.
- J. W. Elam, M. Schuisky, J. D. Ferguson and S. M. George, Thin Solid Films, 2003, 436, 145–156 CrossRef CAS.
- Y. Y. Wu, A. Kohn and M. Eizenberg, J. Appl. Phys., 2004, 95, 6167–6174 CrossRef CAS.
- J. S. Becker, E. Kim and R. G. Gordon, Chem. Mater., 2004, 16, 3497–3501 CrossRef CAS.
- V. Miikkulainen, M. Suvanto, T. A. Pakkanen, S. Siitonen, P. Karvinen, M. Kuittinen and H. Kisonen, Surf. Coat. Technol., 2008, 202, 5103–5109 CrossRef CAS.
- M. Vehkamaki, T. Hatanpaa, M. Ritala and M. Leskela, J. Mater. Chem., 2004, 14, 3191–3197 RSC.
- K. Kukli, M. Ritala, V. Pore, M. Leskelä, T. Sajavaara, R. I. Hegde, D. C. Gilmer, P. J. Tobin, A. C. Jones and H. C. Aspinall, Chem. Vap. Deposition, 2006, 12, 158–164 CrossRef CAS.
- K. Kukli, M. Ritala, T. Pilvi, T. Sajavaara, M. Leskelae, A. C. Jones, H. C. Aspinall, D. C. Gilmer and P. J. Tobin, Chem. Mater., 2004, 16, 5162–5168 CrossRef CAS.
- W.-H. Nam and S.-W. Rhee, Electrochem. Solid-State Lett., 2003, 7, C55–C56 CrossRef.
- W.-H. Nam and S.-W. Rhee, Stud. Surf. Sci. Catal., 2006, 159, 373–376 CrossRef CAS.
- W.-H. Nam and S.-W. Rhee, Chem. Vap. Deposition, 2004, 10, 201–205 CrossRef CAS.
- O. Nilsen, H. Fjellvåg and A. Kjekshus, Thermochim. Acta, 2003, 404, 187–192 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, S. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- K. Refsen, S. J. Clark and P. R. Tulip, Phys. Rev. B, 2006, 73, 155144 Search PubMed.
- X. Ke, A. Kuwabara and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 184107–184113 CrossRef.
- P. Vajeeston, P. Ravindran and H. Fjellvåg, J. Phys. Chem. A, 2011, 115, 10708–10719 CrossRef CAS.
- A. Grzechnik, P. Bouvier and L. Farina, J. Solid State Chem., 2003, 173, 13–19 CrossRef CAS.
- H. Schulz and K. H. Thiemann, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1979, 35, 309–314 Search PubMed.
- M. Ylilammi, Thin Solid Films, 1996, 279, 124–130 CrossRef CAS.
- T. Aaltonen, M. Ritala, Y.-L. Tung, Y. Chi, K. Arstila, K. Meinander and M. Leskelae, J. Mater. Res., 2004, 19, 3353–3358 CrossRef CAS.
- J. Hamalainen, T. Hatanpaa, E. Puukilainen, L. Costelle, T. Pilvi, M. Ritala and M. Leskela, J. Mater. Chem., 2010, 20, 7669–7675 RSC.
- O. Nilsen, H. Fjellvåg and A. Kjekshus, Thin Solid Films, 2004, 450, 240–247 CrossRef CAS.
- Y.-H. Liu, Synlett, 2011, 732–733 CrossRef.
- S. Urgaonkar and J. G. Verkade, Adv. Synth. Catal., 2004, 346, 611–616 CrossRef CAS.
- D. C. Bradley, R. G. Copperthwaite, M. W. Extine, W. W. Reichert and M. H. Chisholm, in Inorganic Syntheses, John Wiley & Sons, Inc., 2007, pp112–120 Search PubMed.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 1021–1023 RSC.
| This journal is © The Royal Society of Chemistry 2012 |