Carbohydrate nitrone and nitrile oxide cycloaddition approach to chiral sulfur heterocycles and nucleosides†
Subhrangshu
Mukherjee
,
Sukhendu B.
Mandal
* and
Anup
Bhattacharjya
*
Chemistry Department, Indian Institute of Chemical Biology (a unit of CSIR), 4, Raja S. C. Mullick Road, Kolkata, 700032, India
First published on 26th July 2012
Abstract
S-Alkenyl and -alkynyl carbohydrate derivatives were prepared from 1,2:5,6-O-diisopropylidene-α-D-allofuranose. Nitrones and nitrile oxides generated from these derivatives led to the formation of 6, 7, and 11-membered chiral sulfur heterocycles fused to isoxazolidine, isoxazoline and isoxazole rings. Some of these sulfur heterocycles were converted to nucleosides. The 11-memebered sulfur compound was found to gelate hydrocarbon solvents.
One of the important methods adopted for the synthesis of enantiomerically pure compounds is the chiral pool strategy, which is based on the use of enantiomerically pure natural products as starting materials. In this context, the preference for using carbohydrates from the chiral pool is generally accepted. Carbohydrate derivatives have been the source of a vast number of chiral molecules of varying nature and structure.1 A few years ago we reported the synthesis of chiral cyclic ethers and amines by the application of O- and N-alkenyl carbohydrate nitrone and nitrile oxide cycloadditions,2 and the cycloaddition strategy proved to be an important method for the synthesis of chiral heterocycles.3 Carbohydrate derived nitrones and nitrile oxides have also been used by others for the synthesis of enantiopure compounds.3c However, the synthesis of chiral sulfur heterocycles by S-alkenyl or S-alkynyl nitrone and nitrile oxide cycloaddition strategies has remained hitherto unaccomplished. It should be mentioned that a large number of sulfur heterocycles are known to have both biological and synthetic uses.4 As examples, sulthiame and brinzolamide are drugs used for epilepsy and glaucoma, respectively, while Oppolzer's sultam is a well-known chiral auxiliary used in synthetic chemistry. We herein reveal the successful realization of the S-alkenyl carbohydrate nitrone and nitrile oxide cycloaddition leading to the synthesis of chiral sulfur heterocycles.
1,2:5,6-O-Diisopropylidene-α-D-allofuranose (1)5 was converted to the 3-thioacetyl glucose derivative 2 by employing the Mitsunobu reaction using thioacetic acid (Scheme 1). Treatment of 2 with NaOMe in the presence of NaBH4 led to removal of the thioacetyl group and concomitant allylation, giving the S-allyl carbohydrate derivative 3 in 70% yield. The addition of NaBH4 was found to be beneficial for preventing the oxidation of the thiolate anion generated during the reaction. The introduction of the allyl group was evident from the characteristic olefinic 1H NMR signals for the vinyl moiety. Removal of the 5,6-isopropylidene group followed by vicinal diol cleavage by NaIO4 afforded the aldehyde 4, in which the sulfide moiety also underwent oxidation to sulfoxide, which was evident from the mass spectrum of the product. Immediately after its formation, 4 was treated with N-PhNHOH in toluene under reflux, and isoxazolidines 6 and 7 fused to thiane and thiepane rings, respectively, were obtained as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture in 74% yield via the intermediate nitrone 5. The high resolution mass spectra of the compounds indicated that the compounds were present as sulfoxides. The appearance of a set of a one-proton doublet at δ 2.23 and a one-proton multiplet at δ 2.08–2.13 in the 1H NMR spectrum was a clear indication of the bridged isoxazolidine nature of 7.2a,3d The stereochemistry of the newly formed carbon chiral centres was based on the similarity of the chemical shifts of the aforementioned bridge methylene protons with those occurring in analogous oxygen heterocycles;2a,3d however, the sulfoxide stereochemistry remained unestablished, although it should be mentioned that only one diastereomer involving sulfoxide chirality was obtained in the reaction. The fused isoxazolidine 6 was characterized by the appearance of a one-photon multiplet at δ 3.42 due to the isoxazolidine ring-juncture proton. The assignment of stereochemistry of the newly formed chiral centres was problematic, and analysis of the NOESY spectrum was of no help. However, it could be settled by analysis of the product obtained by cleavage of the isoxazolidine ring as described below. Again, the stereochemistry of the sulfoxide group in 6, which was obtained as a single sulfoxide diastereomer, remained unestablished. The cycloaddition of the S-allylnitrone 5 behaved differently from the corresponding N-Ph-O-allyl nitrone reported previously.2e,3d Bridged isoxazolidines were obtained predominantly in the case of O-allyl nitrones, whereas the cycloaddition of the nitrone 5 led to both bridged and fused isoxazolidines in equal ratios, and so was not regioselective. The two cycloaddition modes leading to 6 and 7 are depicted in Scheme 1. Previously, it was reported that increasing steric demands favoured the formation of fused isoxazolidines. It is possible in the present case that the presence of the bulkier sulfur atom led to the increased formation of the six-membered ring. Treatment of 6 with Mo(CO)6 resulted in the cleavage of the isoxazolidine rings with concomitant deoxygenation of the sulfoxide moiety giving rise to 8 (92%) (Scheme 2).6,7 The 1H NMR spectrum of 8 exhibited one of the S–C(7)H2 protons as a triplet with J = 13.8 Hz, which indicated the diaxial relationship of this proton with 6-H. Apart from this fact, the absence of any significant coupling for the 5-H signal (broad singlet) pointed to the conformation depicted in Fig. 1 for 8. Thus, the cis-α stereochemistry of 5C–6C in 8 suggested a cis-α ring juncture for its precursor 6 as shown. Treatment of 7 with Mo(CO)6 led to the formation of 9 in 85% yield. The 1H and 13C NMR spectra, as well as the mass spectrum of 9, were in agreement with the deoxygenated ring-opened structure. The C–CH2–C signals appeared as a set of a multiplet and a doublet at δ 1.60–1.71 (m, 1H) and 2.10 (br d, 1H, J = 14.4 Hz), whereas S–CH2 signals appeared as a set of multiplets at δ 2.54 (dd, 1H, J = 10.8, 14.1 Hz) and 3.03 (ddd, 1H, J = 1.8, 4.2, 14.0 Hz). In addition, two methylene carbon signals at δ 39.9 and 40.6 were consistent with the structure of 9.
:1 mixture in 74% yield via the intermediate nitrone 5. The high resolution mass spectra of the compounds indicated that the compounds were present as sulfoxides. The appearance of a set of a one-proton doublet at δ 2.23 and a one-proton multiplet at δ 2.08–2.13 in the 1H NMR spectrum was a clear indication of the bridged isoxazolidine nature of 7.2a,3d The stereochemistry of the newly formed carbon chiral centres was based on the similarity of the chemical shifts of the aforementioned bridge methylene protons with those occurring in analogous oxygen heterocycles;2a,3d however, the sulfoxide stereochemistry remained unestablished, although it should be mentioned that only one diastereomer involving sulfoxide chirality was obtained in the reaction. The fused isoxazolidine 6 was characterized by the appearance of a one-photon multiplet at δ 3.42 due to the isoxazolidine ring-juncture proton. The assignment of stereochemistry of the newly formed chiral centres was problematic, and analysis of the NOESY spectrum was of no help. However, it could be settled by analysis of the product obtained by cleavage of the isoxazolidine ring as described below. Again, the stereochemistry of the sulfoxide group in 6, which was obtained as a single sulfoxide diastereomer, remained unestablished. The cycloaddition of the S-allylnitrone 5 behaved differently from the corresponding N-Ph-O-allyl nitrone reported previously.2e,3d Bridged isoxazolidines were obtained predominantly in the case of O-allyl nitrones, whereas the cycloaddition of the nitrone 5 led to both bridged and fused isoxazolidines in equal ratios, and so was not regioselective. The two cycloaddition modes leading to 6 and 7 are depicted in Scheme 1. Previously, it was reported that increasing steric demands favoured the formation of fused isoxazolidines. It is possible in the present case that the presence of the bulkier sulfur atom led to the increased formation of the six-membered ring. Treatment of 6 with Mo(CO)6 resulted in the cleavage of the isoxazolidine rings with concomitant deoxygenation of the sulfoxide moiety giving rise to 8 (92%) (Scheme 2).6,7 The 1H NMR spectrum of 8 exhibited one of the S–C(7)H2 protons as a triplet with J = 13.8 Hz, which indicated the diaxial relationship of this proton with 6-H. Apart from this fact, the absence of any significant coupling for the 5-H signal (broad singlet) pointed to the conformation depicted in Fig. 1 for 8. Thus, the cis-α stereochemistry of 5C–6C in 8 suggested a cis-α ring juncture for its precursor 6 as shown. Treatment of 7 with Mo(CO)6 led to the formation of 9 in 85% yield. The 1H and 13C NMR spectra, as well as the mass spectrum of 9, were in agreement with the deoxygenated ring-opened structure. The C–CH2–C signals appeared as a set of a multiplet and a doublet at δ 1.60–1.71 (m, 1H) and 2.10 (br d, 1H, J = 14.4 Hz), whereas S–CH2 signals appeared as a set of multiplets at δ 2.54 (dd, 1H, J = 10.8, 14.1 Hz) and 3.03 (ddd, 1H, J = 1.8, 4.2, 14.0 Hz). In addition, two methylene carbon signals at δ 39.9 and 40.6 were consistent with the structure of 9.
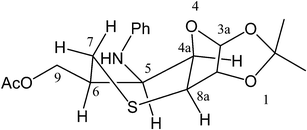 | ||
| Fig. 1 Conformation of 8 consistent with observed J values. | ||
 | ||
| Scheme 1 3-S-Allyl carbohydrate nitrone cycloaddition. | ||
 | ||
| Scheme 2 Cleavage of isoxazolidine ring in 6 and 7. | ||
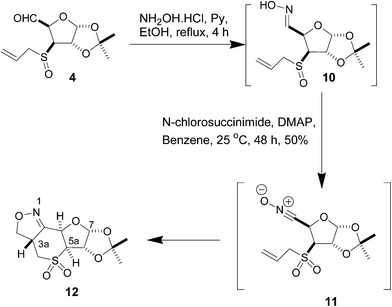
In a different approach to six-membered sulfur heterocycles, nitrile oxide cycloaddition was applied to 3-S-allyl and 3-S-propargyl carbohydrate derivatives. The aldehyde 4, upon treatment with hydroxylamine, furnished the oxime 10, which, without isolation or purification, was treated with N-chlorosuccinimide, and the nitrile oxide 11 formed as an intermediate underwent cycloaddition to lead to the isoxazoline 12 fused to a thian ring as the exclusive product in 50% yield. The sulfoxide moiety in 10 was oxidized to sulfone during the reaction (Scheme 3). The appearance of a quaternary carbon signal at δ 151.5 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N in the 13C NMR spectrum and a one-proton multiplet at δ 4.13–4.22 in the 1H NMR spectrum of 12 was indicative of the isoxazoline structure. The final confirmation of the structure and the stereochemistry of the newly formed chiral centre came from the X-ray crystallographic analysis of 12 (Fig. 2).
N in the 13C NMR spectrum and a one-proton multiplet at δ 4.13–4.22 in the 1H NMR spectrum of 12 was indicative of the isoxazoline structure. The final confirmation of the structure and the stereochemistry of the newly formed chiral centre came from the X-ray crystallographic analysis of 12 (Fig. 2).
 | ||
| Fig. 2 ORTEP diagram of 12. | ||
 | ||
| Scheme 3 3-S-Allyl carbohydrate nitrile oxide cycloaddition. | ||
The 3-S-propargyl carbohydrate derivative 13 was prepared in the same manner as the corresponding allyl derivative 3, and deprotected by acid treatment to the diol 14, which was subjected to vicinal diol cleavage by NaIO4, followed by treatment with hydroxylamine to give the oxime 15 (Scheme 4). Unlike the sulfide 3, 13 did not undergo oxidation to sulfoxide during the reaction, as was evident from the mass spectrum of 15, which did not exhibit any peak corresponding to the oxygenated product. The reason for this difference in reactivity is not known to us. The oxime 15 derived from 14 was treated with N-chlorosuccinimide, and the nitrile oxide intermediate 16 underwent a cycloaddition reaction to afford the isoxazole 17 in 45% yield. The mass spectrum of 17 did not indicate the introduction of any oxygen atom on the sulfur atom. The appearance of a quaternary carbon signal at δ 157.0 in the 13C NMR spectrum and a downfield one-proton singlet at δ 8.28 in the 1H NMR spectrum coupled with the molecular ion at m/z 255 in the mass spectrum was ample evidence for the isoxazole structure of 17.
 | ||
| Scheme 4 3-S-Propargyl carbohydrate nitrile oxide cycloaddition. | ||
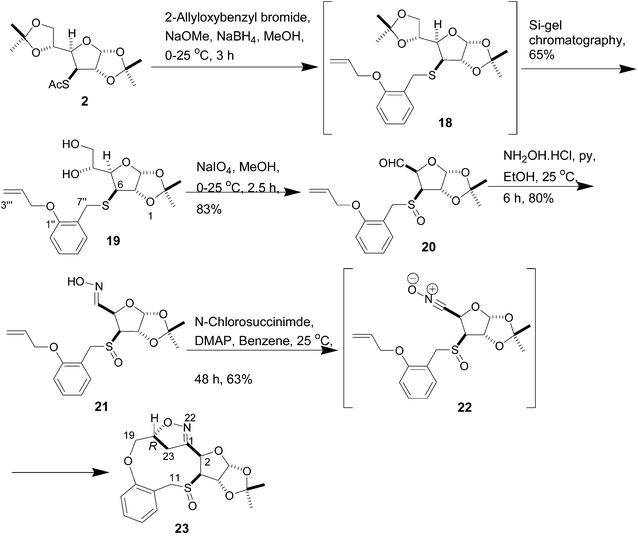
The nitrile oxide cycloaddition strategy was next applied to a tethered-O-alkenyl carbohydrate derivative in order to synthesize sulfur rings larger than six or seven.8 The thioacetyl derivative 2 was treated with NaOMe and NaBH4 in the presence of 2-allyloxybenzyl bromide. The product 18 was selectively deprotected during Si-gel chromatographic purification giving rise to 19, which was cleaved by NaIO4 to give the aldehyde 20. The oxime 21 was prepared from 20 by treatment with hydroxylamine hydrochloride in the presence of pyridine, and was used without purification for the next step. The mass spectrum of 21 exhibited a M + Na peak at m/z 404, which indicated that the sulfur atom in S-alkenyl derivative 14 underwent oxidation to SO during NaIO4 treatment. The oxime 21 was next treated with N-chlorosuccinimide to afford the 11-membered sulfur heterocycle 23 as the exclusive product via the nitrile oxide 22 in 63% yield (Scheme 5). Unlike 10, oxime 21 was not oxidized to the corresponding sulfone during treatment with N-chlorosuccinimide. The presence of the bridged isoxazoline moiety was evident from the large geminal coupling constant (J = 18.0 Hz) of the bridge methylene protons (δ 3.38 and 3.63), which was characteristic of bridge methylene protons present in similar structures.8 The assignment of stereochemistry of the newly formed chiral centre was difficult, because the NOESY spectrum of 23 did not exhibit any cross peak, which could correlate the newly formed isoxazoline chiral center with any existing chiral centers. The formation of crystals required for X-ray diffraction analysis was precluded by the fact that 23 formed an organogel in some of the solvents used for crystallization. Comparison of structures of 23 with R configuration of the newly formed chiral centre and its epimer by molecular modeling using Chem3D revealed that in the case of the alternative S configuration,9 the distances (3.03 Å and 3.45 Å) between the bridge methylene protons 23-H's and the existing chiral centre 2-H are more favourable for noticeable NOE correlation (Fig. 3). Since no such NOE was observed, the newly formed chiral centre in 23 was assigned R configuration. It should be stressed that the lack of NOE data for compound 23 is not conclusive proof of stereochemistry, and so the assigned stereochemistry should be regarded as purely tentative. Due to the formation of 23 as the exclusive product as well as the absence of the epimer of 23, comparison of NOE data of 23 and its epimer was not possible. Instead, a good indication of the NOE of the other epimer was obtained by the modeling study described above. As mentioned earlier, 23 was observed to possess organogelation properties, and was able to gelate hydrocarbon solvents such as pentane, hexane, heptanes, n-octane, i-octane and cyclohexane at room temperature with varying efficiency. Although this property of 23 has not been systematically investigated so far, an AFM image of the gel of 23 in cyclohexane is shown in Fig. 4 as preliminary evidence.
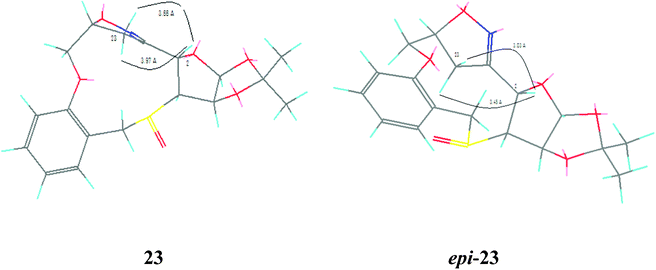 | ||
| Fig. 3 Energy-minimised Chem3D structures of 23; left-R, right-S. | ||
 | ||
| Fig. 4 AFM image of cyclohexane gel of 23 (22 mg mL−1 in cyclohexane). | ||
 | ||
| Scheme 5 Nitrile oxide cycloaddition route to medium-ring sulfur heterocycles. | ||
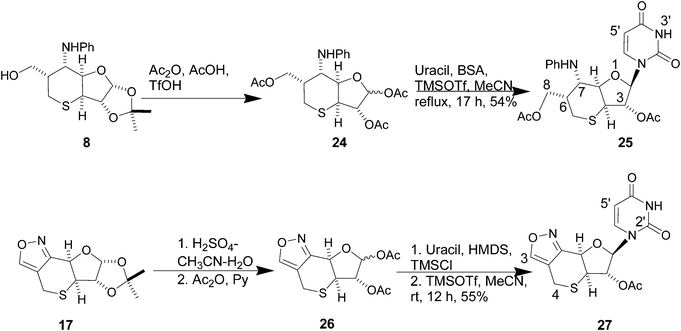
In view of the importance of bicyclic perhydrofuropyran nucleosides10 and other modified bicyclic nucleoside analogues,11 we planned to carry out glycosidation reactions with a pyrimidine base on some of the above furanose-appended sulfur heterocycles, which could generate conformationally restricted potential nucleoside analogues with improved recognition as well as binding to RNA/DNA.12 Towards this end, the six-membered thiane derivative 8 was deprotected in a mixture of trifluoromethane sulfonic acid, acetic acid and acetic anhydride to give the triacetate 24 as an anomeric mixture, as evident from the 1H NMR spectrum of the crude acetates. Application of the Vorbrüggen nucleosidation13 method to 24 resulted in the formation of the uracil nucleoside 25 in 54% yield. The mass spectrum of 25 was consistent with the structure. The 1H NMR spectrum exhibited the uracil olefinic protons as doublets at δ 5.66 and δ 7.81 as expected. The anomeric proton appeared as a doublet at δ 5.60. The 13C NMR spectrum was also in agreement with the structure. The uracil CO carbon signals appeared at δ 150.9 and δ 162.7. Similarly, the nucleoside 27 was prepared (55%) via the acetate 26 from 10 (Scheme 6) and characterized by mass, 1H and 13C NMR spectra. The isoxazole proton signal appeared at δ 8.38, and the uracil olefinic protons appeared as doublets at δ 5.68 and δ 7.46. The characteristic quaternary carbon signals due to the isoxazole CN and CC appeared at δ 150.1 (C) and δ 111.7 respectively, whereas the uracil CO carbon signals appeared at δ 156.2 and δ 162.9.
 | ||
| Scheme 6 Conversion of sulfur heterocycles to nucleosides. | ||
In conclusion, the above work demonstrated that the S-allyl, -propargyl and -tethered allyl carbohydrate nitrone and nitrile oxide cycloaddition strategy is a potentially useful strategy to synthesize chiral six- and seven-membered as well as medium-ring S-heterocycles. The presence of the isopropylidene protected furanoside rings in these heterocycles makes them useful also for the synthesis of novel nucleosides. The present work also established that carbohydrate nitrone and nitrile oxide cycloaddition is an efficient procedure for chiral N-, O- and S-heterocycles in general.
Experimental
General
1H and 13C NMR spectra were recorded at the indicated field strengths using 300 MHz and 600 MHz instruments. Reactions were monitored by thin layer chromatography using a Merck 60 F254 precoated silica gel plate (No. 1.05554). Organic extracts were dried over anhydrous sodium sulphate. For routine column chromatography 60–120 mesh silica gel (SRL, India) and for flash chromatography 230–400 mesh silica gel (Merck, grade 60) were used. Solvents were distilled and dried immediately prior to use. Unless otherwise mentioned petroleum ether refers to a fraction boiling between 60 and 80 °C. Room temperature refers to 25 °C.S-{(3aR,5R,6S,6aS)-5-[(4R)-2,2-Dimethyl-1,3-dioxolan-4-yl]-2,2-dimethyltetrahydro-furo[2,3-d][1,3]dioxol-6-yl} ethanethioate (2)
Diisopropyl azodicarboxylate (DIAD) (15.7 mL, 79.6 mmol) was added to an efficiently stirred solution of triphenylphosphine (17.2 g, 65.7 mmol) in THF (180 mL) at 0 °C. After 30 min stirring, a white precipitate was formed. To this heterogeneous mixture, a solution of a mixture of 1 (9.00 g, 34.6 mmol) and thioacetic acid (4.9 mL, 69.2 mmol) in THF (75 mL) was slowly added over a period of 10 min, and the mixture was stirred for an additional 2 h at 0 °C and 6 h at room temperature. During this time the precipitate dissolved and a clear yellow solution was obtained. The solvent was evaporated in vacuo and the solid residue was dissolved in hot toluene (50 mL), and the solution was kept in a freezer overnight to precipitate out most of the triphenylphosphine oxide formed. The solid was filtered off and washed with cold toluene (25 mL). The solvent toluene was evaporated, and the crude product was purified by silica gel (100–200 mesh) column chromatography using petroleum ether:EtOAc (93:7) as the eluent to give 2 (8.6 g, 78%) as a yellowish viscous oil; [α]25D −41 (c 0.16, CHCl3); IR (neat) νmax: 1698, 1431, 1377, 1213, 1068 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.31 (s, 3H, C–CH3), 1.34 (s, 3H, C–CH3), 1.42 (s, 3H, C–CH3), 1.54 (s, 3H, C–CH3), 2.40 (s, 3H, –COCH3), 3.99–4.02 (m, 1H, H-4′), 4.08–4.11 (m, 2H, H-5′a,5′b), 4.14 (d, 1H, J = 3.9 Hz, H-6), 4.31 (dd, 1H, J = 3.9, 7.5 Hz, H-5), 4.55 (d, 1H, J = 3.6 Hz, H-6a), 5.80 (d, 1H, J = 3.6 Hz, H-3a); 13C NMR (CDCl3, 75 MHz): δ 24.9 (CH3), 26.0 (CH3), 26.3 (CH3), 26.6 (CH3), 30.8 (CH3), 49.9 (CH), 67.3 (CH2), 74.0 (CH), 78.6 (CH), 86.1 (CH), 104.4 (CH), 109.3 (C), 111.9 (C), 192.8 (C). HRMS (ESI, positive ion) calcd for C14H22O6SNa, m/z 341.1035. Found 341.1034.
(3aR,5R,6S,6aS)-5-[(4R)-2,2-Dimethyl-1,3-dioxolan-4-yl]-2,2-dimethyl-6-(prop-2-en-1-ylsulfanyl)tetrahydrofuro[2,3-d][1,3]dioxole (3)
To a solution of the thioacetate 2 (200 mg, 0.63 mmol) in dry MeOH (5 mL) at 0 °C was added NaBH4 (48 mg, 1.26 mmol), portion-wise. After 10 min stirring, allyl bromide (0.082 mL, 0.945 mmol) was added to the mixture by syringe. A methanolic solution of NaOMe (28 wt %) (0.17 mL, 0.693 mmol) was added dropwise to the mixture, which was allowed to stir at room temperature for 3 h. The solvent was evaporated in vacuo, and EtOAc (25 mL) and water (30 mL) were added to the residue. The organic layer was separated and the aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic layer was washed with H2O (2 × 10 mL), dried (Na2SO4) and the solvent was removed in vacuo. The crude product was purified by column chromatography over silica gel (100–200 mesh) using petroleum ether–EtOAc (24:1) as the eluent to obtain 3 (139 mg, 70%) as a yellowish viscous liquid; [α]25D −7.1 (c 0.08, CHCl3); IR (neat) νmax: 1635, 1377, 1251, 1214, 1162, 1065 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.32 (s, 3H, C–CH3), 1.37 (s, 3H, C–CH3), 1.42 (s, 3H, C–CH3), 1.52 (s, 3H, C–CH3), 3.21 (dd, 1H, J = 6.7, 13.6 Hz, H-1′′b), 3.35 (d, 1H, J = 3.6 Hz, H-6), 3.37 (m, 1H, 1′′a), 4.00 (dd, 1H, J = 5.0, 8.6 Hz, H-5′a), 4.13 (dd, 1H, J = 6.0, 8.6 Hz, H-5′b), 4.21 (dd, 1H, J = 3.6, 8.6 Hz, H-5), 4.34–4.40 (m, 1H, H-4′), 4.66 (d, 1H, J = 3.4 Hz, H-6a), 5.16 (d, 1H, J = 8.9 Hz, H-3′′b), 5.21 (d, 1H, J = 15.5, Hz), 5.75–5.89 (m, 1H, H-2′′), 5.82 (d, 1H, J = 3.6 Hz, H-3a); 13C NMR (CDCl3, 75 MHz): δ 25.3 (CH3), 26.2 (CH3), 26.6 (CH3), 26.9 (CH3), 34.6 (CH2), 50.2 (CH), 67.6 (CH2), 73.9 (CH), 80.3 (CH), 85.9 (CH), 104.8 (CH), 109.3 (C), 111.8 (C), 118.2 (CH2), 133.5 (CH). HRMS (ESI, positive ion) calcd for C15H24O5SNa, m/z 339.1242. Found 339.1262
(3aS,5aR,6S,7S,8aR,8bS)- 6,7-Isopropylidene-1-phenyloctahydro-3H-furo[2′,3′:5,6]-thiopyrano[4,3-c][1,2]oxazole-6,7-diol 5-oxide (6) and (1S,2R,4R,8R,9R,12R)-6,6-dimethyl-14-phenyl-3,5,7,13-tetraoxa-10-thia-14-azatetracyclo[10.2.1.02,9.04,8]-pentadecane 10-oxide (7)
A solution of the thioallyl derivative 3 (1.08 g, 3.42 mmol) in a mixture of H2O–HOAc (1:3, 20 mL) was stirred at room temperature for 12 h. The solvent was evaporated in a rotary evaporator and the last trace of acetic acid was removed by azeotropic distillation with toluene. The crude product was purified by column chromatography over silica gel (100–200 mesh) using petroleum ether–EtOAc (3:2) to furnish the expected dihydroxy compound (736 mg, 78%) as a yellowish oil. To a solution of this in methanol (25 mL) at 0 °C, was added a solution of NaIO4 (1.7 g, 7.93 mmol) in H2O (15 mL), with stirring, and stirring was continued for 30 min and then for another 2 h at 25 °C. The mixture was filtered and the filtrate was concentrated in vacuo. The residue was extracted with CHCl3 (2 × 30 mL). The solution was washed with a saturated solution of brine (20 mL), dried (Na2SO4) and removal of the solvent afforded the aldehyde 4 (764 mg, 86%) as a pale yellow syrup, which was immediately used in the next step without further purification; IR (neat) νmax: 1733, 1636, 1380, 1216, 1064, 756 cm−1; ESIMS: m/z 261 (M+H)+, 299 (M+K)+.
To a solution of the aldehyde 4 (800 mg, 3.07 mmol) in dry toluene (40 mL) under a N2 atmosphere were added molecular sieves (3 Å) (5 g) and freshly prepared N-phenyl hydroxylamine (502 mg, 4.60 mmol). The mixture was heated at reflux for 12 h. The solvent was evaporated in vacuo and the residue was extracted with CHCl3 (3 × 20 mL). The combined extract was washed with H2O (2 × 15 mL), dried (Na2SO4) and the solvent was evaporated to a gummy residue, which was purified by column chromatography over silica gel (230–400 mesh), eluting with petroleum ether–EtOAc (1:1) to afford 6 (410 mg, 38%) and 7 (389 mg, 36%) as yellowish white amorphous solids.
6: [α]25D −125 (c 0.11, CHCl3); IR (neat): 1596, 1493, 1380, 1261, 1215, 1163, 1075, 1016 cm−1; 1H NMR (CDCl3, 600 MHz): δ 1.39 (s, 3H, C–CH3), 1.53 (s, 3H, C–CH3), 2.96 (dd, 1H, J = 9.0, 13.8 Hz, H-4B), 3.35 (dd, 1H, J = 4.2, 13.8 Hz, H-4A), 3.39 (d, 1H, J = 4.8 Hz), 3.41–3.43 (m, 1H, H-3a), 4.02 (d, 1H, J = 8.4 Hz), 4.16–4.19 (m, 2H, H-3B, H-8b), 4.90 (dd, 1H, J = 3.0, 4.8 Hz, H-8a), 5.33 (d, 1H, J = 3.6 Hz, H-6), 5.94 (d, 1H, J = 4.2 Hz, H-7), 7.06 (t, 1H, J = 7.8 Hz, Ar–H), 7.10 (d, 2H, J = 7.8 Hz, Ar–H), 7.34 (t, 2H, J = 7.8 Hz, Ar–H); 13C NMR (CDCl3, 150 MHz): δ 26.2 (CH3), 26.8 (CH3), 34.7 (CH2), 45.6 (CH), 56.9 (CH), 63.4 (CH), 72.1 (CH2), 76.1 (CH), 80.6 (CH), 104.8 (CH), 112.1 (C), 115.6 (2 × CH), 123.3 (CH), 129.3 (2 × CH), 150.6 (C). HRMS (ESI, positive ion) calcd for C17H21NO5SNa, m/z 374.1038. Found 374.1082.
7: [α]25D −154 (c 0.42, CHCl3); IR (neat) νmax: 1595, 1489, 1379, 1213, 1073, 1017, 768 cm−1; 1H NMR (CDCl3, 600 MHz): δ 1.38 (s, 3H, C–CH3), 1.60 (s, 3H, C–CH3), 2.08–2.13 (m, 1H, H-15B), 2.23 (d, 1H, J = 13.8 Hz, H-15A), 3.12 (d, 1H, J = 13.8 Hz, H-11B), 3.87 (br d, 1H, J = 2.4 Hz, H-9), 3.91 (dd, 1H, J = 5.4, 13.8 Hz, H-11A), 4.41 (d, 1H, J = 3.6 Hz, H-1), 4.75 (s, 1H, H-2), 4.89 (br dd, 1H, J = 5.4, 9.0 Hz, H-12), 5.30 (d, 1H, J = 3.0 Hz, H-8), 6.02 (d, 1H, J = 3.0 Hz, H-4), 7.03–7.08 (m, 3H, Ar–H), 7.29–7.31 (m, 2H, Ar–H); 13C NMR (CDCl3, 150 MHz): δ 25.9 (CH3), 26.1 (CH3), 30.5 (CH2), 56.4 (CH2), 66.3 (CH), 70.7 (CH), 71.5 (CH), 79.0 (CH), 80.2 (CH), 103.3 (CH), 112.1 (C), 114.8 (2 × CH), 123.4 (CH), 129.2 (2 × CH), 149.8 (C). HRMS (EI, positive ion) calcd for C17H21NO5S, m/z 351.1140. Found 351.1125.
[(3aR,4aR,5S,6S,8aS,8bS)-2,2-Dimethyl-5-(phenylamino)hexahydro-3aH-thiopyrano-[2′,3′:4,5]furo[2,3-d][1,3]dioxol-6-yl]methanol (8)
To a stirred solution of 6 (0.65 g, 1.85 mmol) in 15:1 CH3CN–H2O (16 mL) was added Mo(CO)6 (0.73 g, 2.78 mmol), and the mixture was heated at reflux under N2 for 5 h. The solvent was removed in vacuo and the residue, dissolved in a CH2Cl2–MeOH mixture (15:1), was passed through a bed of neutral alumina. The solvent was evaporated to give 8 (0.574 g, 92%) as a colorless solid, [α]25D −131 (c 0.1, CHCl3); IR (neat) νmax: 1601, 1503, 1076, 790 cm−1; 1H NMR (CDCl3, 600 MHz): δ 1.29 (s, 3H, C–CH3), 1.43 (s, 3H, C–CH3), 2.43–2.47 (m, 2H, H-6, H-7B), 2.80 (t, 1H, J = 13.8 Hz, H-7A), 3.54 (d, 1H, J = 2.4 Hz, H-8a), 3.65 (dd, 1H, J = 5.4, 10.8 Hz, H-9B), 3.71 (dd, 1H, J = 5.4, 10.8 Hz, H-9A), 4.09 (br s, 1H, H-5), 4.34 (d, 1H, J = 3.0 Hz, H-8b), 4.47 (t, 1H, J = 3.6 Hz, H-4a), 5.99 (d, 1H, J = 3.6 Hz, H-3a), 6.72 (d, 2H, J = 7.8 Hz, Ar–H), 6.76 (t, 1H, J = 7.2 Hz, Ar–H), 7.20 (m, 2H, J = 7.2, 8.4 Hz, Ar–H); 13C NMR (CDCl3, 150 MHz): δ 22.9 (CH2), 26.1 (CH3), 26.5 (CH3), 36.8 (CH), 41.3 (CH), 49.6 (CH), 65.2 (CH2), 75.2 (CH), 85.9 (CH), 105.1 (CH), 111.7 (C), 113.6 (2 × CH), 118.7 (CH), 129.6 (2 × CH), 146.6 (C). HRMS (EI, positive ion) calcd for C17H23NO4S, m/z 337.1348. Found 337.1346.
(3aR,4aR,5S,7R,9aS,9bS)-2,2-Dimethyl-5-(phenylamino)octahydrothiepino-[2′,3′:4,5]-furo[2,3-d][1,3]dioxol-7-ol (9)
For cleavages of N–O and SO bond in 7 the same protocol and procedure as described earlier (in the conversion of 6 to 8) was followed to produce 9 (0.530 g, 85%) as an amorphous solid, which turns violet in the presence of DCM and CHCl3, [α]25D + 53 (c 0.16, CHCl3); IR (neat) νmax: 3368, 2987, 2925, 1602, 1503, 1053 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.32 (s, 3H, C–CH3), 1.56 (s, 3H, C–CH3), 1.60–1.71 (m, 1H, H-6B), 1.80 (br s, 1H, OH), 2.10 (br d, 1H, J = 14.4 Hz, H-6A), 2.54 (dd, 1H, J = 10.8, 14.1 Hz, H-8B), 3.03 (ddd, 1H, J = 1.8, 4.2, 14.0 Hz, H-8A), 3.43 (d, 1H, J = 4.2 Hz, H-9A), 3.86 (t, 1H, J = 9.0 Hz, H-5), 4.11–4.20 (m, 1H, H-7), 4.42 (dd, 1H, J = 4.2, 8.1 Hz, H-4a), 4.58 (d, 1H, J = 3.6 Hz, H-9B), 5.86 (d, 1H, J = 3.9 Hz, H-3a), 6.60 (d, 2H, J = 7.8 Hz, Ar–H), 6.74 (t, 1H, J = 7.5 Hz, Ar–H), 7.19 (m, 2H, Ar–H); 13C NMR (CDCl3, 75 MHz): δ 26.2 (CH3), 26.7 (CH3), 39.9 (CH2), 40.6 (CH2), 53.4 (CH), 53.9 (CH), 71.8 (CH), 84.2 (CH), 85.9 (CH), 104.7 (CH), 111.6 (C), 113.9 (2 × CH), 118.1 (CH), 129.3 (2 × CH), 146.7 (C). HRMS (ESI, positive ion) calcd for C17H23NO4SNa, m/z 360.1245. Found 360.1250.
6,7-Isopropylidene-3a,4,5a,6,7,8a-hexahydro-3H-furo[2′,3′:5,6]thiopyrano[4,3-c]-[1,2]oxazole-6,7-diol 5,5-dioxide (12)
To a solution of 4 (520 mg, 2.0 mmol) in dry ethanol (20 mL) containing dry pyridine (0.8 mL), hydroxylamine hydrochloride (173 mg, 2.5 mmol) was added and the mixture was heated at reflux for 4 h. After removal of the solvent, the residue was extracted with CH2Cl2 (2 × 30 mL). The solvent was washed with H2O (10 mL), dried (Na2SO4) and concentrated in vacuo. The residual pyridine was azeotropically removed with toluene to obtain 10 (385 mg, 70%) as a syrupy liquid. A solution of a mixture of this crude oxime (1.40 mmol), N-chlorosuccinimide (187 mg, 1.40 mmol) and DMAP (171 mg, 1.40 mmol) in dry benzene (20 mL) was stirred at 25 °C for 48 h. The solvent was evaporated in vacuo. The residue was dissolved in CH2Cl2 (30 mL), which was successively washed with 1 M aq. HCl, saturated aqueous NaHCO3 solution, H2O and dried (Na2SO4). Removal of the solvent afforded a syrupy residue, which was purified by column chromatography on silica gel (100–200 mesh size) (eluting solvent: petroleum ether–EtOAc, 9:1) to furnish the isoxazoline 12 (202 mg, 50%) as a colorless solid, [α]25D −45 (c 8.0, CHCl3); IR (neat) νmax: 1723, 1379, 1316, 1219, 1069 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.38 (s, 3H, C–CH3), 1.57 (s, 3H, C–CH3), 3.18 (t, 1H, J = 13.5 Hz, H-4b), 3.37 (dd, 1H, J = 4.2, 13.8 Hz, H-4a), 3.76 (d, 1H, J = 3.9 Hz, H-5a), 4.13–4.22 (m,1H, H-3a), 4.29 (m, , 1H, J = 6.3, 8.7 Hz, H-3b), 4.62 (t, 1H, J = 9.6 Hz, H-3A), 5.25 (d, 1H, J = 2.7 Hz, H-6), 5.45 (d, 1H, J = 3.9 Hz, H-8a), 6.10 (d, 1H, J = 2.4 Hz, H-7); 13C NMR (CDCl3, 75 MHz): δ 26.0 (CH3), 26.5 (CH3), 42.7 (CH), 55.1 (CH2), 68.7 (CH), 71.1 (CH), 73.6 (CH2), 79.0 (CH), 106.5 (CH), 113.0 (C), 151.5 (C). HRMS (EI, positive ion) calcd for C11H15NO6S, m/z 289.0620. Found 289.0658.
(3aR,5R,6S,6aS)-5-[(4R)-2,2-Dimethyl-1,3-dioxolan-4-yl]-2,2-dimethyl-6-(prop-2-yn-1-ylsulfanyl)tetrahydrofuro[2,3-d][1,3]dioxole (13)
NaBH4 (62 mg, 1.64 mmol) was added to a stirred solution of 2 (260 mg, 0.82 mmol) in dry MeOH (5 mL) at 0 °C. After 15 min, propargyl bromide (80% w/w in toluene, 0.14 mL) was added to the mixture and then a methanolic solution of NaOMe (28 wt %) (0.22 mL, 0.90 mmol) was added, and the mixture was allowed to stir at room temperature for 3 h. Work up and purification (as described in the preparation of 3 from 2) afforded 13 (180 mg, 70%) as a yellowish viscous liquid, [α]25D −11 (c 0.13, CHCl3); IR (neat) νmax: 2115, 1380, 1313, 1215, 1060, 1014, 925, 864 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.33 (s, 3H, C–CH3), 1.36 (s, 3H, C–CH3), 1.43 (s, 3H, C–CH3), 1.53 (s, 3H, C–CH3), 2.30 (t, 1H, J = 2.1 Hz, H-3′′), 3.35 (dd, 1H, J = 2.4, 16.8 Hz, H-1′′b), 3.51 (dd, 1H, J = 2.4, 16.8 Hz, H-1′′a), 3.63 (d, 1H, J = 3.3 Hz, H-6), 4.01 (dd, 1H, J = 4.8, 8.7 Hz, H-5′b), 4.13 (dd, 1H, J = 5.7, 8.4 Hz, H-5′a), 4.25 (dd, 1H, J = 3.3, 8.4 Hz, H-5), 4.30–4.34 (m, 1H, H-4′), 4.81 (d, 1H, J = 3.3 Hz, H-6a), 5.84 (d, 1H, J = 3.0 Hz, H-3a); 13C NMR (CDCl3, 75 MHz): δ 19.7 (CH2), 25.2 (CH3), 26.2 (CH3), 26.6 (CH3), 26.8 (CH3), 51.8 (CH), 67.6 (CH2), 71.8 (CH), 73.9 (CH), 79.4 (C), 80.1 (CH), 85.5 (CH), 104.7 (CH), 109.4 (C), 111.9 (C); ESIMS, m/z: 337 (M+Na)+.(1R)-1-[(3aR,5R,6S,6aS)-2,2-Dimethyl-6-(prop-2-yn-1-ylsulfanyl)tetrahydrofuro[2,3-d][1,3]dioxol-5-yl]ethane-1,2-diol (14)
Following the procedure described in the transformation of 3 to 4, the thiopropargyl derivative 13 (1.10 g, 3.50 mmol) was treated with aqueous AcOH (70%, 20 mL) at room temperature for 12 h to afford the corresponding 5,6-diol 14 (720 mg, 75%) as a yellowish oil, [α]25D −65 (c 0.3, CHCl3); IR (neat): 3433, 3287, 2116, 1380, 1215, 1062, 1013 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.33 (s, 3H, C–CH3), 1.53 (s, 3H, C–CH3), 2.35 (t, 1H, J = 2.4 Hz, H-3′′), 2.62 (br m, 1H, OH), 2.99 (br m, 1H, OH), 3.40 (m, 2H, H-1′′a, H-1′′b), 3.66 (d, 1H, J = 3.9 Hz, H-6), 3.73 (dd, 1H, J = 5.7, 11.4 Hz, H-2′b), 3.85–3.93 (m, 2H, H-2′a, H-5′), 4.34 (dd, 1H, J = 3.9, 9.0 Hz, H-5), 4.80 (d, 1H, J = 3.6 Hz, H-6a), 5.89 (d, 1H, J = 3.3 Hz, H-3a); 13C NMR (CDCl3, 75 MHz): δ 19.3 (CH2), 26.2 (CH3), 26.5 (CH3), 51.7 (CH), 64.2 (CH2), 70.5 (CH), 72.1 (CH), 78.7 (CH), 79.5 (C), 85.3 (CH), 105.0 (CH), 112.0 (C). HRMS (EI, positive ion) calcd for C12H18O5S, m/z 274.0875. Found 274.0861.6,7-Isopropylidene-(5aR,8aR)-5a,6,7,8a-tetrahydro-4H-furo[2′,3′:5,6]thiopyrano[4,3-c][1,2]oxazole-6,7-diol (17)
The diol 14 (700 mg, 2.55 mmol) in methanol (25 mL) was converted to an aldehyde (556 mg, 90%) using NaIO4 (1.64 g, 7.65 mmol) in H2O (15 mL) following the procedure earlier described (in the preparation of 4). To a solution of the above aldehyde (500 mg, 2.07 mmol) in dry ethanol (20 mL) containing dry pyridine (0.6 mL), hydroxylamine hydrochloride (200 mg, 2.88 mmol) was added and the mixture was heated at reflux for 4 h. The usual work-up afforded a syrupy liquid, which was purified by column chromatography on silica gel (100–200 mesh), eluting with petroleum ether–EtOAc (9:1) to afford 15 (393 mg, 74%) as a mixture of isomers, 1H NMR (CDCl3, 300 MHz): δ 1.35 (s), 1.54 (s), 2.32 (br d, J = 2.1 Hz), 3.33 (s), 3.66 (br d, J = 3.0 Hz), 3.99 (br d, J = 3.0 Hz), 4.84 (br s), 5.01 (br t, J = 4.8 Hz), 5.31 (s), 5.41 (br s), 5.95 (br s), 6.96 (br s), 7.53 (d, J = 6.6 Hz). ESIMS, m/z: 280 (M+Na)+.
The oxime 15 (0.5 g, 1.95 mmol) in dry benzene (20 mL) at 25 °C was treated with N-chlorosuccinimide (0.26 g, 1.95 mmol) and DMAP (0.24 g, 1.95 mmol) according to the procedure described earlier (in the preparation of 12 from 10). The usual work-up and purification afforded 17 (223 mg, 45%) as a colorless oil, [α]25D +63 (c 0.65, CHCl3); IR (neat) νmax: 1724, 1690, 1607, 1429, 1380, 1069 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.37 (s, 3H, C–CH3), 1.60 (s, 3H, C–CH3), 3.62 (d, 1H, J = 15.3 Hz, H-4B), 3.73 (d, 1H, J = 15.3 Hz, H-4A), 3.76 (br s,1H, H-5a), 4.67 (d, 1H, J = 3.3 Hz, H-6), 5.46 (d, 1H, J = 3.3 Hz, H-8a), 6.01 (d, 1H, J = 3.0 Hz, H-H-7), 8.28 (s, 1H, H-3); 13C NMR (CDCl3, 75 MHz): δ 20.2 (CH2), 26.1 (CH3), 26.5 (CH3), 48.5 (CH), 68.6 (CH), 84.4 (CH), 105.3 (CH), 112.2 (C), 112.2 (C), 152.6 (CH), 157.0 (C). HRMS (EI, positive ion) calcd for C11H13NO4S, m/z 255.0565. Found 255.0574.
(R)-1-(3aR,5R,6S,6aS)-[6-(2-Allyloxy-benzylsulfanyl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]- dioxol-5-yl]ethane-1,2-diol (19)
To a solution of the thioacetate 2 (1.50 g, 4.72 mmol) dissolved in dry MeOH (25 mL) at 0 °C, NaBH4 (357 mg, 9.44 mmol) was added portion-wise. 2-(Allyloxy)benzyl bromide (1.6 g, 7.08 mmol) was added to the mixture through a syringe. A methanolic solution of NaOMe (28 wt%, 1.27 mL, 5.19 mmol) was added dropwise to the mixture and the reaction was allowed to stir at room temperature for 3 h. The usual work-up followed by purification on silica gel using petroleum ether–EtOAc (24:1) as the eluent, as described in the preparation of 3, however, yielded 19 (1.28 g, 71%) as a yellowish viscous liquid, [α]25D −44 (c 0.25, CHCl3); IR (neat) νmax: 3451, 1644, 1595,1491, 1059,1012 cm−1; 1H NMR (CDCl3, 600 MHz): δ 1.30 (s, 3H, C–CH3), 1.48 (s, 3H, C–CH3), 2.35 (br m, 1H, OH), 2.67 (br s, 1H, OH), 3.37 (d, 1H, J = 4.2 Hz, H-6), 3.65 (dd, 1H, J = 5.4, 10.8 Hz, H-7′′B), 3.80 (br d, 1H, J = 11.4 Hz, H-7′′A), 3.86–3.88 (m, 3H, H-1′, H-2′A, H-2′B), 4.25 (dd, 1H, J = 4.2, 9.0 Hz, H-5), 4.59 (m, 2H, H-8′′A, H-8′′B), 4.69 (d, 1H, J = 3.6 Hz, H-6a), 5.29 (d, 1H, J = 10.8 Hz, H-10′′B), 5.44 (d, 1H, J = 16.8 Hz, H-10′′A), 5.86 (d, 1H, J = 3.0 Hz, H-3a), 6.06–6.11 (m, 1H, H-9′′), 6.88 (d, 1H, J = 8.4 Hz, ArH), 6.93 (t, 1H, J = 7.2 Hz, ArH), 7.22–7.27 (m, 2H, ArH); 13C NMR (CDCl3, 75 MHz): δ 26.3 (CH3), 26.7 (CH3), 30.4 (CH2), 51.4 (CH), 64.3 (CH2), 68.9 (CH2), 70.7 (CH), 78.9 (CH), 85.7 (CH), 105.3 (CH), 111.9 (C), 112.1 (CH), 117.6 (CH2), 120.9 (CH), 125.9 (C), 128.9 (CH), 130.4 (CH), 133.1 (CH), 156.2 (C). HRMS (ESI, positive ion) calcd for C19H26O6SNa, m/z 405.1348. Found 405.1385.
(12E,14Z,16E)-6,6-Dimethyl-3,5,7,18,21-pentaoxa-10-thia-22-azabicyclo-[18.2.1.012,17.02,9.04,8]tricosa-1(22),12,14,16-tetraene 10-oxide (23)
Compound 19 (1.1 g, 2.88 mmol) in MeOH (110 mL) at 0 °C was converted to 20 (0.874 g, 83%) as a pale yellow syrup using NaIO4 (1.85 g, 8.64 mmol) in H2O (15 mL) according to the procedure used in the preparation of 4. The aldehyde was sufficiently pure to be used in the next step, IR (neat) νmax: 1731, 1595, 1493, 1379, 1055 cm−1.The oxime 21 was prepared from 20 in 80% yield (670 mg) using the following protocol (as described in the preparation of 15): hydroxylamine hydrochloride (0.23 g, 3.27 mmol), 20 (0.8 g, 2.19 mmol), ethanol (80 mL) and pyridine (1.6 mL), and the mixture was allowed to stir at room temperature for 6 h. After removal of the solvent in rotary evaporator, the residue was extracted with CH2Cl2 (2 × 20 mL). Work-up and chromatography purification furnished the oxime 21 (0.67 g, 80%) as mixture of isomers, IR (neat) νmax: 3235, 1595, 1492, 1456, 1380, 1249, 1018 cm−1; 1H NMR (CDCl3, 300 MHz): δ 1.34 (s), 1.52 (s), 3.43 (d, J = 5.4 Hz), 3.80 (d, J = 5.1 Hz), 4.15 (dd, J = 9.0, 12.3 Hz), 4.30 (dd, J = 7.8, 12.6 Hz), 4.57–4.61 (m), 5.04 (dd, J = 5.4, 10.0 Hz), 5.14 (d, J = 3.9 Hz), 5.20 (d, J = 3.6 Hz), 5.27–5.44 (m), 5.88 (d, J = 3.6 Hz), 6.01–6.12 (m), 6.87–6.98 (m), 7.11 (d, J = 3.3 Hz). 7.24–7.33 (m), 7.63 (d, J = 6.9 Hz), 8.12 (br s), 8.18 (br s); 13C NMR (CDCl3, 75 MHz): δ 26.1 (CH3), 26.2 (CH3), 26.7 (CH3), 53.1(CH2), 53.3 (CH2), 63.3 (CH), 65.2 (CH), 69.1 (CH2), 74.9 (CH), 79.2 (CH), 79.3 (CH), 105.3 (CH), 105.6 (CH), 111.5 (C), 111.8 (C), 111.9 (C), 112.1 (CH), 117.9 (CH2), 118.1 (CH2), 120.9 (CH), 121.1 (CH), 130.0 (CH), 130.3 (CH), 132.1 (CH), 132.8 (CH), 132.9 (CH), 148.9 (C), 156.5 (C), 156.6 (C); ESIMS, m/z: 404 (M+Na)+.
The oxime 21 (0.5 g, 1.31 mmol) in benzene (20 mL) was converted to the isoxazole 23 (0.313 g, 63%) by reaction with N-chlorosuccinamide (0.175 g, 1.31 mmol) in the presence of DMAP (0.16 g, 1.31 mmol) followed by work-up and purification (as described in the preparation of 12) as a colorless viscous liquid, [α]25D +95 (c 1.16, CHCl3); IR (neat) νmax: 1713, 1599, 1493, 1455, 1381, 1257, 1039 cm−1; 1H NMR (CDCl3, 600 MHz): δ 1.37 (s, 3H, C–CH3), 1.57 (s, 3H, C–CH3), 3.06 (d, 1H, J = 16.8 Hz, H-23B), 3.37 (dd, 1H, J = 10.8, 17.4 Hz, H-23A), 3.63 (d, 1H, J = 12.6 Hz, H-11B), 3.82 (d, 1H, J = 9.6 Hz, H-19A), 4.02 (d, 1H, J = 3.6 Hz, H-9), 4.27 (d, 1H, J = 9.6 Hz, H-19B), 4.39 (d, 1H, J = 12.6 Hz, H-11A), 4.99 (d, 1H, J = 10.8 Hz, H-20), 5.30 (d, 1H, J = 3.6 Hz, H-8), 5.53 (d, 1H, J = 3.0 Hz, H-2), 6.04 (d, 1H, J = 3.0 Hz, H-4), 6.74 (d, 1H, J = 7.8 Hz, ArH), 6.96 (t, 1H, J = 7.2 Hz, ArH), 7.32 (m, 2H, ArH); 13C NMR (CDCl3, 150 MHz): δ 26.0 (CH3), 26.5 (CH3), 38.9 (CH2), 61.6 (CH2), 70.1 (CH2), 70.4 (CH), 73.3 (CH), 77.5 (CH), 79.8 (CH), 104.4 (CH), 109.5 (CH), 112.1 (C), 120.9 (C), 121.2 (CH), 130.3 (CH), 131.0 (CH), 155.7 (C), 155.8 (C). HRMS (EI, positive ion) calcd for C18H21NO6S, m/z 379.1090. Found 379.1082.
(2R,3S,3aS,6R,7R,7aR)-2-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-6-actoxymethyl-7-phenylamino-hexahydro-2H-thiopyrano[3,2-b]furan-3-yl acetate (25)
Ac2O (0.68 mL, 7.2 mmol) was added to 8 (0.4 g, 1.2 mmol) in HOAc (24 mL). The mixture was cooled to 0 °C, triflic acid (0.0035 mL, 0.04 mmol) was added to it and stirred for 2 h at room temperature. The reaction was quenched with cold saturated NaHCO3 solution (10 mL) and the mixture was extracted with CH2Cl2 (3 × 25 mL). The solvent was dried (Na2SO4), and evaporated to give 24 as a syrupy liquid, which was dried via co-evaporation with anhydrous CH3CN (2 × 15 mL). Uracil (0.18 g, 1.63 mmol) and N,O-bis(trimethylsilyl)acetamide (0.83 mL, 3.4 mmol) were added to a solution of the above material in CH3CN (20 mL) and the mixture was heated at reflux for 45 min until the suspension became a clear solution. The reaction mixture was cooled to 0 °C, TMSOTf (0.31 mL, 1.7 mmol) was added dropwise and the mixture was heated at reflux for 17 h. CH3CN was evaporated under reduced pressure to a residue, to which was added a saturated ammonium chloride solution (10 mL). The mixture was extracted with CH2Cl2 (2 × 25 mL). The organic layer was dried (Na2SO4), evaporated, and the product was purified by column chromatography over silica gel (100–200 mesh size) using 0.2% methanol in CH2Cl2, v/v) to give 25 (0.304 mg, 54%) as a colorless amorphous solid.25: [α]25D +75 (c 0.28, CHCl3); IR (neat) νmax: 3381, 3204, 1689, 1602, 1375,1231,1041 cm−1; 1H NMR (CDCl3, 600 MHz): δ 2.05 (s, 6H,C–CH3), 2.58 (d, 1H, J = 13.8 Hz, H-5B), 2.63 (m, 1H, H-6), 2.71 (d, 1H, J = 13.2 Hz, H-5A), 3.53 (br s, 1H, H-3a), 3.53–3.62 (m, 1H, H-7), 3.97 (br d, 1H, NH), 4.07 (s, 1H, H-7a), 4.15–4.21 (m, 2H, H-8A, H-8B), 4.52 (t, 1H, J = 4.2 Hz, H-3), 5.60 (d, 1H, J = 1.8 Hz, H-2), 5.66 (d, 1H, J = 7.8 Hz, H-5′), 6.65 (d, 2H, J = 7.8 Hz, Ar–H), 6.79 (t, 1H, J = 7.2 Hz, Ar–H), 7.21 (t, 2H, J = 7.8 Hz, Ar–H), 7.81 (d, 1H, J = 7.8 Hz, H-6′), 8.65 (s, 1H, 3′-NH); 13C NMR (CDCl3, 75 MHz): δ 20.9 (2 × CH), 23.9 (CH2), 31.9 (CH), 35.5 (CH), 42.9 (CH), 49.3 (CH), 64.5 (CH2), 80.9 (CH), 101.9 (CH), 113.3 (2 × CH), 118.7 (CH), 129.6 (2 × CH), 140.2 (CH), 146.2 (C), 151.2 (C), 163.7 (C), 170.8 (2 × C); 13C NMR (CDCl3, 150 MHz): δ 20.9 (2 × CH3), 24.9 (CH2), 31.4 (CH), 35.6 (CH), 43.3 (CH), 50.2 (CH), 64.0 (CH2), 81.2 (CH), 94.0 (CH), 101.9 (CH), 113.4 (2 × CH), 119.0 (CH), 129.7 (2 × CH), 139.5 (CH), 145.8 (C), 150.9 (C), 162.7 (C), 170.8 (2 × C). HRMS (EI, positive ion) calcd for C22H25N3O7S, m/z 475.1413. Found 475.1407.
(5aS,6S,7R,8aR)-7-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-5a,6,7,8a-tetrahydro-4H-furo[2′,3′:5,6]thiopyrano[4,3-c][1,2]oxazol-6-yl acetate (27)
Compound 17 (245 mg, 0.96 mmol) was dissolved in 10 mL H2SO4–CH3CN–H2O (1:18:6) and stirred at room temperature for 14 h. The solution was neutralized by portion-wise addition of solid CaCO3. The precipitate was filtered off and the filtrate was evaporated in vacuo to obtain a gummy mass. This was dried over P2O5. It was then treated with pyridine (3 mL) and Ac2O (1 mL), and the mixture was stirred at room temperature for 10 h. The solvent was evaporated, and the sticky material was dried in vacuo to furnish 26 (0.22 g) as a syrupy liquid, which was subsequently used without purification. A solution of uracil (256 mg, 2.28 mmol) in hexamethyldisilazane (10 mL) and chlorotrimethylsilane (2 drops) was heated at 135–140 °C under N2 for 8 h. The solvent was distilled off under vacuo, and a solution of the residue in CH3CN (7 mL) was added to a stirred solution of the above material in CH3CN (5 mL) containing TMSOTf (0.21 mL, 1.17 mmol). The solution was stirred at room temperature for 12 h under N2, when TLC showed complete disappearance of the starting material. The reaction was neutralised with solid NaHCO3, H2O (2–3 drops) was added to it, and the solvent was evaporated to furnish a residue, which was extracted with 98:2 CHCl3–MeOH (3 × 30 mL). The combined organic layer was washed with brine (2 × 20 mL), dried (Na2SO4) and concentrated to a gummy residue. The crude product was purified by column chromatography using petroleum ether–EtOAc mixture (2:3) to afford 27 (185 g, 55%) as a colorless solid, [α]25D +73 (c 0.59, CHCl3); IR (neat) νmax: 3299, 1689,1460, 1382, 1225, 1049, 756 cm−1; 1H NMR (CDCl3, 300 MHz): δ 2.20 (s, 3H, C–CH3), 3.67–3.81 (m, 3H, H-4A, H-4B, H-5a), 5.17 (s, 1H, H-8a), 5.46 (d, 1H, J = 4.5 Hz, H-6), 5.68 (d, 1H, J = 8.1 Hz, H-5′), 6.15 (d, 1H, J = 2.7 Hz, H-7), 7.46 (d, 1H, J = 8.1 Hz, H-6′), 8.34 (br s, 1H, NH), 8.38 (s, 1H, H-3); 13C NMR (CDCl3, 75 MHz): δ 20.0 (CH2), 20.7 (CH3), 46.6 (CH), 69.8 (CH), 79.1 (CH), 89.6 (CH), 102.7 (CH), 111.7 (C), 139.7 (CH), 150.1 (C), 153.5 (CH), 156.2 (C), 162.9 (C), 169.6 (C). HRMS (ESI, positive ion) calcd for C14H13N3O6SNa, m/z 374.0423. Found 374.0455.
Acknowledgements
AB is grateful to CSIR, India for Emeritus Scientist Fellowship. The award of Senior Research Fellowship to SM by CSIR, India is gratefully acknowledged. SBM thanks CSIR for financial support from Network Project (No. NWP0036).References
- S. Hannesian, Total Synthesis of Natural Products: The “Chiron” Approach, Pergamon, Oxford, 1983 Search PubMed.
- (a) A. Bhattacharjee, S. Datta, P. Chattopadhyay, N. Ghoshal, A. P. Kundu, A. Pal, R. Mukhopadhyay, S. Chowdhury, A. Bhattacharjya and A. Patra, Tetrahedron, 2003, 59, 4623 CrossRef CAS; (b) S. Majumdar, A. Bhattacharjya and A. Patra, Tetrahedron Lett., 1997, 38, 8581 CrossRef CAS; (c) S. Majumdar, A. Bhattacharjya and A. Patra, Tetrahedron, 1999, 55, 12157 CrossRef CAS; (d) J. Sengupta, R. Mukhopadhyay, A. Bhattacharjya, M. M. Bhadbhade and G. V. Bhosekar, J. Org. Chem., 2005, 70, 8579 CrossRef CAS; (e) S. Datta, M. Nath, S. Chowdhury and A. Bhattacharjya, Tetrahedron Lett., 2008, 49, 2935 CrossRef CAS.
- (a) K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS; (b) M. Frederickson, Tetrahedron, 1997, 53, 403 CrossRef CAS; (c) H. M. I. Osborn, N. Gemmell and L. M. Harwood, J. Chem. Soc., Perkin Trans. 1, 2002, 1, 2419 RSC; (d) A. Chatterjee and P. Bhattacharya, J. Org. Chem., 2006, 71, 345 CrossRef CAS.
- M. D. McReynolds, J. M. Dougherty and P. R. Hanson, Chem. Rev., 2004, 104, 2239 CrossRef CAS and references cited therein.
- O. T. Schmidt, Methods in Carbohydrate Chemistry, Academic, New York, 1963, vol. 11, pp. 318–325 Search PubMed.
- (a) A. Patra, M. Bandyopadhyay and D. Mal, Tetrahedron Lett., 2003, 44, 2355 CrossRef CAS; (b) S. Iyer and G. M. Kulkarni, Synth. Commun., 2004, 34, 721 CrossRef CAS; (c) C. J. Moody, Chem. Commun., 2004, 1341 RSC; (d) A. Guarna, A. Guidi, A. Goti, A. Brandi and F. De Sarlo, Synthesis, 1989, 175 CrossRef CAS.
- B. W. Yoo, M. S. Song and M. C. Park, Bull. Korean Chem. Soc., 2007, 28, 171 CrossRef CAS.
- S. Majumdar, R. Mukhopadhyay and A. Bhattacharjya, Tetrahedron, 2000, 56, 8945 CrossRef CAS.
- Minimum energy analysis was done by using Chem3D (version Ultra 6.0). The structure was subjected to molecular dynamics operation followed by minimum energy calculation using MM2. The structures with the smallest minimum energy values obtained after several iterations are presented in Fig. 3.
- (a) S. Hanessian, D. M. Dixit and T. J. Liak, Pure Appl. Chem., 1981, 53, 129 CrossRef CAS; (b) S. Danishefsky and R. Hungate, J. Am. Chem. Soc., 1986, 108, 2486 CrossRef CAS; (c) S. Hanessian, J. Kloss and T. Sugawara, J. Am. Chem. Soc., 1986, 108, 2758 CrossRef CAS; (d) S. Hanessian, G. Huang, C. Chenel, R. Machaalani and O. Loiseleur, J. Org. Chem., 2005, 70, 6721 CrossRef CAS.
- (a) P. Crowley, T. Wagner and S. Hanessian, J. Org. Chem., 2007, 72, 6353–6363 CrossRef; (b) L. Kværnø, R. H. Wightman and J. Wengel, J. Org. Chem., 2001, 66, 5106–5109 CrossRef; (c) P. Nielsen, H. M. Pfundheller, C. E. Olsen and J. Wengel, J. Chem. Soc., Perkin Trans. 1, 1997, 1, 3423–3434 RSC; (d) P. Srivastava, J. Barman, W. Pathmasiri, O. Plash-Kevych, M. Wenska and J. Chattopadhyaya, J. Am. Chem. Soc., 2007, 129, 8362–8379 CrossRef CAS; (e) M. Freitag, H. Thomasen, N. K. Christensen, M. Petersen and P. Nielsen, Tetrahedron, 2004, 60, 3775–3786 CrossRef CAS; (f) K. I. Shaikh, S. Kumar, L. Lundhus, A. D. Bond, P. K. Sharma and P. Nielsen, J. Org. Chem., 2009, 74, 1557–1566 CrossRef CAS; (g) P. Russ, P. Schelling, L. Scapozza, G. Folkers, E. De Clercq and V. E. Marquez, J. Med. Chem., 2003, 46, 5045–5054 CrossRef CAS and references cited therein; (h) M. Meldgaard and J. Wengel, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 3539–3554 RSC and references cited therein; (i) S. Tripathi, B. G. Roy, M. G. B. Drew, B. Achari and S. B. Mandal, J. Org. Chem., 2007, 72, 7427–7430 CrossRef CAS; (j) R. Ghosh, J. K. Maity, B. Achari and S. B. Mandal, J. Org. Chem., 2010, 75, 2419–2422 CrossRef CAS.
- (a) C. Mathé and C. Périgaud, Eur. J. Org. Chem., 2008, 1489–1505 CrossRef; (b) J. Lebreton, J.-M. Escudier, L. Arzel and C. Len, Chem. Rev., 2010, 110, 3371–3418 CrossRef CAS.
- (a) U. Niedballa and H. Vorbrüggen, J. Org. Chem., 1974, 39, 3654 CrossRef CAS; (b) U. Niedballa and H. Vorbrüggen, J. Org. Chem., 1974, 39, 3660 CrossRef CAS; (c) H. Vorbrüggen, K. Krolikewiez and B. Bennua, Chem. Ber., 1981, 114, 1234 CrossRef; (d) H. Vorbrüggen and G. Höfle, Chem. Ber., 1981, 114, 1256 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference number 861102. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20689g |
| This journal is © The Royal Society of Chemistry 2012 |