Iron(II) catalyzed dehydrative etherification of alcohols: a convenient route to ferrocenylmethanol-ethers†
Luigi
Busetto
,
Rita
Mazzoni
,
Mauro
Salmi
,
Stefano
Zacchini
and
Valerio
Zanotti
*
Dipartimento di Chimica Fisica e Inorganica, Università di Bologna, Viale Risorgimento 4, I-40136, Bologna, Italy. E-mail: valerio.zanotti@unibo.it; Fax: 0039-0512093690; Tel: 0039-0512093695
First published on 21st May 2012
Abstract
The iron complex [Fp][OTf] (Fp+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) [Fe(CO)2(Cp)]+, OTf−SO3CF3−) was found to act as efficient catalyst for the dehydrative etherification of ferrocenylmethanol [HOCH2–Fc] with a variety of alcohols (ROH), providing a valuable route for the formation of ferrocenylmethanol-ethers [ROCH2–Fc]. The complex [Fp][OTf] also catalyzes etherification of propargyl alcohols with other alcohols. The advantages of the method are associated with the use of a non toxic and easily available transition metal as catalyst, and the dehydrative synthetic approach, which produces water as the only by-product.
[Fe(CO)2(Cp)]+, OTf−SO3CF3−) was found to act as efficient catalyst for the dehydrative etherification of ferrocenylmethanol [HOCH2–Fc] with a variety of alcohols (ROH), providing a valuable route for the formation of ferrocenylmethanol-ethers [ROCH2–Fc]. The complex [Fp][OTf] also catalyzes etherification of propargyl alcohols with other alcohols. The advantages of the method are associated with the use of a non toxic and easily available transition metal as catalyst, and the dehydrative synthetic approach, which produces water as the only by-product.
Introduction
We have recently reported on a new approach to the synthesis of functionalized ferrocenes based on the [3 + 2] cycloaddition of alkynes with bridging C3 ligands in diiron complexes.1 A general representation, shown in Scheme 1, evidences the peculiar feature of the synthetic method, which affords ferrocenes containing different substituents and functional groups in only one of the two Cp rings.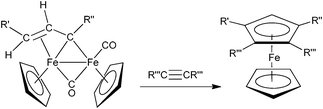 | ||
| Scheme 1 | ||
These polysubstituted ferrocenes are valuable scaffolds for the construction of functional molecules with potential application in catalysis, material science and medicinal chemistry.2
In a related reaction, more recently reported, the bridging vinyliminium complexes of type 1 were found to react with propargyl alcohol to form ferrocene complexes containing a propargylic pendant chain (Scheme 2).3 In this case two propargyl units are involved: one undergoes the [3 + 2] cycloaddition with the bridging vinyliminium ligand, whereas a second alkynol gives rise to the pendant propargyl chain, by –OH substitution. The reaction has been proposed to occur according to the sequence shown in Scheme 2, with I as intermediate species.3 It was evidenced that ferrocenylmethanol-propargyl alcohol etherification step is catalyzed by the complex [Fp][OTf] (Fp+[Fe(CO)2(Cp)]+ and OTf−SO3CF3−), generated upon fragmentation of the parent dinuclear complex 1.
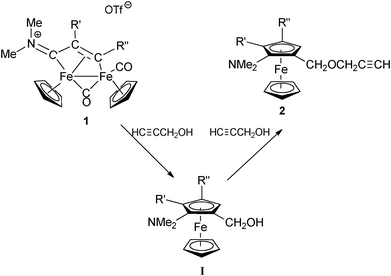 | ||
| Scheme 2 | ||
However, the precise mechanism by which [Fp][OTf] promotes OH displacement was not established; in particular it was not determined whether ether formation occurs by OH displacement at the propargyl alcohol, or at the ferrocenylmethanol. Nevertheless, the observation that [Fp][OTf] acts as etherification catalyst is of great interest, since dehydrative etherification is not easy to accomplish due to poor leaving group ability of the OH group. Indeed, classic methods, including Williamson ether synthesis, require preliminary conversion of alcohols to halides, tosylates or other species containing better leaving groups.4 Therefore, straightforward synthetic methods based on dehydrative coupling of alcohols are more attractive and environmentally sustainable, in that water is the only by-product, but this approach requires activation. Indeed, in recent years, a growing number of catalytic methods have been developed, which mostly make use of Lewis acids5 or transition metal compounds,6 in particular complexes of ruthenium7 and palladium.8 A possible catalytic activity of [Fp][OTf] in dehydrative etherification of alcohols is particularly desirable in that iron is cheap and non toxic, and few catalysts based on iron have been so far employed in the activation of this reaction.5d,6b The present report describes investigations that involve a range of alcohols and is aimed at determining whether the catalytic behaviour of [Fp][OTf] in dehydrative etherification of alcohols is general, possibly providing a better understanding of the activation mechanism.
Results and discussion
Investigation on catalytic dehydrative etherification started from the reactions of ferrocenylmethanol (3) with a few propargyl alcohols (4a–e), in that these resemble the etherification step shown in Scheme 2. The conditions under which these reactions have been carried out involve the use of a slight excess of propargyl alcohols respect to ferrocenylmethanol (3) (about 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio), in the presence of [Fp][OTf] (1% mol to 3), in toluene solution at room temperature. Reactions lead to the formation of propargyl ferrocenylmethyl ethers 5 in good yields, as shown in Scheme 3. The iron catalyst was freshly generated before use by reacting [Fe2(Cp)2(CO)4] with AgOTf according to a published procedure.9 Reactions lead to the formation of propargyl ferrocenylmethyl ethers in good yields, as shown in Scheme 3.
:1 molar ratio), in the presence of [Fp][OTf] (1% mol to 3), in toluene solution at room temperature. Reactions lead to the formation of propargyl ferrocenylmethyl ethers 5 in good yields, as shown in Scheme 3. The iron catalyst was freshly generated before use by reacting [Fe2(Cp)2(CO)4] with AgOTf according to a published procedure.9 Reactions lead to the formation of propargyl ferrocenylmethyl ethers in good yields, as shown in Scheme 3.
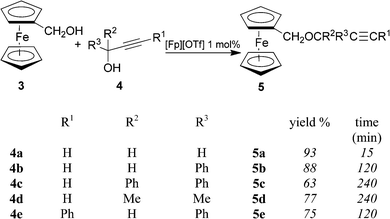 | ||
| Scheme 3 | ||
The use of [Fp][PF6] in place of [Fp][OTf] has been also checked in the reaction of the ferrocenylmethanol (3) with 4a in order to evidence possible effects due to the counteranion. No difference with the corresponding reaction with [Fp][OTf] was found.
Compounds 5a–e have been characterized by NMR spectroscopy, ESI-MS spectrometry and elemental analysis. Moreover, the molecular structure of 5b has been determined by X-ray diffraction (Fig. 1 and Table 1).
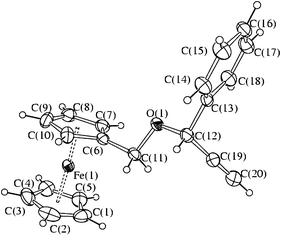 | ||
| Fig. 1 ORTEP diagram of 5b. Thermal ellipsoids are drawn at 30% probability level. | ||
| a Cp(1) is defined by atoms C(1), C(2), C(3), C(4) and C(5). b Cp(2) is defined by atoms C(6), C(7), C(8), C(9) and C(10). | |||
|---|---|---|---|
| Fe(1)–Cp(1) a | 2.022(5)–2.034(4) average 2.028(9) | C–C Cp(1) a | 1.387(6)–1.411(8) average 1.398(16) |
| Fe(1)–Cp(2) b | 2.029(3)–2.047(3) average 2.036(8) | C–C Cp(2) b | 1.410(7)–1.430(5) average 1.419(13) |
| C(6)–C(11) | 1.485(5) | C(11)–O(1) | 1.448(4) |
| C(12)–O(1) | 1.415(4) | C(12)–C(13) | 1.518(5) |
| C(12)–C(19) | 1.471(6) | C(19)–C(20) | 1.174(7) |
| Sum angles Cp(1) a | 539.9(11) | Sum angles Cp(2) b | 539.9(9) |
| O(1)–C(11)–C(6) | 108.1(3) | C(12)–O(1)–C(11) | 113.1(3) |
| O(1)–C(12)–C(13) | 107.5(3) | O(1)–C(12)–C(19) | 112.8(3) |
| C(13)–C(12)–C(19) | 111.2(3) | C(12)–C(19)–C(20) | 179.1(5) |
5b is a mono-substituted ferrocene with the two Cp-rings almost parallel [the angle between the least squares mean planes of the five-membered rings is 2.6°]. The Fe–C interactions with the substituted C5-ring [average 2.036(8) Å] are sensibly longer than those with the unsubstituted Cp-ring [average 2.028(9) Å]. The C(6)–C(11) [1.485(5) Å], C(11)–O(1) [1.448(4) Å], C(12)–O(1) [1.415(4) Å] and C(1)–C(19) [1.471(6) Å] interactions are typical for single bonds, whereas C(19)–C(20) [1.74(7) Å] is essentially a triple bond.
The results shown in Scheme 3 are remarkable in consideration of the very mild reaction conditions (i.e. temperature and amount of catalyst). Conversions and reaction times are comparable to the excellent performance in propargylic substitution obtained by diruthenium catalysts developed by Nishibayashi,7a,7b,10 as well as the catalysts designed by Gimeno and coworkers, based on mononuclear ruthenium complexes.7c,11 However, a difference has to be outlined: Nishibayashi's catalysts preferentially works with secondary propargyl alcohols (such as 4b), and Gimeno's catalysts are substantially limited to the tertiary 1,1-diphenyl-2-propynol 4c. Conversely, in our case primary, secondary and also tertiary alkynols (i.e.4a, 4b and 4c) undergo dehydrative coupling with ferrocenylmethanol, although with decreasing yields, respectively. Comparison with ruthenium catalysts is appropriate, in that our reactions might also proceed through catalytic displacement of OH from the propargyls. In the case of the above mentioned ruthenium catalysts, there is strong evidence that propargylic substitution occurs via vinylidene and allenylidene intermediates. Also iron bound propargyls can be transformed into vinylidene and allenylidene complexes,12 but the “allenylidene mechanism” appears unrealistic in our case in that etherification takes place also with an internal propargylic alcohol, such as 4e, whereas allenylidene intermediates can be formed only with primary propargylic alcohols. Having excluded allenylidene intermediates does not necessarily rule out a possible role of [Fp][OTf] in propargylic activation. For example, Dixneuf and co-workers have recently developed mononuclear ruthenium catalysts (i.e. [RuCl(PR3)(p-cymene)][OTf] and [RuCl(CO)(PR3)(p-cymene)][OTf]) which are active in propargylic substitution without implying allenylidene intermediates.7d,13 Moreover, several other complexes, containing cobalt,14 rhenium,15 molybdenum,16 and gold17 and also Lewis acids, such as BiCl3,18 InBr3,19 CuBr2,20 and FeCl321 promote or catalyze propargylic substitution, indicating that the reaction can be assisted through a number of different mechanisms. On the other hand, we cannot exclude the possibility that the [Fp][OTf] catalytic effect concerns OH displacement from ferrocenyl alcohol, rather than propargyl alcohols. This hypothesis is reasonable in consideration of the relative stability of ferrocenylcarbenium cations, which can be generated from ferrocenyl alcohols upon treatment of with Brønsted acids, Lewis acids or transition metal compounds.22
A remarkable aspect of the reaction is the selectivity: only unsymmetrical etherification (i.e. involving two different alcohols 3 and 4) takes place, except in one case (the reaction of 3 with 4c), in which small amounts of the ferrocenylmethyl ether 6 (7% yield) was found as by-product. Interestingly, we have found that [Fp][OTf] is able to catalyze the self condensation of ferrocenylmethanol 3, affording 6 (Scheme 4), but not the self condensation of propargyl alcohols. Indeed, it was necessary a 5% catalyst loading and a reaction time of 16 h, in order to obtain a good conversion of 3 to 6 (84% yield), whereas under the conditions used for obtaining propargyl ferrocenylmethyl ethers (Scheme 3), the self condensation of ferrocenylmethanol is sluggish and only 15% yield of 6 is obtained after 240 min. These observations, together with those above reported about the limited influence of the type of alkynol over the reaction outcome indicate that [Fp][OTf] activation is predominantly directed to ferrocenylmethanol, rather than propargyl alcohols.
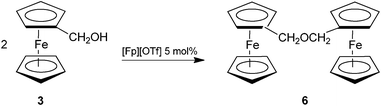 | ||
| Scheme 4 | ||
Beside mechanistic considerations, which will be further developed in the following paragraphs, the results shown in Scheme 2 are interesting in that they provide a straightfoward approach to ferrocene functionalization. In particular, the method gives easy access to ferrocenes containing a pendant propargylic chain, which can be efficiently used to connect the ferrocenyl unit to a variety of molecular fragments, via covalent or coordinative bonds. Examples have been shown in a previous work.3
Further investigation has been focused on the dehydrative etherification of propargyl alcohols with phenols and aliphatic alcohols, using [Fp][OTf] as catalyst.
The latter reactions required an increased catalyst loading (10% molar ratio to 4), the use of an excess of the alcohols 7 respect to the alkynols 4 (molar ratio 4:7 = 1:3) and a reaction time of 16 h. Results, reported in Scheme 5, indicate a lower catalytic activity compared to the reactions of propargyl alcohols with 3 previously discussed: dehydrative etherification of alkynols with a variety of different alcohols generally occurs in moderate yields. Indeed, we have examined all possible combinations of the propargyl alcohols 4a–c with 7a–f, and also with other alcohols (i.e. R = p-ClC6H4, p-CH3C6H4, CH2CHCH2), which have not been reported in Scheme 5 simply because they were unreactive. In particular no reaction has been observed in the case of the propargyl alcohol 4a. Moreover, also in this case self-etherification of propargyl alcohols was not observed.
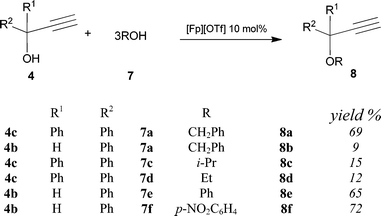 | ||
| Scheme 5 | ||
We have also found that reactions of 4c to yield the ethers 8a, 8c and 8d are accompanied by formation of small amounts (<10% yield) of the α,β-unsaturated aldehyde CHOCHCPh2, which is the enal isomeric form of 4c. The aldehyde results from the formal isomerization of the alkynol 4c, according to the well known Meyer–Schuster-type rearrangements.23 It should be remarked that the Meyer–Schuster transformation of propargylic alcohols into enals is usually performed in strong acidic media under much more drastic reaction conditions compared to those we used.24 A somewhat similar situation has been described for the ruthenium complexes [(p-cymene)Ru(OTf)(PCy3)(CO)][OTf]7d and [Ru(η3-2-C3H4Me)(CO)(dppf)][SbF6] (dppf = 1,10-bis(diphenylphosphino)ferrocene)25 which behave as efficient catalyst both in propargyl substitution and in Meyer–Schuster isomerization. The analogy with the above examples also concern the fact that the Meyer–Schuster-type rearrangement shows a marked preference for tertiary propargyl alcohols, such as the 1,1-diphenyl-2-propyn-1-ol 4c.
Since results indicates that [Fp][OTf] is a more effective catalyst in dehydrative etherification of propargyl alcohols with ferrocenylmethanol, than with other alcohols, one should argue that it preferentially promotes OH displacement from the ferrocenylmethanol 3. In order to clarify the point, a third series of reactions has been examined, combining 3 and various alcohols (Scheme 6). In particular we have considered the reactions between ferrocenylmethanol and the alcohols previously investigated in combination with propargyl alcohols (i.e. ethanol, allyl alcohol, isopropyl alcohol, benzyl alcohol and phenol). All the reactions shown in Scheme 6 were carried out for 16 h.
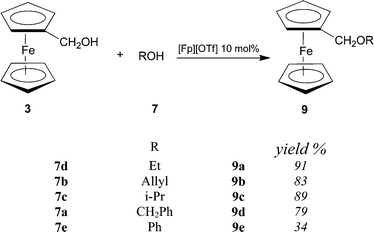 | ||
| Scheme 6 | ||
Again, the results are remarkable both in terms of conversion and selectivity: only unsymmetric ethers are formed in good yields. Etherification takes place between 3 and activated alcohols, such as allyl alcohol, and benzyl alcohol, which are more frequently involved in catalytic dehydrative coupling, but also aliphatic alcohols were found to be reactive.
Only phenol gave moderate yield, and formation of the corresponding ether 9e was accompanied by the formation, in comparable amounts (nearly 40% yield), of the symmetric ether 6. Results shown in Scheme 6 can be compared with those of a recent work describing the formation of unsymmetric ferrocenyl ethers by reaction of ferrocenyl alcohols with various aliphatic alcohols in the presence of catalytic amounts of Yb(OTf)3.5b In that case, conversions are excellent only when aliphatic alcohols are used as solvent, otherwise formation of symmetric ferrocenyl ethers prevails.
Conclusions
The iron complex [Fp][OTf] (Fp+[Fe(CO)2(Cp)]+, OTf−SO3CF3−) has proved to be an excellent catalyst in the dehydrative etherification of ferrocenylmethanol with alcohols, in particularly with propargyls.
Several factors make the method particularly convenient for the synthesis of ferrocenylmethanol-ethers: a straightforward approach with formation of water as only by-product, general applicability and good selectivity, mild reaction conditions and, more importantly, the use of a simple iron organometallic complex as an efficient catalyst.
Beside the formation of ferrocenyl ethers, [Fp][OTf] also proved to be interesting as a dehydrative etherification catalyst in the reaction of alkynols with other alcohols.
Thus, our result deliver a small contribution to the ongoing effort in the design of a sustainable bond forming process based on non toxic, readily available and inexpensive transition metal, such as iron.26
Experimental section
General
All reactions were routinely carried out under a nitrogen atmosphere, using standard Schlenk techniques. Solvents were distilled immediately before use under nitrogen from appropriate drying agents. Chromatography separations were carried out on columns of deactivated alumina (4% w/w water). Glassware was oven-dried before use. Elemental analyses were performed on a ThermoQuest Flash 1112 Series EA Instrument. ESI MS spectra were recorded on Waters Micromass ZQ 4000 with samples dissolved in CH3CN. All NMR measurements were performed on Mercury Plus 400 instrument. The chemical shifts for 1H and 13C were referenced to internal TMS. The spectra were fully assigned via DEPT experiments and 1H,13C correlation through gs-HSQC and gs-HMBC experiments. All NMR spectra were recorded at 298 K. NOE measurements were recorded using the DPFGSE-NOE sequence. NMR resonances due to a second isomeric form, when it was possible to detect them, are italicized. All the reagents were commercial products (Aldrich) of the highest purity available and used as received.Due to its high reactivity, the iron complex [Fp][OTf] was freshly prepared immediately before each reaction, by reacting [Fe2(Cp)2(CO)4] with AgOTf. A typical procedure is reported as follows.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CH] (5a).
A portion of the solution of [Fp][OTf] (2.0 mL, 0.01 mmol of catalyst) was added to a solution of ferrocenylmethanol (3) (216 mg, 1.0 mmol) and propargyl alcohol (0.09 mL, 1.5 mmol) in toluene (20 mL). The resulting mixture was stirred at room temperature. NMR spectrum of the reaction crude showed that the reaction was complete within 15 min. Solvent removal and chromatography of the residue on alumina, using petroleum ether as eluent, gave a yellow fraction, corresponding to 5a. Yield: 235 mg, 93%. Anal. C14H14FeO: C, 66.17; H, 5.55. Found: C, 66.09; H, 5.62. 1H NMR (CDCl3) δ 4.40 (s, 2H, Cp–CH2O–); 4.26 (m, 2H, Cp); 4.17 (m, 2H, Cp); 4.16 (s, 5H, Cp); 4.13 (d, 2H, 4JHH = 2.40 Hz, –OCH2–C
CH] (5a).
A portion of the solution of [Fp][OTf] (2.0 mL, 0.01 mmol of catalyst) was added to a solution of ferrocenylmethanol (3) (216 mg, 1.0 mmol) and propargyl alcohol (0.09 mL, 1.5 mmol) in toluene (20 mL). The resulting mixture was stirred at room temperature. NMR spectrum of the reaction crude showed that the reaction was complete within 15 min. Solvent removal and chromatography of the residue on alumina, using petroleum ether as eluent, gave a yellow fraction, corresponding to 5a. Yield: 235 mg, 93%. Anal. C14H14FeO: C, 66.17; H, 5.55. Found: C, 66.09; H, 5.62. 1H NMR (CDCl3) δ 4.40 (s, 2H, Cp–CH2O–); 4.26 (m, 2H, Cp); 4.17 (m, 2H, Cp); 4.16 (s, 5H, Cp); 4.13 (d, 2H, 4JHH = 2.40 Hz, –OCH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH); 2.45 (t, 1H, 4JHH = 2.40 Hz, –CCH). 13C NMR (CDCl3) δ 82.5 (Cipso Cp); 80.2 (CCH); 74.6 (CCH); 69.8, 68.9 (Cp); 68.7 (Cp free); 67.8 (–CH2O–); 56.7 (–OCH2–). ESI MS 254 [M]+; 277 [M + Na]+; 293 [M + K]+.
CH); 2.45 (t, 1H, 4JHH = 2.40 Hz, –CCH). 13C NMR (CDCl3) δ 82.5 (Cipso Cp); 80.2 (CCH); 74.6 (CCH); 69.8, 68.9 (Cp); 68.7 (Cp free); 67.8 (–CH2O–); 56.7 (–OCH2–). ESI MS 254 [M]+; 277 [M + Na]+; 293 [M + K]+.
Compounds 5b–e were prepared by the same procedure described for 5a, by reacting 3 with the propargyl alcohols 4b–e, respectively. Crystals of 5b suitable for X-ray analysis were obtained by slow evaporation from a CH2Cl2 solution.
5b Yield: 293 mg, 88%. Anal. Calcd. for C20H18FeO: C, 74.75; H, 5.49. Found: C, 74.67; H, 5.54. 1H NMR (CDCl3) δ 7.54–7.30 (m, 5H, Ph); 5.23 (d, 1H, 4JHH = 2.10 Hz, –OCH(Ph)–); 4.52, 4.46 (d, 2H, 2JHH = 11.2 Hz, Cp–CH2O–); 4.31 (m, 1H, Cp); 4.27 (m, 1H, Cp); 4.18 (m, 2H, Cp); 4.16 (s, 5H, Cp); 2.68 (d, 1H, 4JHH = 2.10 Hz, –CCH). 13C NMR (CDCl3) δ 138.6 (Cipso Ph); 128.7, 128.6, 127.6 (CPh); 83.0 (Cipso Cp); 82.1 (CCH); 75.8 (CCH); 69.9, 69.8 (Cp); 69.6 (–OCH(Ph)–); 68.9 (Cp); 68.7 (Cp free); 68.6 (Cp); 66.5 (–CH2O–). ESI MS 330 [M]+; 353 [M + Na]+; 369 [M + K]+.
5c Yield: 255 mg, 63%. Anal. Calcd. for C26H22FeO: C, 76.86; H, 5.46. Found: C, 76.94; H, 5.38. 1H NMR (CDCl3) δ 7.63–7.24 (m, 10H, Ph); 4.28 (m, 4H, Cp–CH2O– and Cp); 4.15 (br s, 7H, Cp and Cp free); 2.95 (s, 1H, –CCH). 13C NMR (CDCl3) δ 143.6 (Cipso Ph); 128.4, 127.9, 126.9 (CPh); 85.0 (Cipso Cp); 83.8 (CCH); 80.2 (–OC(Ph)2–); 77.9 (CCH); 68.9 (Cp free); 68.8, 68.3 (Cp), 63.4 (–CH2O–). ESI MS 406 [M]+; 429 [M + Na]+; 445 [M + K]+.
The synthesis of 5c is accompanied by the formation of a small amount of the ferrocenylmethyl ether [Fc–CH2–O–CH2–Fc], 6, as a by-product. Complex 6 was separated from 5c by alumina chromatography, using petroleum ether as eluent. 5c was eluted as a first yellow-orange fraction, while 6 was isolated as a second orange fraction.
6 Yield: 28 mg, 7%. Anal. Calcd. for C22H22Fe2O: C, 63.81; H, 5.35. Found: C, 63.94; H, 5.38. 1H NMR (CDCl3) δ 4.29 (br s, 4H, CH2); 4.25 (m, 4H, Cp); 4.16 (m, 4H, Cp); 4.14 (s, 10H, Cp free). 13C NMR (CDCl3) δ 84.0 (Cipso Cp); 69.6 (Cp); 68.7 (Cpfree); 68.6 (Cp); 68.2 (CH2). ESI MS 414 [M]+; 437 [M + Na]+.
5d Yield: 216 mg, 77%. Anal. Calcd. for C16H18FeO: C, 68.11; H, 6.43. Found: C, 68.19; H, 6.40. 1H NMR (CDCl3) δ 4.38 (s, 2H, Cp–CH2O–); 4.25 (m, 2H, Cp); 4.16 (s, 5H, Cp); 4.13 (m, 2H, Cp); 2.49 (s, 1H, –CCH); 1.51 (s, 6H, Me). 13C NMR (CDCl3) δ 86.6 (CCH); 84.8 (Cipso Cp); 72.3 (CCH); 70.2 (–OC(Me)2–); 69.3 (Cp); 68.7 (Cp free); 68.5 (Cp); 62.9 (–CH2O–); 29.2 (Me). ESI MS 282 [M]+; 305 [M + Na]+; 321 [M + K]+.
5e Yield: 304 mg, 75%. Anal. Calcd. for C26H22FeO: C, 76.86; H, 5.46. Found: C, 76.77; H, 5.51. 1H NMR (CDCl3) δ 8.30–7.33 (m, 10H, Ph); 5.49 (s, 1H, –OCH(Ph)–); 4.60, 4.56 (d, 2H, 2JHH = 11.2 Hz, Cp–CH2O–); 4.37 (m, 1H, Cp); 4.32 (m, 1H, Cp); 4.21 (m, 2H, Cp); 4.18 (s, 5H, Cp). 13C NMR (CDCl3) δ 139.2–123.0 (CPh); 87.8, 87.6 (CC); 83.3 (Cipso Cp); 70.6 (–OCH(Ph)–); 69.9 (Cp); 69.6 (Cp); 68.9 (Cp); 68.8 (Cp free); 68.7 (Cp); 66.6 (–CH2O–). ESI MS 406 [M]+; 429 [M + Na]+; 445 [M + K]+.
Compound 6 can be obtained also reacting 3 in the presence of a catalytic amount of Fp+, as follows.
A solution of 3 (216 mg, 1.0 mmol) in toluene (20 mL) was treated with the catalyst (20 mL of a toluene solution containing 0.1 mmol of Fp+). The resulting mixture was stirred overnight at room temperature. Solvent removal and chromatography of the residue on alumina, using petroleum ether as eluent, gave an orange fraction, corresponding to 6. Yield: 174 mg, 84%.
CH] (8a).
The catalyst (20 mL of a toluene solution containing 0.1 mmol of Fp+) was added to a solution of 4c (208 mg, 1.0 mmol) and benzyl alcohol (0.3 mL, 3.0 mmol) in toluene (20 mL). The resulting mixture was stirred overnight at room temperature. Solvent removal and chromatography of the residue on silica gel, using hexane as eluent, gave a pale yellow fraction, corresponding to 8a. Yield: 206 mg, 69%. Anal. C22H18O: C, 88.56; H, 6.08. Found: C, 88.39; H, 5.99. 1H NMR (CDCl3) δ 7.71–7.25 (m, 15H, Ph); 4.62 (br s, 2H, Ph–CH2O–); 2.98 (br s, 1H, –CCH). 13C NMR (CDCl3) δ 143.2–126.6 (CPh); 83.3 (CCH); 80.2 (–OC(Ph)2–); 77.9 (CCH); 66.8 (–CH2O–). ESI MS 298 [M]+.
The synthesis of 8a is accompanied by the formation, as a by product, of a small amount of the α,β-unsaturated aldehyde CHOCHCPh2, according to the Meyer–Schuster-type rearrangement. This latter compound was separated from 8a by chromatography on silica gel, using a mixture 10:1 (v/v) hexane/ethyl acetate. Yield: 23 mg, 11%.
Compounds 8b–f were prepared by the same procedure described for 8a, by reacting the propargyl alcohols 4b and 4c (1.0 mmol) with the corresponding alcohols 7a–f (3.0 mmol), respectively.
The reactions involving the tertiary alcohol are accompanied by the formation of the Meyer–Schuster rearrangement product in low amount (<10%). This latter compound was separated from the products 8c and 8d by chromatography on silica gel, using hexane as eluent for the ethers and a mixture 10:1 (v/v) hexane/ethyl acetate for the aldehyde.
8b Yield: 20 mg, 9%. Anal. C16H14O: C, 86.45; H, 6.35. Found: C, 86.58; H, 6.19. 1H NMR (CDCl3) δ 7.65–7.28 (m, 10H, Ph); 5.22 (d, 1H, 4JHH = 2.1 Hz, –OCH(Ph)–); 4.74, 4.66 (d, 2H, 2JHH = 11.7 Hz, Ph–CH2O–); 2.69 (d, 1H, –CCH). 13C NMR (CDCl3) δ 133.4–127.6 (CPh); 81.3 (CCH); 76.1 (CCCH); 69.6 (–OCH(Ph)–); 66.9 (–CH2O–). ESI MS 222 [M]+.
8c Yield: 38 mg, 15%. Anal. C18H18O: C, 86.36; H, 7.24. Found: C, 86.21; H, 7.19. 1H NMR (CDCl3) δ 7.60–7.22 (m, 10H, Ph); 4.05 (m, 1H, 3JHH = 6.2 Hz, –CH(CH3)2); 2.86 (br s, 1H, –CCH); 1.15 (d, 6H, 3JHH = 6.2 Hz, –CH(CH3)2). 13C NMR (CDCl3) δ 144.6, 128.2, 127.7, 127.3 (CPh); 84.8 (CCH); 79.5 (–OC(Ph)2–); 77.4 (CCH); 68.7 (–CH(CH3)2); 24.1 (–CH(CH3)2). ESI MS 250 [M]+.
8d Yield: 28 mg, 12%. Anal. C17H16O: C, 86.40; H, 6.82. Found: C, 86.54; H, 6.69. 1H NMR (CDCl3) δ 7.60–7.22 (m, 10H, Ph); 3.56 (q, 2H, 3JHH = 6.8 Hz, –CH2–); 2.87 (s, 1H, –CCH); 1.28 (t, 3H, 3JHH = 6.8 Hz, –CH3). 13C NMR (CDCl3) δ 143.6, 128.4, 127.8, 126.7 (CPh); 83.8 (CCH); 80.1 (–OC(Ph)2–); 77.4 (CCH); 60.6 (–CH2–); 15.6 (–CH3). ESI MS 236 [M]+.
8e Yield: 135 mg, 65%. Anal. C15H12O: C, 86.51; H, 5.81. Found: C, 86.38; H, 5.89. 1H NMR (CDCl3) δ 7.63–7.04 (m, 10H, Ph); 5.66 (d, 1H, 4JHH = 2.1 Hz, –OCH(Ph)–); 2.67 (d, 1H, –CCH). 13C NMR (CDCl3) δ 140.6–127.4 (CPh); 84.9 (CCH); 73.4 (CCH). ESI MS 208 [M]+.
8f Yield: 182 mg, 72%. Anal. C15H11NO3: C, 71.14; H, 4.38. Found: C, 71.25; H, 4.30. 1H NMR (CDCl3) δ 7.67–7.17 (m, 9H, Ph); 5.94, 5.68, 5.28 (d, 1H, 4JHH = 2.1 Hz, –OCH(Ph)–); 2.79, 2.73, 2.68 (d, 1H, 4JHH = 2.1 Hz, –CCH). 13C NMR (CDCl3) δ 142.4–126.1 (CPh); 81.7, 81.3, 79.9 (CCH); 78.1, 76.6, 76.2 (CCH); 70.5, 69.6, 69.1 (–OCH(Ph)–). ESI MS 253 [M]+.
Compounds 9b–e were prepared by the same procedure described for 9a, by reacting ferrocenylmethanol with the alcohols 7b–e, respectively.
9b Yield: 213 mg, 83%. Anal. Calcd. for C14H16FeO: C, 65.65; H, 6.30. Found: C, 65.67; H, 6.44. 1H NMR (CDCl3) δ 5.98–5.87 (m, 1H, –CH); 5.33–5.17 (m, 2H, CH2); 4.30 (s, 2H, Cp–CH2O–); 4.25 (m, 2H, Cp); 4.16 (m, 2H, Cp); 4.14 (s, 5H, Cp); 3.99 (m, 2H, –OCH2–). 13C NMR (CDCl3) δ 135.2 (–CH); 117.1 (–CH); 83.7 (Cipso Cp); 71.0 (–OCH2–); 69.7 (Cp); 68.7 (Cp free); 68.5 (Cp–CH2O–). ESI MS 256 [M]+; 279 [M + Na]+; 295 [M + K]+.
9c Yield: 229 mg, 89%. Anal. Calcd. for C14H18FeO: C, 65.14; H, 7.03. Found: C, 65.27; H, 6.94. 1H NMR (CDCl3) δ 4.27 (s, 2H, Cp–CH2O–); 4.24 (m, 2H, Cp); 4.12 (s, 5H, Cp); 4.08 (m, 2H, Cp); 3.66 (m, 1H, 3JHH = 6.2 Hz, –CH(CH3)2); 1.16 (d, 6H, 3JHH = 6.2 Hz, –CH(CH3)2). 13C NMR (CDCl3) δ 84.6 (Cipso Cp); 70.6 (–CH(CH3)2); 69.5, 68.8 (Cp); 68.6 (Cp free); 66.5 (–CH2O–); 22.4 (–CH(CH3)2). ESI MS 258 [M]+; 281 [M + Na]+; 297 [M + K]+.
9d Yield: 242 mg, 79%. Anal. Calcd. for C18H18FeO: C, 70.61; H, 5.93. Found: C, 70.47; H, 5.94. 1H NMR (CDCl3) δ 7.74–7.35 (m, 5H, Ph); 4.63 (br s, 2H, Ph–CH2O–); 4.45 (s, 2H, Cp–CH2O–); 4.36 (m, 2H, Cp); 4.26 (m, 2H, Cp); 4.22 (s, 5H, Cp). 13C NMR (CDCl3) δ 134.2–125.3 (CPh); 84.2 (Cipso Cp); 72.0 (Ph–CH2O–); 69.6, 68.7 (Cp); 68.6 (Cp free); 68.3 (Cp–CH2O–). ESI MS 306 [M]+; 329 [M + Na]+; 345 [M + K]+.
9e Yield: 98 mg, 34%. Anal. Calcd. for C17H16FeO: C, 69.89; H, 5.52. Found: C, 70.01; H, 5.44. 1H NMR (CDCl3) δ 7.54–7.04 (m, 5H, Ph); 4.46 (s, 2H, Cp–CH2O–); 4.13 (s, 5H, Cp); 4.08 (m, 2H, Cp); 3.98 (m, 2H, Cp). 13C NMR (CDCl3) δ 135.1–126.9 (CPh); 85.1 (Cipso Cp); 68.9 (Cp free); 68.5, 68.1 (Cp); 67.1 (Cp–CH2O–). ESI MS 292 [M]+; 315 [M + Na]+; 331 [M + K]+.
| Formula | C20H18FeO |
|---|---|
| Fw | 330.19 |
| λ/Å | 0.71073 |
| T/K | 295(2) |
| Crystal system | Monoclinic |
| Space group | P21 |
| a/Å | 7.418(2) |
| b/Å | 10.583(3) |
| c/Å | 10.003(3) |
| β (°) | 98.473(3) |
| Cell volume/Å3 | 776.7(4) |
| Z | 2 |
| D c/g cm−3 | 1.412 |
| μ/mm−1 | 0.969 |
| F(000) | 344 |
| θ limits (°) | 2.06–26.00 |
| Reflections collected | 7282 |
| Independent reflections | 3028 (Rint = 0.0425) |
| Data/restraints/parameters | 3028/133/200 |
| Goodness on fit on F2 | 0.990 |
| R 1 (I > 2σ(I)) | 0.0385 |
| wR2 (all data) | 0.0920 |
| Absolute structure parameter | 0.05(2) |
| Largest diff. peak and hole, e Å−3 | 0.801 and −0.308 |
Acknowledgements
We thank the Ministero dell'Università e della Ricerca Scientifica e Tecnologica (M.U.R.) and the University of Bologna for financial support.References
- (a) L. Busetto, F. Marchetti, R. Mazzoni, M. Salmi, S. Zacchini and V. Zanotti, Organometallics, 2009, 28, 3465–3472 CrossRef CAS; (b) L. Busetto, F. Marchetti, R. Mazzoni, M. Salmi, S. Zacchini and V. Zanotti, Chem. Commun., 2010, 46, 3327–3329 RSC.
- (a) Ferrocenes - Ligands Materials and Biomolecules, ed. P. Štěpnička, John Wiley & Sons, West Sussex, 2008 Search PubMed; (b) Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science, ed. A. Togni, T. Hayashi, Wiley-VCH, Weinheim, Germany, 1995 Search PubMed.
- L. Busetto, R. Mazzoni, M. Salmi, S. Zacchini and V. Zanotti, Organometallics, 2012, 31, 2667–2674 CrossRef CAS.
- (a) A. W. Williamson, J. Chem. Soc., 1852, 4, 229 Search PubMed; (b) N. Baggett, in Comprehensive Organic Synthesis, ed. D. Barton and W. D. OllinsPergaman, Oxford, 1979, vol. 1 Search PubMed.
- (a) K. Ohta, E. Koketsu, Y. Nagase, N. Takahashi, H. Watanabe and M. Yoshimatsu, Chem. Pharm. Bull., 2011, 59, 1133–1140 CrossRef CAS; (b) R. Jiang, Y. Shen, X. Xu, Y. Zhang, J. Shao and S. Ji, Chin. J. Chem., 2011, 29, 1887–1893 CrossRef CAS; (c) A. Corma and M. Renz, Angew. Chem., Int. Ed., 2007, 46, 298–300 CrossRef CAS; (d) G. V. M. Sharma, T. R. Prasad and A. K. Mahalingam, Tetrahedron Lett., 2001, 42, 759–761 CrossRef CAS.
- (a) J. Muzart, Tetrahedron, 2008, 64, 5815–5849 CrossRef CAS; (b) V. V. Namboodiri and R. S. Varma, Tetrahedron Lett., 2002, 43, 4593–4595 CrossRef CAS; (c) T. Mitsudome, T. Matsuno, S. Sueoka, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2012, 14, 610–613 RSC.
- (a) Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11020 CrossRef CAS; (b) Y. Nishibayashi, M. D. Milton, Y. Inada, M. Yoshikawa, I. Wakiji, M. Hidai and S. Uemura, Chem.–Eur. J., 2005, 11, 1433–1451 CrossRef CAS; (c) V. Cadierno, J. Díez, S. E. García-Garrido and J. Gimeno, Chem. Commun., 2004, 2716–2717 RSC; (d) E. Bustelo and P. H. Dixneuf, Adv. Synth. Catal., 2007, 349, 933–942 CrossRef CAS; (e) J. A. van Rijn, M. C. Guijt, D. de Vries, E. Bouwman and E. Drent, Appl. Organomet. Chem., 2011, 25, 212–219 CrossRef CAS; (f) J. A. van Rijn, M. Lutz, L. S. von Chrzanowski, A. L. Spek, E. Bouwman and E. Drent, Adv. Synth. Catal., 2009, 351, 1637–1647 CrossRef CAS; (g) J. A. van Rijn, M. A. Siegler, A. L. Spek, E. Bouwman and E. Drent, Organometallics, 2009, 28, 7006–7014 CrossRef CAS.
- (a) H. A. Kalviri, C. F. Petten and F. M. Kerton, Chem. Commun., 2009, 5171–5173 RSC; (b) Y. Kayaki, T. Koda and T. Ikariya, J. Org. Chem., 2004, 69, 2595–2597 CrossRef CAS; (c) S. Bouquillon, F. Henin and J. Muzart, Organometallics, 2000, 19, 1434–1437 CrossRef CAS.
- (a) M. Rosenblum, A. Bucheister, T. C. T. Chang, M. Cohen, M. Marsi, S. B. Samuels, D. Scheck, N. Sofen and J. C. Watkins, Pure Appl. Chem., 1984, 56, 129–136 CrossRef CAS; (b) D. L. Reger and C. Coleman, J. Organomet. Chem., 1977, 131, 153–162 CrossRef CAS.
- (a) Y. Nishibayashi, Y. Inada, M. Hidai and S. Uemura, J. Am. Chem. Soc., 2002, 124, 7900–7901 CrossRef CAS; (b) Y. Nishibayashi, H. Imajima, G. Onodera, M. Hidai and S. Uemura, Organometallics, 2004, 23, 26–30 CrossRef CAS; (c) Y. Miyake, S. Endo, M. Yuki, Y. Tanabe and Y. Nishibayashi, Organometallics, 2008, 27, 6039–6042 CrossRef CAS.
- V. Cadierno, S. E. García-Garrido, J. Gimeno and N. Nebra, Inorg. Chim. Acta, 2010, 363, 1912–1934 CrossRef CAS.
- (a) A. I. F. Venancio, M. F. C. G. da Silva, L. M. D. R. S. Martins, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Organometallics, 2005, 24, 4654–4665 CrossRef CAS and references therein; (b) J. P. Selegue, Coord. Chem. Rev., 2004, 248, 1543–1563 CrossRef CAS; (c) C. Gauss, D. Veghini, O. Orama and H. Berke, J. Organomet. Chem., 1997, 541, 19–38 CrossRef CAS.
- E. Bustelo and P. H. Dixneuf, Adv. Synth. Catal., 2005, 347, 393–397 CrossRef CAS.
- (a) K. M. Nicholas, Acc. Chem. Res., 1987, 20, 207–214 CrossRef CAS; (b) J. R. Green, Curr. Org. Chem., 2001, 5, 809–826 CrossRef CAS; (c) B. J. Teobald, Tetrahedron, 2002, 58, 4133–4170 CrossRef CAS.
- (a) B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076–6077 CrossRef CAS; (b) M. R. Luzung and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 15760–15761 CrossRef CAS; (c) J. J. Kennedy-Smith, L. A. Young and F. D. Toste, Org. Lett., 2004, 6, 1325–1327 CrossRef CAS; (d) R. V. Ohri, A. T. Radosevich, K. J. Hrovat, C. Musich, D. Huang, T. R. Holman and F. D. Toste, Org. Lett., 2005, 7, 2501–2504 CrossRef CAS.
- M. Zhang, H. Yang, Y. Cheng, Y. Zhu and C. Zhu, Tetrahedron Lett., 2010, 51, 1176–1179 CrossRef CAS.
- (a) M. Georgy, V. Boucard and J. M. Campagne, J. Am. Chem. Soc., 2005, 127, 14180–14181 CrossRef CAS; (b) M. Georgy, V. Boucard, O. Debleds, C. D. Zotto and J. M. Campagne, Tetrahedron, 2009, 65, 1758–1766 CrossRef CAS.
- Z. P. Zhan, W. Z. Yang, R. F. Yang, J. L. Yu, J. P. Li and H. J. Liu, Chem. Commun., 2006, 3352–3354 RSC.
- J. S. Yadav, B. V. S. Reddy, K. V. R. Rao and G. G. K. S. N. Kumar, Synthesis, 2007, 3205–3210 CrossRef CAS.
- H. H. Hui, Q. Zhao, M. Y. Yang, D. B. She, M. Chen and G. Huang, Synthesis, 2008, 191–196 CAS.
- Z. P. Zhan, J. L. Yu, H. J. Liu, Y. Y. Cui, R. F. Yang, W. Z. Yang and J. P. Li, J. Org. Chem., 2006, 71, 8298–8301 CrossRef CAS.
- See for example: (a) R. Jiang, Y. Zhang, Y. C. Shen, X. Zhu, X. P. Xu and S. J. Ji, Tetrahedron, 2010, 66, 4073–4078 CrossRef CAS; (b) P. Vicennati and P. G. Cozzi, Eur. J. Org. Chem., 2007, 2248–2253 CrossRef CAS; (c) R. Jiang, X.-Q. Chu, X.-P. Xu, B. Wu and S.-J. Ji, Aust. J. Chem., 2011, 64, 1530–1537 CrossRef CAS.
- K. H. Meyer and K. Schuster, Ber. Dtsch. Chem. Ges., 1922, 55, 819–823 CrossRef.
- S. Swaminathan and K. V. Narayanan, Chem. Rev., 1971, 71, 429–438 CrossRef CAS.
- V. Cadierno, J. Díez, S. E. García-Garrido and J. Gimeno, Chem. Commun., 2004, 2716–2717 RSC.
- Selected examples include (a) Iron Catalysis in Organic Chemistry, ed. B. Plietker, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (c) B. D. Sherry and A. Fürstner, Acc. Chem. Res., 2008, 41, 1500–1511 CrossRef CAS; (d) R. M. Bullock, Angew. Chem., Int. Ed., 2007, 46, 7360–7363 CrossRef CAS; (e) G. Cahiez, V. Habiak, C. Duplais and A. Moyeux, Angew. Chem., Int. Ed., 2007, 46, 4364–4366 CrossRef CAS; (f) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS.
- G. M. Sheldrick, SADABS, Program for empirical absorption correction, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, SHELX97, Program for crystal structure determination, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † CCDC reference number 874558. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20708g |
| This journal is © The Royal Society of Chemistry 2012 |