A green, mild and efficient one-pot method for the synthesis of sulfonamides from thiols and disulfides in water†
Ahmad
Reza Massah
*ab,
Safura
Sayadi
a and
Sara
Ebrahimi
a
aShahreza Branch, Islamic Azad University, Shahreza, Isfahan 86145-311, Iran
bRazi Chemistry Research Center, Shahreza Branch, Islamic Azad University, Shahreza, Isfahan 86145-311, Iran. E-mail: Massah@iaush.ac.ir
First published on 15th May 2012
Abstract
A general, mild, convenient and environmentally benign method was developed for the synthesis of various N-aryl and N-alkyl sulfonamides in water. Trichloroisocyanuric acid (TCCA) was used for the oxidative chlorination of disulfides and thiols to produce the corresponding sulfonyl chloride, which reacted in situ with different amines in the absence of organic bases, to furnish sulfonamides in good to excellent yields. The isolation of the products involves simple experimental conditions and a product isolation procedure (only filtration) in the absence of organic solvents, which makes this protocol potentially useful in the development of a green strategy for the synthesis of sulfonamides.
Introduction
Sulfonamides are a very important class of compounds in medicinal chemistry as a result of their potent biological activities.1 A large number of pharmaceutically active compounds contain arylsulfonamide motifs, which are employed in the prevention and treatment of bacterial infections, diabetes mellitus, oedema, hypertension and gout.2 Recently, approved drugs with a sulfonamide structure have found widespread use in a number of pharmacological and medicinal applications.3 Recently, several synthetic methods, such as the cross-coupling of sulfonamides with aryl halides4 and alcohols,5 the hydrosulfonamidation of homo allylic alcohols,6 the indium catalyzed synthesis of sulfonamides,7 the amidation and oxidation of methyl sulfinates,8 the conversion of iodides to sulfonamides9 and the reaction of activated sulfonate esters with amines,10 have been focused on the development of novel sulfonamide syntheses. Clearly, these methods are efficient and have proven helpful; however, they suffer from some drawbacks, such as the creation of unwanted side-products, the use of toxic solvents, catalysts and reagents and difficult product separations. In spite of the great number of available methods, the vast majority of sulfonamides are prepared by the reaction of ammonia (a primary amine) or a secondary amine with a sulfonyl chloride in the presence of a base.11 However, the use of sulfonyl chlorides causes severe storage and handling problems as well as significant waste generation.12 In addition, the reactions are usually performed in organic solvents and employ organic amine bases to scavenge the generated hydrogen chloride.13 Furthermore, this approach is limited by the formation of undesired disulfonamides by use of primary amines and the need for harsh reaction conditions for less nucleophilic amines, such as anilines.14 As a consequence, considerable effort has recently been devoted to the development of milder routes to sulfonylchlorides, several of which are based on the oxidative chlorination of sulfur compounds. This protocol employs chlorine gas, which is quite hazardous and requires careful handing.15 These drawbacks make it less desirable for routine use, especially for large-scale syntheses. Recently, numerous methods for the oxidative chlorination have been reported. These include dual-function agents, such as NaOCl16 and NCS17, under an acidic and aqueous medium. Also, a combination of oxidant–chlorinating agents, such as KNO3–TMSCl,18 KNO3–SO2Cl219 H2O2–SOCl2,20 oxone–SOCl2,21 or Br2–Cl3PO,22 have been used and this has allowed for milder reaction conditions; however, they are still too acidic for acid-sensitive functionalities to withstand. In this context, Bonk et al. have used trichloroisocyanuric acid (TCCA) as an oxidant in combination with/BnMe3NCl as a chlorinating agent for the conversion of thiols to sulfonyl chloride and, subsequently, to sulfonamides using 6.5 equivalents of various amines in organic solvents.23 Although all these methods have proved to be of great utility for the synthesis of sulfonamides, the development of a synthetic protocol that is environmentally friendly, simple, efficient, cost effective and straight forward remains an ever challenging objective.In recent years, organic reactions that can proceed in water have attracted great interest because of the significant environmental and economical advantages over those occurring in organic solvents.24 In continuation of our studies on the development of green methods for the synthesis of sulfonamides and their acylated derivatives,25 we report here an efficient, facile and environmentally benign in situ method for the synthesis of sulfonamides directly from thiols and disulfides in water (Scheme 1). The oxidation, chlorination and amination steps were carried out in water without the need to isolate sulfonyl chloride. This method eliminates the use of organic solvents and amine bases and the desired sulfonamides were easily isolated in high to excellent yields and purities by a simple filtration technique, which makes it ideal for green chemistry applications.
 | ||
| Scheme 1 The direct conversion of disulfides and thiols to sulfonamides. | ||
Results and discussion
Given that most of the problems associated with the synthesis of sulfonamides are related to the use of sulfonic acid derivatives, we reasoned that easily available disulfide and thiol derivatives can be used as precursors of sulfonamides without the need to isolate the sufonyl chlorides. As a starting point for this work, the reaction was optimized in order to find the best conditions, with a particular emphasis on a green chemistry route (Table 1). The reaction of diphenyldisulfide was chosen as a model substrate and TCCA was used as an oxidative chlorination agent, which is safer in comparison with other reagents.26 The reaction was carried out in various polar and non-polar solvents and it was found that the product was obtained in good to high yield with dichloromethane, acetonitrile and water (Table 1, entries 1–5). Water was chosen as the best green solvent for further study. The reaction in this step is extremely exothermic, so the temperature was kept below 4 °C. Under these conditions, the reaction was completed in 15 min according to TLC. With reference to the product separated following this step, for each equivalent of disulfide, two equivalents of sulfonylchloride is produced in 98% yield. Following this, the reaction was repeated and the amount of TCCA was varied (Table 1, entries 5–8). The best result was obtained when 0.5 g of TCCA was used. The effect of the amount of water was studied in the range of 1–4 mL (Table 1, entries 5 and 9–11). The yield increased from 64 to 89% when the amount of water was decreased from 1 to 4 mL. This decrease in the yield may be because water can hydrolyze the sulfonyl chloride. The second step, the sulfonamide synthesis, was carried out after the first step without the need to separate the sulfonyl chloride. In the literature, it is usual for organic bases or an excess of amine (up to 8 equivalents) to be used to scavenge the generated hydrogen chloride; however, according to our previous experience, it was found that potassium carbonate can be used as a green base in this step. The effect of the amount of potassium carbonate on the reaction was also investigated. The optimum amount of base (6 mmol) was determined from experiments corresponding to entries 5 and 12–15 in Table 1. It is interesting that the maximum amount of amine used is 2.4 equivalents for every one equivalent of disulfide (or 2 equivalents of the generated sulfonyl chloride). The amine should be added slowly because the reaction in this step is exothermic, akin to the first step. For aliphatic amines, the reaction was completed after 15 min; whereas for aromatic amines the reaction time was found to be 30 min. The sulfonamides were separated from the reaction mixture in high yield and purity after a very easy work-up, which involved the addition of water, a simple filtration and the washing of the filtrate with additional water. The benzenethiols were converted to the corresponding sulfonamides following the same procedure. Sulfonamides derived from benzenethiols and aromatic amines contained some impurities and were purified by column chromatography or recrystallized from ethyl acetate.| Entry | Solvent | K2CO3 (mmol) | TCCA (g) | Time (min) | Yield (%)a |
|---|---|---|---|---|---|
| a Isolated yield. b H2O: 1.0 mL. c H2O: 1.5 mL. d H2O: 2.0 mL. e H2O: 4.0 mL. | |||||
| 1 | Acetone | 6 | 0.5 | 270 | 30 |
| 2 | CH3CN | 6 | 0.5 | 15 | 83 |
| 3 | CH2Cl2 | 6 | 0.5 | 60 | 79 |
| 4 | THF | 6 | 0.5 | 150 | 58 |
| 5 | H2Ob | 6 | 0.5 | 30 | 89 |
| 6 | H2O | 6 | 0.125 | 55 | 45 |
| 7 | H2O | 6 | 0.250 | 45 | 50 |
| 8 | H2O | 6 | 0.625 | 30 | 68 |
| 9 | H2Oc | 6 | 0.5 | 30 | 81 |
| 10 | H2Od | 6 | 0.5 | 30 | 77 |
| 11 | H2Oe | 6 | 0.5 | 30 | 64 |
| 12 | H2O | 2 | 0.5 | 50 | 40 |
| 13 | H2O | 4 | 0.5 | 40 | 59 |
| 14 | H2O | 8 | 0.5 | 30 | 77 |
| 15 | H2O | 10 | 0.5 | 35 | 68 |
These pleasing results prompted us to apply this new protocol to a variety of amines. The results, which are summarized in Tables 2 and 3, indicate that various functional groups are well tolerated and the desired sulfonamides are obtained in good to excellent yields in relatively short reaction times. It was observed that electronic and steric factors play a significant role in these reactions. Both aliphatic and aromatic amines gave high yields of the corresponding sulfonamides. As may be expected, primary amines gave excellent yields of sulfonamides, except for primary amines with long chains (Table 2 and 3, entries 3 and 6). Not surprisingly, a moderate yield of sulfonamide was obtained with sterically hindered amines (Table 2 and 3, entry 2). Diisopropylamine did not react via this method due to the high steric bulk effect from the substituents. With the excellent results achieve for the alkyl amines in hand, we turned our attention to the less nucleophilic anilines. With aniline itself, 80 and 85% yields of sulfonamide were afforded (Table 2 and 3, entry 8). Anilines with poor electron-withdrawing groups, such as chloro and bromo groups, showed similar reactivity to aniline, whereas strong electron-withdrawing groups, such as the nitro group, reduced the rate and the yield of the reaction substantially. Aromatic amines substituted with electron-donating groups reacted faster and provided sulfonamides in higher yields. The negative influence of steric hindrance upon the yield of the products obtained from anilines was observed, with the exception of anilines with electron donating groups at the ortho position (Table 2, entries 10 and 12). The extension of this new method for the synthesis of sulfonamide containing heterocyclic groups is anticipated. When several commercially available heterocyclic amines, such as histamine (Table 2, entry 20) and primary amines derived from pyridine (Table 2, entry 21, 22, 24 and 25) as well as piperazine (Table 2, entry 23) and 2-amino thiazol (Table 2, entry 27), were reacted, the corresponding sulfonamides were formed in good to high yields in less than 1.5 h. These results indicate that this one-pot two-step synthesis of aliphatic and aromatic sulfonamides is likely to be generally applicable. It should be mentioned that several efforts to use this method for the sulfonation of alcohols and phenols for the synthesis of sulfonate esters were not successful.

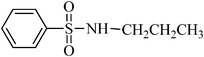
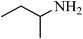
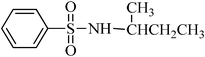

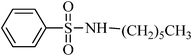

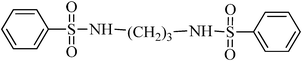
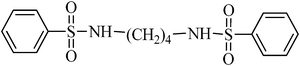
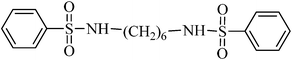
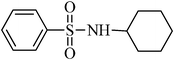
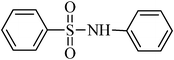
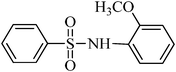
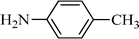
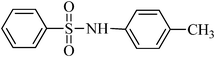
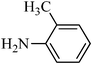
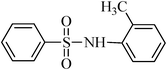
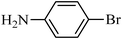
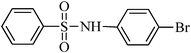
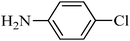
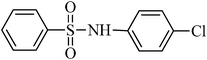
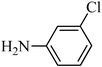
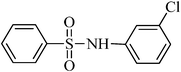
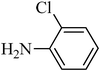
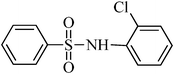
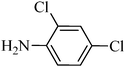
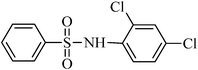
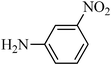
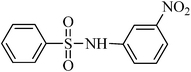
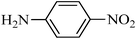
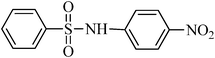
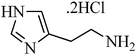
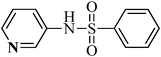
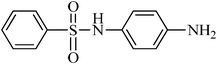
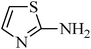
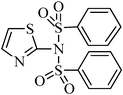

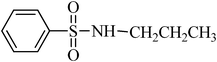
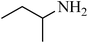
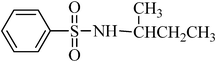

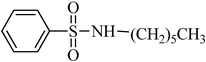

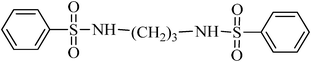
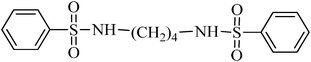
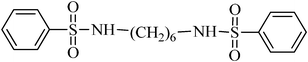
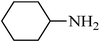
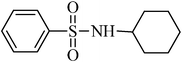
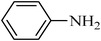
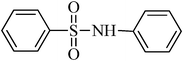
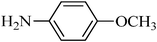
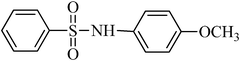
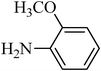
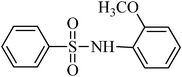
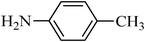
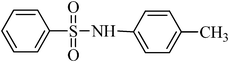
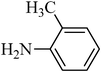
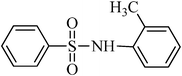
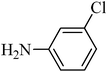
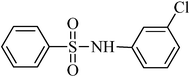
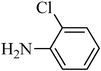
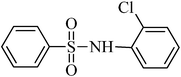
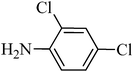
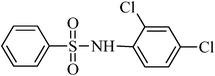
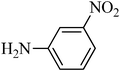
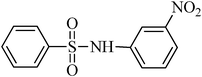
The plausible mechanism for the direct conversion of thiols and disulfides to sulfonamides with TCCA in water is shown in Scheme 2. Water as the media promotes the conversion of TCCA to a super electrophilic species (A, B, C and HOCl) causing “Cl+” transfer to the disulfide.27 Then, the successive oxidation of both sulfur atoms of the disulfide molecule by hypochlorous acid produces the intermediate (D) that undergoes rapid isomerization to the thiosulfonate (E), which can easily furnish the sulfonyl chloride and thiol. The produced thiol gives the corresponding symmetric disulfide through the corresponding sulfenic acid (F).28
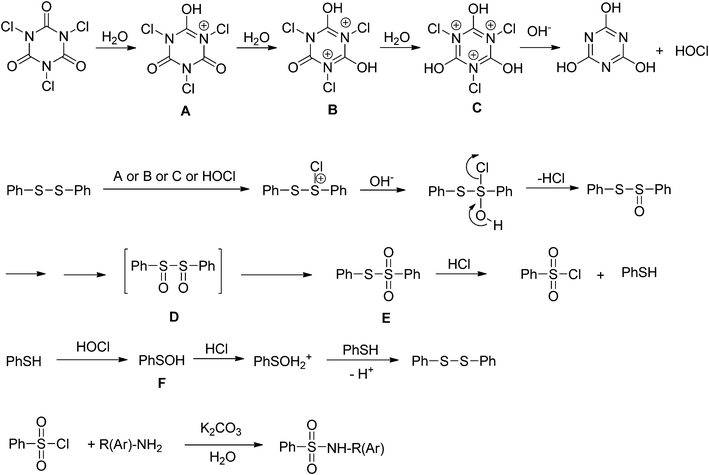 | ||
| Scheme 2 A plausible mechanism for the oxidative chlorination and amination of thiol and disulfide with TCCA in water. | ||
With regards to the reagents, solvent, base and reaction time, a comparative study was performed comparing some of the reported methods for the synthesis of sulfonamides from thiol and disulfide to our method employing TCCA in water (Table 4). It is noteworthy that, in all of the reported methods, the preparation of sulfonamides is performed in organic solvents. In addition, the use of organic amine bases, such as pyridine or an excess of amines, to scavenge the generated hydrogen chloride is another disadvantage of these methods. Furthermore, in most of the methods, sulfonamides were synthesized in two steps and it is necessary to separate the sulfonyl chloride from the reaction mixture in the first step. In the new synthetic protocol reported here, oxidation, chlorination and amination are carried out in water without the need to isolate the sulfonyl chloride. This method eliminates the use of organic solvents and amine bases and the desired sulfonamides are obtained after a simple work-up procedure.
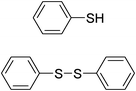
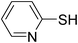
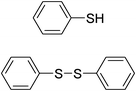
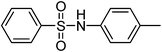
| Entry | Thiol or disulfide | Product | Reagent | Solvent | Time (min) | Base | Ref. |
|---|---|---|---|---|---|---|---|
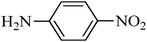
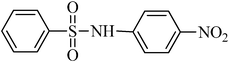
| 1 |
|
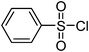
|
TMSCl–KNO3 | CH2Cl2 | 60 | 18 | |
| 360 | |||||||
| 2 |
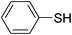
|
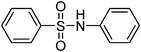
|
H2O2–SOCl2 | CH3CN | 2 | Pyridine | 28 |
| 3 |
|
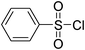
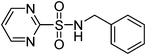
|
|
CH3CN–HOAc–H2O | 60 | 30 | |
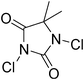
| 4 |
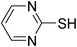
|
|
Cl2 or NaOCl, HCl | CH2Cl2 | 55 | BnNH2 (excess) | 16 |
| 5 |
|
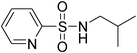
|
TCCA, BnMe3NCl | CH3CN | 130 | Et3N | 23 |
| 6 |
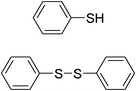
|
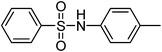
|
H2O2–ZrCl4 | CH3CN | 2 | Pyridine | 29 |
| 7 |
|
|
TCCA | H2O | 45 | K2CO3 | This work |
Chemoselectivity and large scale use
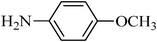
The importance of selectivity in organic chemistry encouraged us to consider the selectivity of sulfonation using the different amines. Several reactions were carried out and, surprisingly, excellent selectivity was found (Scheme 3). For example, aromatic amines with electron donating groups, such as p-anisidine, can be converted into sulfonamides in the presence of p-nitroaniline, an amine with an electron withdrawing group. On the other hand, aliphatic amines were sulfonated selectively in the presence of aromatic amines. Furthermore, the reactions of the amines were so fast in comparison to those of the alcohols and phenols that the selective sulfonation of an amine in the presence of an alcohol or phenol appeared to be a distinct possibility. To show another advantage of the present method and the importance of the scale up ability for laboratory and industrial purposes, a selection of amines were sulfonated on a large scale successfully. For example, aniline and 4-methyl aniline were converted to the corresponding benzene sulfonamides in 20 mmol scales as well as 1 mmol scales.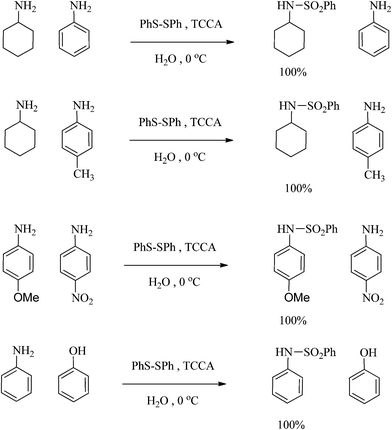 | ||
| Scheme 3 The selective sulfonation of amines. | ||
Conclusions
In conclusion, we have developed a new environmentally friendly strategy for the direct synthesis of sulfonamides from disulfides and thiols. This procedure is quite general for a wide range of amines, including less nucleophilic and sterically hindered anilines, and is easily scalable. The key feature is the one-pot conversion of disulfides and thiols in the presence of TCCA, which was utilized as an oxidative chlorination reagent, and K2CO3 as a base in water. This protocol has advantages in terms of its low cost and the availability of the reagents, its short reaction times, high chemoselectivity, operational simplicity and easy work-up. Thus, this method obeys several key principles of green chemistry and could be widely applicable in the synthesis of various alkyl and aryl sulfonamides.Experimental
All chemicals were purchased from Merck and Fluka chemical companies. Infrared spectra were recorded on a Perkin–Elmer V IR spectrophotometer. 1HNMR and 13C NMR spectra were recorded on a Bruker (400 MHz) FT spectrometer in CDCl3 and DMSO-d6. Column chromatography was performed using silica gel 60 (230–400 mesh). All reactions were conducted open to the atmosphere and the yields refer to the isolated products.General procedure for the synthesis of sulfonamides
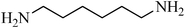
To a stirred mixture of disulfide or thiol (1 mmol) in H2O (1 mL), TCCA (0.5 g, 2.15 mmol) was added in portions over 1–2 min at 0–4 °C. The reaction was exothermic and the color turned yellow, then pink–yellow and, after 15 min, provided a light yellow solution. The reaction was shown to be completed in this step by TLC. K2CO3 (6–8 mmol) was then added to the mixture over 1–2 min and, after that, the amines [aliphatic amines (2.2 mmol), aromatic amines (2.4 mmol) or diamines (2.2 mmol)] were added to the mixture over 1–2 min. The addition was performed in this manner to avoid the temperature increasing too suddenly. In this step, H2O (0.5 mL) was added to the mixture to achieve a more homogenous solution with stirring. The reactions were completed in 30–90 min as indicated by TLC. The desired sulfonamide was easily isolated in high to excellent yields by simple a filtration procedure and washing with additional H2O (3 × 10 mL). The purity of the products obtained from the disulfide was very high, which was assessed by comparison of their melting point, NMR and IR spectra with authentic samples. Some sulfonamides were obtained from thiols purified with a short column chromatography method or recrystallized from ethyl acetate.N-hexylbenzenesulfonamide, (Table 2, entry 3) mp 87–89 °C; Rf 0.45 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3285 (N–H), 1325, 1161(SO2), 587 (C–N); δH (400 MHz; CDCl3) 0.83 (3H, t, J 7.2), 1.12–1.28 (6H, m), 1.45 (2H, quint., J 7.2 Hz), 2.93 (2H, td, J 6.8, 7.2 Hz), 5.16 (1HN–H, t, J 5.6), 7.49–7.60 (3Harom, m), 7.89 (2Harom, d, J 7.2); δC (100 MHz; CDCl3) 17.6, 24.0, 26.1, 29.7, 32.2, 42.0, 129.1, 130.7, 134.0, 140.0.
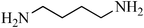
N,N′-(propane-1,3-diyl)dibenzenesulfonamide, (Table 2, entry 4) mp 88–90 °C; Rf 0.23 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3277(N–H), 1332, 1173 (SO2), 728 (C–N); δH (400 MHz; DMSO-d6) 1.52 (2H, quint., J 6.8 Hz), 2.72 (4H, td, J 6.4, 6.8 Hz), 7.57–7.68 (6Harom, 2HN–H, m), 7.75–7.80 (4Harom, m); δC (100 MHz; DMSO-d6) 29.9, 39.9, 126.9, 129.7, 132.8, 140.8.
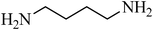
N,N′-(butane-1,4-diyl)dibenzenesulfonamide, (Table 2, entry 5) mp 120–122 °C; Rf 0.26 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3269 (N–H), 1326, 1161 (SO2),885 (C–N); δH (400 MHz; DMSO-d6) 1.30–1.42 (4H, m, br), 2.60–2.74 (4H, m, br), 7.54–7.67 (6Harom, 2HN–H, m), 7.78 (4Harom, d, J 6.8); δC (100 MHz; DMSO-d6) 26.9, 40.2, 126.9, 129.7, 132.8, 144.8.
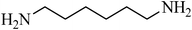
N,N′-(hexane-1,6-diyl)dibenzenesulfonamide, (Table 2, entry 6) mp 142–144 °C; Rf 0.26 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3292 (N–H), 1324, 1157 (SO2), 919 (C–N); δH (400 MHz; DMSO-d6) 1.01–1.14 (4H, m), 1.21–1.36 (4H, m), 2.82 (4H, q, J 6.4), 7.56–7.68 (6Harom, 2HN–H, m), 7.78–7.83 (4Harom,m); δC (100 MHz; DMSO-d6) 28.9, 29.9, 40.4, 126.9, 129.7, 132.8, 140.8.
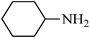
N-cyclohexylbenzenesulfonamide,(Table 2, entry 7) mp76–78 °C; Rf 0.45 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3267 (N–H), 2934 (C–Haliph), 1329, 1158 (SO2), 881 (C–N); δH (400 MHz; DMSO-d6) 0.90–1.29 (5H, m), 1.35–1.69 (5H, m), 2.80–3.12 (1H, m), 7.45–7.69 (4Harom, m), 7.70–7.81 (2 H, m), δC (100 MHz; DMSO-d6) 24.9, 25.9, 33.9, 39.9, 126.9, 129.7, 132.8, 142.8.
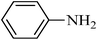
N-phenylbenzenesulfonamide, (Table 2, entry 8) mp 100–102 °C; Rf 0.39 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3214 (N–H), 1330, 1157 (SO2), 928 (C–N); δH (400 MHz; DMSO-d6) 6.99–7.03 (1Harom, m), 7.09–7.13 (2Harom, m), 7.20–7.24 (2Harom, m), 7.51–7.62 (3 Harom, m), 7.76–7.81 (2Harom, m), 10.30 (1 HN–H, s); δC (100 MHz; DMSO-d6) 120.5, 124.4, 127.1, 129.6, 129.7, 133.2, 138.4, 140.2.
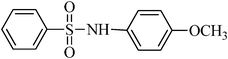
N-(4-methoxyphenyl)benzenesulfonamide, (Table 2, entry 9) mp 88–90 °C; Rf 0.26 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3256 (N–H), 1334, 1154 (SO2), 581 (C–N); δH (400 MHz; CDCl3) 3.81 (3H,s), 6.78 (2Harom, d, J 9.2 Hz), 7.05 (2Harom, d, J 9.2 Hz), 7.11(1HN–H, s), 7.41–7.53 (3Harom, m), 7.72 (2Harom, d, J 7.6); δC (100 MHz; CDCl3) 56.6, 115.0, 127.2, 129.7, 130.1, 133.3, 133.9, 140.0, 154.5.
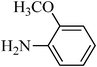
N-(2-methoxyphenyl)benzenesulfonamide, (Table 2, entry 10) mp 86–88 °C; Rf 0.27 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3256 (N–H), 1337, 1168 (SO2), 577 (C–N); δH (400 MHz; CDCl3) 3.76 (3H,s), 6.77 (2Harom, d, J 9.0 Hz), 7.02 (2Harom, d, J 9.2 Hz), 7.08(1HN–H, s), 7.41–7.44 (2Harom, m), 7.55 (1Harom, t, J 8.0), 7.75 (2Harom, d, J 7.6); δC (100 MHz; CDCl3) 53.6, 115.4, 121.4,122.0, 127.0, 128.6, 132.2, 135.2, 137.6, 139.2, 147.1.
N-p-tolylbenzenesulfonamide, (Table 2, entry 11) mp 110–112 °C; Rf 0.45 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3270 (N–H), 1129, 1156 (SO2), 911 (C–N); δH (400 MHz; DMSO-d6) 2.17 (3H,s), 6.97–7.05 (4Harom, m), 7.51–7.62 (3Harom, m), 7.73–7.77 (2 Harom, m), 10.16 (1 HN–H, s); δC (100 MHz; DMSO-d6) 20.7, 121.1, 127.1, 129.6, 130.0, 133.2, 133.9, 135.5, 140.0.
N-o-tolylbenzenesulfonamide, (Table 2, entry 12) mp 109–111 °C; Rf 0.42 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3273 (N–H), 1327, 1165 (SO2),758 (C–N); δH (400 MHz; DMSO-d6) 2.00 (3H,s), 6.97–7.00 (1Harom, m), 7.08–7.15 (3Harom, m), 7.53–7.57 (2 Harom, m), 7.62–7.68 (3Harom, m), 9.62 (1 H, s, N–H); δC (100 MHz; DMSO-d6) 18.0, 126.8, 126.9, 127.0, 127.0, 129.6, 131.2, 133.2, 134.6, 135.2, 141.1.
N-(3-chlorophenyl)benzenesulfonamide, (Table 2, entry 15) mp 109–111 °C; Rf 0.37 (20% ethyl acetate, 80% n-hexane); Vmax (KBr)/cm−1: 3204 (N–H), 1317, 1157 (SO2), 941 (C–N), 731 (C–Cl); δH (400 MHz; DMSO-d6) 7.05–7.16 (3Harom, m), 7.23–7.29 (1Harom, m), 7.54–7.66 (3Harom, m), 7.78–7.84 (2 Harom, m), 10.65 (1 HN–H, s); δC (100 MHz; DMSO-d6) 118.5, 119.5, 124.2, 127.1, 129.9, 131.4, 133.6, 133.9, 139.6, 139.8.
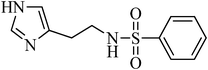
N-(2-(1H-imidazol-4-yl)ethyl)benzenesulfonamide, (Table 2, entry 20); Rf 0.24 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3129 (N–H), 1379, 1178 (SO2), 596 (C–N); δH (400 MHz; CDCl3) 2.66 (2H, t, J 10 Hz), 3.09 (2H,td, J 10.8, 8 Hz), 7.67–7.86 (6Harom, 1HN–H, m), 8.01 (1Harom, d, J 8.0), 13.03 (1HN–H, br); δC (100 MHz; CDCl3) 27.6, 40.4, 120.5, 129.4, 132.1, 134.6, 134.7, 138.2, 143.4, 145.2.
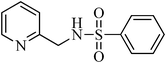
N-(pyridine-2-ylmethyl)benzenesulfonamide, (Table 2, entry 21) mp 77–80 °C; Rf 0.27 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3064 (N–H), 1329, 1160 (SO2), 750 (C–N); δH (400 MHz; CDCl3) 4.26 (2H, d, J 5.6 Hz), 6.60 (1HN–H, br), 7.11–7.20 (2Harom, m), 7.40–7.62 (4Harom, m), 7.81 (2Harom, d, J 7.6 Hz), 8.42 (1Harom, d, J 4.0 Hz); δC (100 MHz; CDCl3).47.1, 121.5, 125.4, 129.1, 132.1, 133.2, 140.2, 145.0, 149.0, 156.0.
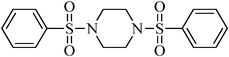
1,4-bis(phenylsulfonyl)piperazine, (Table 2, entry 23) mp 260–262 °C; Rf 0.28 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3166 (N–H), 1347, 1172 (SO2),576 (C–N); δH (400 MHz; CDCl3) 2.8–3.2 (8H, m), 7.51–7.62 (6Harom, m), 7.72–7.77 (4Harom, m); δC (100 MHz; CDCl3) 45.2, 127.9, 129.7, 132.6, 137.8.
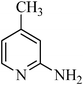
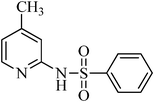
N-(4-methylpyridin-2-yl)benzenesulfonamide, (Table 2, entry 24) mp 180–185 °C; Rf 0.32 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3133 (N–H), 1374, 1177 (SO2), 581 (C–N); δH (400 MHz; CDCl3) 2.05 (3H, s), 6.53 (1Harom, s), 6.70 (1Harom, d, J 8.0 Hz), 7.18–7.34 (3Harom, m), 7.63 (2Harom, d, J 8.0), 8.11 (1Harom, d, J 10.5), 11.50 (1HN–H, S); δC (100 MHz; CDCl3) 21.5, 112.6, 121.2, 127.1, 129.9, 131.4, 139.6, 139.8, 151.4, 153.9.
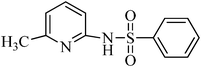
N-(6-methylpyridin-2-yl)benzenesulfonamide, (Table 2, entry 25) mp 128–129 °C; Rf 0.34 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3171 (N–H), 1371, 1171 (SO2), 1089 (C–N); δH (400 MHz; CDCl3) 2.52 (3H, s), 6.60 (1Harom, d, J 7.2 Hz), 7.11 (1Harom, d, J 9.6 Hz), 7.44–7.61 (4Harom, m), 7.95 (2Harom, d, J 7.2 Hz), 11.03 (1HN–H , s); δC (100 MHz; CDCl3) 24.0, 106.0, 119.5, 127.1, 129.9, 131.4, 138.6, 139.8, 152.5, 157.7.
N-(4-aminophenyl)benzenesulfonamide, (Table 2, entry 26) mp 168–171 °C; Rf 0.18 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 3431 (N–H2), 3267 (N–H), 1328, 1159 (SO2), 784 (C–N); δH (400 MHz; CDCl3) 3.67 (2HN–H,br), 6.33 (1HN–H, s), 6.54–6.57 (2Harom, m), 6.82–6.87 (2Harom, m), 7.45 (2Harom, t, J 8.0 Hz), 7.55 (1Harom, t, J 8.0), 7.71 (2Harom, d, J 8.2); δC (100 MHz; CDCl3) 118.5, 118.6, 127.2, 127.7, 129.8, 131.9, 138.6, 139.1.
N-(phenylsulfonyl)-N-(thiazol-2-yl)benzenesulfonamide, (Table 2, entry 27) mp 127–131 °C; Rf 0.48 (40% ethyl acetate, 60% n-hexane); Vmax (KBr)/cm−1: 1318,1152 (SO2), 907 (C–N); δH (400 MHz; CDCl3), 6.47 (1Harom, d, J 5.2), 7.41–7.52 (5Harom, m), 7.95 (2Harom, d, J 7.2 Hz), δC (100 MHz; CDCl3) 112.2, 127.7, 129.0, 131.9, 136.2, 138.7, 171.8.
Acknowledgements
Support from the Shahreza Branch, Islamic Azad University (IAUSH) Research council is gratefully acknowledged.References
- (a) W. G. Harter, H. Albrect, K. Brady, B. Caprathe, J. Dunbar, J. Gilmore, S. Hays, C. R. Kostlan, B. Lunney and N. Walker, Bioorg. Med. Chem. Lett., 2004, 14, 809 CrossRef CAS; (b) N. S. Reddy, M. R. Mallireddigari, K. G. Cosenza, S. C. Bell, E. P. Reddy and M. V. R. Reddy, Bioorg. Med. Chem. Lett., 2004, 14, 4093 CrossRef CAS; (c) B. R. Stranix, J. F. Lavallee, G. Sevigny, J. Yelle, V. Perron, N. Leberre, D. Herbart and J. J. Wu, Bioorg. Med. Chem. Lett., 2006, 16, 3459 CrossRef CAS.
- (a) N. K. U. Koehler, C. Y. Yang, J. Varady, Y. Lu, X. W. Wu, M. Liu, D. Yin, M. Bartels, B. Y. Xu, P. P. Roller, Y. Q. Long, P. Li, M. Kattah, M. L. Cohn, K. Moran, E. Tilley, J. R. Richert and S. Wang, J. Med. Chem., 2004, 47, 4989 CrossRef CAS; (b) D. C. Cole, W. J. Lennox, S. Lombardi, J. W. Ellingboe, R. C. Bernotas, G. J. Tawa, H. Mazandarani, D. L. Smith, G. Zhang, J. Coupet and L. E. Schechter, J. Med. Chem., 2005, 48, 353 CrossRef CAS; (c) M. Banerjee, A. Poddar, G. Mitra, A. Surolia, T. Owa and B. Bhattacharyya, J. Med. Chem., 2005, 48, 547 CrossRef CAS.
- (a) C. O. Wilson, O. Gisvold, O. Block, J. H. Block, inWilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry, ed. J. H. Block and J. M. Beale, Lippincott Williams & Wilkins, Philadelphia, 11th edn, 2004 Search PubMed; (b) W. D. Miller, A. H. Fray, J. T. Quatroche and C. D. Sturgill, Org. Process Res. Dev., 2007, 11, 359 CrossRef CAS; (c) S. Oh, H. I. Moon, I.-H. Son and J. C. Jung, Molecules, 2007, 12, 1125 CrossRef CAS; (d) M. Lopez, L. F. Bornaghi, A. Innocenti, D. Vullo, S. A. Charman, C. T. Supuran and S. A. Poulsen, J. Med. Chem., 2010, 53, 2913 CrossRef CAS; (e) W. J. Zuercher, R. G. Buckholz, N. Campobasso, J. L. Collins, C. M. Galardi, R. T. Gampe, S. M. Hyatt, S. L. Merrihew, J. T. Moore, J. A. Oplinger, P. R. Reid, P. K. Spearing, T. B. Stanley, E. L. Stewart and T. M. Willson, J. Med. Chem., 2010, 53, 3412 CrossRef CAS; (f) H. Hu, H. Liao, J. Zhang, W. Wu, J. Yan, Y. Yan, Q. Zhao, Y. Zou, X. Chai, S. Yu and Q. Wu, Bioorg. Med. Chem. Lett., 2010, 20, 3094 CrossRef CAS.
- (a) A. Shaabani, E. Soleimani and A. H. Rezayan, Tetrahedron Lett., 2007, 48, 2185 CrossRef CAS; (b) W. Deng, L. Liu, C. Zhang, M. Liu and Q. X. Guo, Tetrahedron Lett., 2005, 46, 7295 CrossRef CAS; (c) G. Burton, P. Cao, G. Li and R. Rivero, Org. Lett., 2003, 5, 4373 CrossRef CAS.
- F. Shi, M. K. Tse, S. Zhou, M. M. Pohl, J. Radnik, S. Hubner, K. Jahnisch, A. Bruckner and M. Beller, J. Am. Chem. Soc., 2009, 131, 1775 CrossRef CAS.
- B. Sreedhar, V. Ravi and D. Yada, Tetrahedron Lett., 2011, 52, 1208 CrossRef CAS.
- J.-G. Kim and D. O. Jang, Synlett, 2007,(16), 2501 CAS.
- J. L. G. Ruano, A. Parra, F. Yuste and V. M. Mastranzo, Synthesis, 2008,(2), 311 CrossRef CAS.
- D. K. H. Ho, L. Chan, A. Hooper and P. E. Brennan, Tetrahedron Lett., 2011, 52, 820 CrossRef CAS.
- S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024 CrossRef CAS.
- (a) M. Jafarpour, A. Rezaeifard and M. Aliabadi, Appl. Catal., A, 2009, 358, 49 CrossRef CAS; (b) X. Deng and N. S. Mani, Green Chem., 2006, 8, 835 RSC; (c) G. A. Meshram and V. D. Patil, Tetrahedron Lett., 2009, 50, 1117 CrossRef CAS; (d) J. G. Kang, J. H. Hur, S. J. Choi, G. J. Choi, K. Y. Cho, L. N. Ten, K. H. Park and K.Y. Kang, Biosci., Biotechnol., Biochem., 2002, 66, 2677 CrossRef CAS.
- (a) S. Caddick, J. D. Wilden, S. J. Wadman, H. D. Bush and D. B. Judd, Org. Lett., 2002, 4, 2549 CrossRef CAS; (b) S. Caddick, D. Hamza, S. J. Wadman and J. D. Wilden, Org. Lett., 2002, 4, 1775 CrossRef CAS.
- T. J. J. Holmes and R. G. Lawton, J. Org. Chem., 1983, 48, 3146 CrossRef CAS.
- A. Yasuhara, M. Kameda and T. Sakamoto, Chem. Pharm. Bull., 1999, 47, 809 CrossRef CAS.
- (a) R. J. Watson, D. Batty, A. D. Baxter, D. R. Hannah, D. A. Owen and J. G. Montana, Tetrahedron Lett., 2002, 43, 683 CrossRef CAS; (b) V. Percec, T. K. Bera, B. B. De, Y. Sanai, J. Smith, M. N. Holerca, B. Barboiu, R. B. Grubbs and J. M. J. Fréchet, J. Org. Chem., 2001, 66, 2104 CrossRef CAS; (c) W. T. Caldwell and E. C. Kornfeld, J. Am. Chem. Soc., 1942, 64, 1695 CrossRef CAS.
- S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080 CrossRef CAS.
- A. Nishiguuchi, K. Maeda and S. Miki, Synthesis, 2006, 4131–4134 CrossRef.
- G. K. S. Prakash, T. Mathew, C. Panja and G. Olah, J. Org. Chem., 2007, 72, 5847 CrossRef CAS.
- Y. J. Park, H. H. Shin and Y. H. Kim, Chem. Lett., 1992, 1483 CrossRef CAS.
- J. Humljan and S. Gobec, Tetrahedron Lett., 2005, 46, 4069 CrossRef CAS.
- L. Kværnø, M. Werder, B. A. Hauser and E. M. Carreira, Org. Lett., 2005, 7(6), 1145 CrossRef.
- A. Meinzer, A. Breckel, B. A. Thaher, N. Manicone and H. H. Helv. Otto, Helv. Chim. Acta, 2004, 87(1), 90 CrossRef CAS.
- J. D. Bonk, D. T. Amos and S. J. Olson, Synth. Commun., 2007, 37, 2039 CrossRef CAS.
- (a) C. J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS; (b) C. I. Herrerias, X. Yao, Z. Li and C.J. Li, Chem. Rev., 2007, 107, 2546 CrossRef CAS; (c) V. K. Rai, P. K. Rai, S. Bajaj and A. Kumara, Green Chem., 2011, 13, 1217 RSC; (d) X. W. Sun, M. Liu, M. H. Xu and G. Q. Lin, Org. Lett., 2008, 10, 1259 CrossRef CAS; (e) Y. Y. Peng, J. Liu, X. Lei and Z. Yin, Green Chem., 2010, 12, 1072 RSC; (f) N. Ma, B. Jiang, G. Zhang, S-J. Tu, W. Wever and G. Li, Green Chem., 2010, 12, 1357 RSC.
- (a) F. Kazemi, A. R. Massah and H. Javaherian, Tetrahedron, 2007, 63, 5083 CrossRef CAS; (b) A. R. Massah, D. Azadi, H. Aliyan, A. R. Momeni, H. Javaherian Naghash and F. Kazemi, Monatsh. Chem., 2008, 139, 233 CrossRef CAS; (c) A. R. Massah, B. Asadi, M. Hoseinpour, A. Molseghi, R. J. Kalbasi and H. J. Naghash, Tetrahedron, 2009, 65, 7696 CrossRef CAS; (d) A. R. Massah, M. Dabagh, M. Afshar, A. R. Momeni, H. Aliyan and H. J. Naghash, Turk J. Chem., 2007, 31, 611 CAS.
- U. Tilstam and H. Weinmann, Org. Process Res. Dev., 2002, 6, 384 CrossRef CAS.
- G. F. Mendonça, R. R. Magalhães, M. C. S. de Mattos and P. M. Esteves, J. Braz. Chem. Soc., 2005, 16, 695 CrossRef.
- K. Bahrami, M. M. Khodaei and M. Soheilizad, J. Org. Chem., 2009, 74, 9287 CrossRef CAS.
- K. Bahrami, M. M. Khodae and M. Soheilizad, Tetrahedron Lett., 2010, 51, 4843 CrossRef CAS.
- Y. M. Pu, A. Christesen and Y.Y. Ku, Tetrahedron Lett., 2010, 51, 418 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20418e/ |
| This journal is © The Royal Society of Chemistry 2012 |