DOI:
10.1039/C2RA20651J
(Paper)
RSC Adv., 2012,
2, 5418-5423
The opposite effects of Cu(II) and Fe(III) on the assembly of glucagon amyloid fibrils†
Received
10th April 2012
, Accepted 11th April 2012
First published on 14th May 2012
Abstract
A few transition metal ions are strongly implicated as co-factors in modulating the aggregation of amyloid peptides, which is believed to be a key factor in regulating the cytotoxicity of peptides. In this paper, we explored the effects of Cu(II) and Fe(III) on the aggregation/fibrillation of glucagon peptides using various biophysical techniques. AFM analysis demonstrated that Cu(II) could promote the conversion of glucagon peptides into amyloid fibrils, while the formation of fibrils was profoundly suppressed in the presence of Fe(III). Strikingly, at higher Cu(II) concentration (200 μM), spherical assemblies were predominant with abundant fibrils protruding from spherical cores. However, only globular aggregates of several nanometers size were observed when the concentration of Fe(III) was increased beyond 100 μM. In addition, it was also found that the FTIR and CD spectra of glucagon co-incubated with Cu(II) or Fe(III) remarkably differed from that in the absence of ions. These results strongly suggested that Cu(II) and Fe(III) could dramatically modify the morphologies as well as the secondary structures of aggregates during the spontaneous fibrillation of glucagon. Our study could shed light on how the metal ions regulate the amyloid aggregation of glucagon peptide, and might provide a controllable means for the synthesis of amyloid nanostructures for future technological applications.
1. Introduction
The formation of amyloid aggregates/fibrils by misfolded proteins is considered to be a hallmark of more than 30 major neurogenerative diseases such as Alzheimer's (AD) and Parkinson's (PD).1–4 The morphologies and structures of amyloid fibrils can vary dramatically at the nanoscale as revealed by transmission electron microscopy (TEM) and atomic force microscopy (AFM),5,6 although most of them share the characteristic β-sheet structures with an orientation perpendicular to the fibril axis. Increasing evidence suggests that the exhibition of structural heterogeneity among amyloid fibrils is closely associated with the environmental parameters (e.g. pH value, temperature, nanoparticles and various metal ions).7–13 The structural heterogeneity has been proposed to be an important feature of amyloid fibrils, and it is therefore strongly suggested to be key to understanding the role of fibrils in diseases.
Among external factors influencing amyloid fibril formation are transition-metal ions such as Fe(III), Cu(II) and Zn(II), which are able to specifically bind to many peptides and have been proposed as factors or co-factors involved in the etiopathogenesis of neurodegenerative disorders.14–19 An extensively studied case is Aβ peptides associated with AD due to the well-known fact that AD patients show abnormally high levels of specific metal ions present in the Aβ plaques (Cu(II): 400 μM and Fe(III): 1 mM).20 To date, substantial efforts have been devoted to studying the effects of different metal ions on the aggregation of various amyloidogenic proteins. On the one hand, in vitro studies have shown that a few metal ions serve as modulators of amyloid peptide aggregation.21–24 For example, Dong et al. reported that Zn(II) can specifically control the rate of self assembly and dramatically regulate amyloid morphologies via distinct coordination environments.25 Very recently, Liu et al. found that Fe(III) could promote the toxicity of Aβ by delaying the formation of mature and well ordered aggregates,26 though the Fe(III) itself does not incorporate into amyloid fibrils, indicating that the mature amyloid fibrils are not the prime cytotoxic agents and the most toxic Aβ aggregates are small, soluble species. On the other hand, it is suggested that some redox-active metal ions (e.g. Cu(II) and Fe(III)) probably act as co-factors by binding with amyloid peptides in the generation of reactive oxygen species (ROS) via the fenton reaction,16,17,27–29 which subsequently causes damage to a variety of biomolecules thus leading eventually to neuronal cell death. In spite of these intensive studies, however, the detailed molecular mechanism to explain the interactions between metal ions with various amyloid peptides and to describe the exact binding sites is still a matter of intense debate in the literature.
Glucagon is a secreted 29-residue peptide, which has been extensively used as an emergency treatment of insulin-induced hypoglycemia. It exhibits at least three main different conformations: α-helical crystals, random-coil monomer, and β-sheet-rich in fibrils.30 Several studies have suggested that, similar to other amyloid peptides, the misfolding of glucagon peptides generates amyloidogenic fibrils,31,32 leading to cytotoxicity mediated by the activation of the apoptotic enzyme caspase-3 in vitro. The cytotoxicity of glucagon aggregates/fibrils even exceeds that of other peptide hormones and the Aβ peptides.33,34 There is a growing sense that the peptide toxicity depends greatly on the structures of the amyloid aggregates,35,36 implying that the toxicity could arise from a particularly toxic type of glucagon aggregates/fibrils formed under various conditions. Recent studies indicated that some anions (e.g. SO42−) can have a disproportionate influence on the kinetics of fibrillation and the accompanying secondary structures even at low concentrations.37 However, there are few reports on the effects of cations on the aggregations of glucagon. In our previous work, we have studied the kinetics of glucagon fibrils formation by in situ AFM observation.38 Here, we further studied the influence of the essential bio-metals (Cu(II) and Fe(III)) on the aggregation of glucagon peptides. It was found that Cu(II) could catalyze the fibril formation. In contrast, the formation of glucagon fibrils was gradually suppressed with an increasing concentration of Fe(III). The FTIR and CD spectra further indicated that the secondary structure of glucagon aggregates/fibrils was also significantly changed in the presence of Fe(III) or Cu(II) ions.
2. Materials and methods
Materials
Glucagon powder (purity ≥ 70%) and Thioflavine (thioflavin T, ThT) were purchased from Sigma-Aldrich Company (Shanghai, China). Before experiments, the frozen glucagon powder was dissolved with Milli-Q water (18.2 MΩ·cm) to a temporary concentration of 2.0 mg ml−1, then the solution was stored at −20 °C. Thioflavine was diluted with phosphate buffer (pH 7.0) to a concentration of 1 mM and stored at 4 °C. FeCl3, FeCl2, CuCl2·4H2O and H2O2 were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). All these chemicals were used in our experiments without further purification.
Glucagon peptide aggregation
To study the effect of metal ions on the aggregation/fibrillation of glucagon peptides, various metal ions including Fe(III), Cu(II) and Fe(II) were added respectively to a desired metal ion concentration and a final fixed glucagon concentration (1 mg ml−1). Then, samples were incubated at 37 °C for a desired period of time.
A Multimode Nanoscope IIIa SPM (Veeco, Santa Barbara, CA) was used for the measurements of the morphology of the assembled peptide fibrils. The probes used were NSC11 (MikroMash company) with a nominal spring constant of 40 N m−1 and resonance frequency of 300–330 kHz. Tapping mode images were obtained with a scan speed of 1.0–1.5 Hz. Analysis of all the images was carried out using the Nanoscope software (version V530r3sr3). All images were shown without any image processing except the flattening. AFM sample preparation was carried out as follows: About 5 μl glucagon fibril solution was put on a freshly cleaved mica surface. After incubation for 3–5 min, the residual solution was rinsed away gently with Milli-Q water. Then the sample was dried carefully by a stream of nitrogen gas. All the AFM images were collected under ambient conditions at room temperature.
ThT fluorescence measurement
The kinetics of glucagon peptide aggregation was monitored using a dye ThT, the fluorescence of which was dependent on the formation of amyloid aggregates/fibrils. ThT fluorescence measurement was performed at room temperature using an F-4600 fluorescence spectrophotometer (Hitachi Company, Japan). The excitation and emission wavelengths were 440 and 484 nm, respectively. All samples were in quiescence during the incubation. Fluorescence was measured immediately after the mixture was made with the reaction mixture containing 10 μM ThT. At least 3 different samples were independently measured to get the average of fluorescence intensity.
Micro-Fourier transform infrared spectrometry (FTIR)
The secondary structures of glucagon fibrils were monitored at room temperature by a Nicolet Continuum Infrared Microscope (Nicolet 6700) equipped with a liquid-nitrogen cooled MCT detector and continuum infrared microscope accessory. A drop of solution (2–3 μl) was deposited on a diamond sample holder and dried under infrared light at room temperature. The absorbance spectra of different samples were collected with a resolution of 8 cm−1, and total 200 scans were added for a better signal/noise ratio. The Gaussian fittings were performed in Fig. 4.
Circular dichroism (CD)
Experiments were performed at ambient temperature on a Jasco J-815 spectropolarimeter (Jasco, Tokyo, Japan) equipped with a thermostated cell holder. Measurements of the far-UV CD spectra (260–190 nm) were made using a bandwidth of 1 nm with a 1.0 mm path-length cuvette. The buffer spectra were subtracted from all reported spectra. The spectral scan was repeated at least three times and their average results were presented.
3. Results and discussion
Time-course AFM observation of glucagon fibrils formed with and without metal ions
AFM is a non-invasive scanning technique for imaging and manipulating soft materials at nanoscale resolution,39–41 which can be operated in air or liquid environments. Indeed, it has been widely employed to investigate the morphology of amyloid fibrils as well as the kinetics of fibril formation. Fig. 1 shows the typical time-course AFM images of glucagon aggregates/fibrils formed at different time periods of incubation in the absence and presence of Fe(III) and Cu(II) ions, respectively. Clearly, the self-assembled structures of glucagon depended significantly on the salt species used. In the absence of salt, a few big aggregates with heights in the range of 6–9 nm as well as some small ones with heights of 3–4 nm were initially (5.0 h) observed on mica surface. On prolonging the incubation time, fibrillar structures were developed (Fig. 1 a2–a3). In agreement with our previous reports,38 two types of fibrillar structures were mainly observed: filaments and protofibrils. The filaments were thinner and smoother, while the protofibrils were thicker and shorter, which are shown in Fig. 1 a2. In the presence of ions, the growth of glucagon aggregates/fibrils varied significantly with respect to different ions. When glucagon was co-incubated with 100 μM Cu(II), the protofibrils elongated about 2–3 times faster than that of without ions, as illustrated in Fig. S1 (ESI†) where the average fibril length was plotted versus incubation time. Amazingly, we found that numerous amyloid fibrils congregated together (Fig. 1 b1–b3), producing spherical assemblies similar to the phenomena reported by Yagi et al., where Aβ supramolecular assemblies appeared when sodium dodecyl sulfate was introduced.42 In our case, it is likely that in the presence of Cu(II), salt bridges play a prominent role in the specific combination and stabilization of His residue of glucagon thus forming nuclei rapidly.13 The radial pattern of fibril growth suggested the formation of largely clustered seeds in the solution. When the incubation was prolonged to three days, the spherical assemblies become much bigger and remarkably long fibrils were observed protruding from the spherulitic cores (Fig. S2b, ESI†). In contrast, only globular nanoparticles were observed when glucagon was incubated with 100 μM Fe(III) (Fig. 1 c1–c3). The size of these nanoparticles was about 3–5 nm, which did not change significantly upon prolonging the incubation time. Even after three days of incubation, only sparse fibrils with very short length were detected on the mica surface (Fig. S2c, ESI†), indicating that the formation of glucagon fibrils was apparently suppressed in the presence of Fe(III). These results highlight that Cu(II) and Fe(III) have crucial roles in the modulation of glucagon aggregation.
 |
| | Fig. 1 AFM images of the aggregates/fibrils of glucagon formed in the absence and presence of metal ions for the different time periods of incubation. (a1–a3): without ions; (b1–b3): Cu(II); (c1–c3): Fe(III). The incubation time periods for three sets samples were 5, 10 and 24 h, respectively. The final concentration of glucagon in all these samples was 1 mg ml−1, and the concentration of Cu(II) and Fe(III) was both 100 μM. “Protofibrils” and “filaments” are marked with colored arrows, respectively. Scan size: 6 × 6 μm2. | |
ThT fluorescence of glucagon fibrils formed with and without metal ions
It is known that ThT specifically binds with amyloid fibrils having cross-β-sheet structures, generating a new emission peak around 482 nm.43 Therefore, it has been widely applied in studying the kinetics of amyloid fibril formation. Fig. 2 shows the ThT fluorescence signals from different glucagon fibrils produced in the absence and presence of Fe(III) (100 μM) and Cu(II) (100 μM), respectively. According to Fig. 2, there is a hardly detectable increase of ThT fluorescence even after a long incubation in the presence of Fe(III), indicating that the fibril formation was heavily suppressed. As expected, the intensity of ThT fluorescence signals from glucagon in the absence and presence of Cu(II) remarkably enhanced with the increase of incubation time. These results are consistent with the above AFM analysis (Fig. 1). It should also be noted that, Cu(II) only induced a slight increase in ThT fluorescence intensity compared to that in the absence of ions, although the AFM results mentioned above suggested that glucagon fibrils in the presence of Cu(II) grew much faster than glucagon alone. The plausible interpretation for this phenomena is that the addition of Cu(II) has profoundly altered the structures of fibrils, as was reported that the intensity of ThT fluorescence was strongly depend on the different types (structures) of glucagon fibrils.44,45
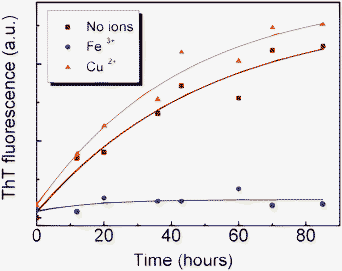 |
| | Fig. 2 ThT fluorescence of aggregates/fibrils of glucagon alone and with a concentration of 100 μM Fe(III) and Cu(II), respectively. The curves in color are the three best-fit curves. | |
Concentration-dependent effects of Cu(II) and Fe(III)
To further explore the effects of Cu(II) and Fe(III), which may provide an alternative means to the aggregation pathway and morphology of aggregates, we studied the concentration-dependent effects of these ions during glucagon aggregation by either co-incubating the glucagon monomers with Fe(III) or Cu(II) in different concentrations (25, 50 and 200 μM) at 37 °C. The morphologies of aggregate/fibrils of different samples were analyzed by AFM. In the case of Cu(II), the rate of fibril elongation become distinctly faster with an increasing amount of metal ions in the incubation solution (Fig. 3 a1–a3). At higher Cu(II) concentration (200 μM), we observed again the densely packed spherical assemblies with numerous long and extruding fibrils. In contrast, the fibrosis progress of glucagon was gradually suppressed with an increase of Fe(III) concentration as illustrated in Fig. 3 b1–b3. At a concentration of 25 μM, both filaments and protofibrils could be still found in addition to a few small aggregates. While at a higher concentration (200 uM), only large aggregates were observed, the size of these aggregates were about twice as big as those observed at lower concentration, probably due to the increasing electrostatic interaction.46 These findings strongly indicate that the morphologies of glucagon aggregates/fibrils heavily relied on the concentration of metal ions.
 |
| | Fig. 3 AFM images of glucagon aggregates/fibrils formed in the presence of Fe(III) or Cu(II) at different concentration. The incubation time for all these samples was 24 h. (a1–a3): Cu(II); (b1–b3): Fe(III); The concentrations of Cu(II) and Fe(III) were 25, 50 and 200 μM, respectively. The final concentration of glucagon in all these samples was 1 mg ml−1. Scan size: 6 × 6 μm2. | |
Secondary structures of glucagon fibrils as revealed by FTIR and CD spectra
We also monitored the secondary structures of protein during the fibrosis progress using micro-FTIR, a spectroscopic technique appropriate for the detection of β-sheet structures and a mixture of unordered and helical structures. Fig. 4 shows the representative micro-FTIR spectra of 1 mg ml−1 glucagon peptides co-incubated with Cu(II) and Fe(III) in different concentrations (50, 100, 200 μM) for 48 h incubation, respectively. In the presence of Cu(II), two broad peaks centered at around 1626 cm−1 and 1665 cm−1 were detected (Fig. 4a), which are attributed to the existence of the β-sheet structures and turns or coils in fibrils, respectively.44,45 Interestingly, the ratio of peak intensity at 1626 cm−1 to that at 1665 cm−1 increased with an increase of Cu(II) concentration, suggesting the augment of the amount of β-sheet structures and the descending of the turns or coils in fibrils. Quite distinct from Cu(II), the spectra of Fe(III) induced fibrils are a little more complicated. A subtle shift in peak position from 1635 to 1655 cm−1 was observed with the increase of Fe(III) concentration (Fig. 4b). In combination with the above AFM data, one can speculate that glucagon fibrils displayed the co-existence of β-sheet and α-helix structures in the presence of lower concentration of Fe(III) (50 μM), whereas the higher Fe(III) concentrations (>100 μM) yielded predominantly α-helix structures. The appearance of a new peak at around 1680 cm−1 in Fig. 4b was probably due to the formation of beta-turn structures.44 We further employed CD spectroscopy to test the conformational changes of glucagon aggregates/fibrils in the presence of metal ions, as shown in Fig. 5. Similarly to a previous report,44 the CD spectra of 1 mg ml−1 glucagon without any ions has a positive CD peak around 191 nm and a negative broad peak around 205–220 nm, suggesting the presence of β-sheet-rich structures. With an addition of 200 μM Cu(II), the β-sheet structures of fibrils became more distinctive. However, the glucagon aggregation formed at 200 μM Fe(III) shows a sharp peak at 205 nm, indicating that they primarily consist of the α-helix structures and possess less of the other structures, if any.
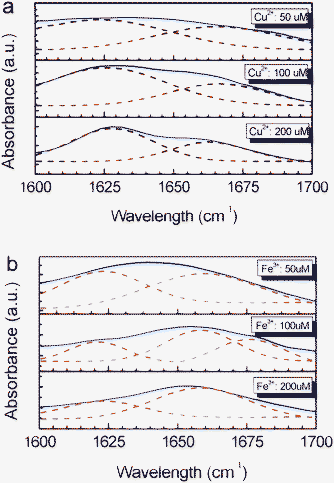 |
| | Fig. 4 FTIR spectra of glucagon aggregates/fibrils induced in the presence of Cu(II) and Fe(III) at different concentrations (50, 100 and 200 μM), respectively. (a): Cu(II); (b): Fe(III). The incubation time was 48 h for all these samples. The dashed curves in the figure are Gaussian fitting to the data. | |
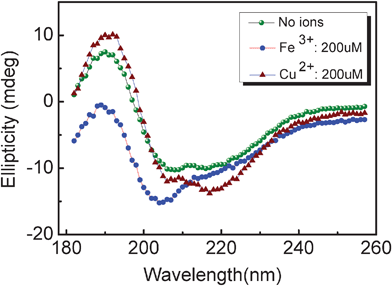 |
| | Fig. 5 CD spectra of the aggregates/fibrils formed by incubating glucagon in the absence and presence of 200 μM ions for 48 h, indicating that primary structures of Fe(III) and Cu(II) induced aggregates/fibrils were α-helix and β-sheet-rich structures, respectively. | |
Possible mechanism by which Cu(II) and Fe(III) interact with glucagon peptides
Copper and iron are essential bio-metals, which are very likely involved in the etiopathogenesis of many neurodegenerative disorders. Our AFM observation indicated that Cu(II) can facilitate the formation of glucagon fibrils, which was also supported by the ThT fluorescence studies. In addition, the analysis of FTIR (Fig. 4a) revealed that the amount of cross β-sheet structure of glucagon fibrils was distinctly dependent on the concentration of Cu(II). We speculate that Cu(II) accelerated glucagon fibril formation probably by specifically capturing intermolecular His-metal-His coordination.13 Our hypothesis is supported by the evidence that a similar conformation was previously determined for the well characterized Aβ(10–35) peptides, where both Zn(II) and Cu(II) associated with two His-13 residues from adjacent peptides arranged along the growing β-sheet surface,25,47 increasing the growth rate and stability of the growing fibril. To our surprise, at higher Cu(II) concentration, spherical assemblies were observed with abundant fibrils sticking out from cores. We postulate that salt bridges may play a central role in the stabilization and congregation of the clustered seeds or precursors at higher Cu(II) concentration, which probably initiated the fibril growth in a radial pattern.42
Unlike Cu(II), the fibrillation of glucagon was remarkably suppressed in the presence of Fe(III). Only small aggregates with size around 3–5 nm were observed. To understand how Fe(III) interacted with glucagon peptide, we co-incubated glucagon with Fe(II) in different concentrations (50, 100 and 200 μM) for 48 h as comparison. Interestingly, many fibrils were present on the mica surface as shown in Fig. S3 (ESI†), the morphologies of which were similar to that obtained in the absence of ions (Fig. 1a3). There was no distinct difference in fibril morphology with an increase in Fe(II) concentration, which was obviously different from the effect of Fe(III) on the fibril formation (Fig. 1c3). We hypothesize that it is likely that the oxidation ability of Fe(III) plays an important role in glucagon aggregation, as it has been proposed that the oxidation of methionine (Met) amino acid residue was catalyzed by Fe(III) bound amyloid peptides.48,49 Further support for our explanation comes from the studies on aggregation of PrP peptides.50 It is well accepted that Met residue has a strong tendency to be oxidized and thus believed to serve as scavengers for various oxidants. The oxidation of Met can have a drastic effect on protein structure due to a much higher polarity of methionine sulfoxide and thus inhibit the formation of PrP amyloid fibrils.50 To further test our hypothesis, we treated glucagon peptide with H2O2 (1 mg ml−1 glucagon, 10 mM H2O2), which is a common method to specifically oxidize methionine residues.51 Over long time incubation (72 h), only a few fibrils were present on the mica surface for the H2O2 treated glucagon sample (data not shown), indicating that the oxidation of Met might have tremendous impact on the glucagon fibrils formation.
4. Conclusions
In summary, we have investigated the effects of Cu(II) and Fe(III) during the fibrillation of glucagon peptides. Our results show that Cu(II) could be one of key elements to accelerate the formation of glucagon fibrils and simultaneously regulate their morphologies. Interestingly, it was found that the spherical assemblies were predominant at higher Cu(II) concentration. In several neurodegenerative diseases, radial and spherical aggregates of amyloid fibrils are often found in tissue deposits;52 therefore, Cu(II) may play dominant roles in the formation of amyloid fibrils and their deposition in vivo. In contrast, fibril formation was gradually suppressed with an increasing concentration of Fe(III) in the solution. We speculate that these metal ions may interfere with β-sheet formation via their specific binding to the glucagon peptides. There has been a growing sense that different amyloid supramolecular assemblies will have distinct biological impacts on the development as well as transmission of amyloidosis. Our results could contribute to a better understanding of the mechanisms involved in the interactions between amyloid peptides with metal ions. Future experiments are naturally required to test the cell toxicity of the aggregates of glucagon induced by various metal ions.
Acknowledgements
This work is funded by the National Natural Science Foundation of China (No. 11074137, 10975175, 90923002 and 21073222), the Qianjiang Talent Project of Zhejiang Province (No. 2011R10084), Ningbo Natural Science Foundation and the K. C. Wong Magna Fund in Ningbo University. Y. Zhang thanks the Max Planck Society for support of a partner group.
References
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS.
- C. X. Wang, A. H. Yang, X. Li, D. H. Li, M. Zhang, H. W. Du, C. Li, Y. Guo, X. B. Mao, M. Dong, F. Besenbacher, Y. Yang and C. Wang, Nanoscale, 2012, 4, 1895–1909 RSC.
- O. S. Makin, E. Atkins, P. Sikorski, J. Johansson and L. C. Serpell, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 315–320 CrossRef CAS.
- D. J. Selkoe, Nat. Cell Biol., 2004, 6, 1054–1061 CrossRef CAS.
- X. Z. Zhou, Y. H. Chen, B. Li, G. Lu, F. Y. C. Boey, J. Ma and H. Zhang, Small, 2008, 4, 1324–1328 CrossRef CAS.
- A. K. Chamberlain, C. E. MacPhee, J. Zurdo, L. A. Morozova-Roche, H. A. Hill, C. M. Dobson and J. J. Davis, Biophys. J., 2000, 79, 3282–3293 CrossRef CAS.
- L. Xiao, D. Zhao, W. H. Chan, M. M. Choi and H. W. Li, Biomaterials, 2010, 31, 91–98 CrossRef CAS.
- Y. Kusumoto, A. Lomakin, D. B. Teplow and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12277–12282 CrossRef CAS.
- R. Paparcone, S. W. Cranford and M. J. Buehler, Nanoscale, 2011, 3, 1748–1755 RSC.
- V. L. Colvin and K. M. Kulinowski, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8679–8680 CrossRef CAS.
- B. Morel, L. Varela, A. I. Azuaga and F. Conejero-lara, Biophys. J., 2010, 99, 3801–3810 CrossRef CAS.
- L. Huang, P. Manandhar, P. B. Chase and S. Hong, Langmuir, 2006, 22, 8635–8638 CrossRef CAS.
- D. P. Smith, D. G. Smith, C. C. Curtain, J. F. Boas, J. R. Pilbrow, G. D. Ciccotosto, T. L. Lau, D. J. Tew, K. Perez, J. D. Wade, A. I. Bush, S. C. Drew, F. Separovic, C. L. Masters, R. Cappai and K. L. Barnham, J. Biol. Chem., 2006, 81, 15145–54 CrossRef.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 8, 1517–1549 CrossRef.
- Y. Miller, B. Y. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra, D. M. Romano, M. A. Hartshorn, R. E. Tanzi and A. I. Bush, J. Biol. Chem., 1998, 273, 12817–12826 CrossRef CAS.
- R. A. Himes, G. Y. Park, A. N. Barry, N. J. Blackburn and K. D. Karlin, J. Am. Chem. Soc., 2007, 129, 5352–5353 CrossRef CAS.
- S. Laurent, M. R. Ejtehadi, M. Rezaei, P. G. Kehoe and M. Mahmoudi, RSC Adv., 2012 10.1039/c2ra01374f.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, J. Neurol. Sci., 1998, 158, 47–52 CrossRef CAS.
- D. P. Smith, G. D. Ciccotosto, D. J. Tew, M. T. Fodero-Tavoletti, T. Johanssen, C. L. Masters, K. J. Barnham and R. Cappai, Biochemistry, 2007, 46, 2881–1891 CrossRef CAS.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS.
- C. Ha, J. Ryu and C. B. Park, Biochemistry, 2007, 46, 6118–6125 CrossRef CAS.
- Y. Peng, C. S. Wang, H. H. Xu, Y. N. Liu and F. Zhou, J. Inorg. Biochem., 2010, 104, 365–370 CrossRef CAS.
- C. J. Frederickson, J.-Y. Koh and A. I. Bush, Nat. Rev. Neurosci., 2005, 6, 449–462 CrossRef CAS.
- J. Dong, J. E. Shokes, R. A. Scott and D. G. Lynn, J. Am. Chem. Soc., 2006, 128, 3540–3542 CrossRef CAS.
- B. N. Liu, A. Moloney, S. Meehan, K. Morris, S. E. Thomas, L. C. Serpell, R. Hider, S. J. Marciniak, D. A. Lomas and D. C. Crowther, J. Biol. Chem., 2010, 286, 4248–4256 CrossRef.
- H. R. Lucas, S. Debeer, M. S. Hong and J. C. Lee, J. Am. Chem. Soc., 2010, 132, 6636–6637 CrossRef CAS.
- X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi and A. I. Bush, Biochemistry, 1999, 38, 7609–7616 CrossRef CAS.
- L. E. Scott and C. Orvig, Chem. Rev., 2009, 109, 4885–4910 CrossRef CAS.
- J. S. Pedersen, J. Diabetes. Sci. Technol., 2010, 4, 1357–1367 Search PubMed.
- K. L. De Jong, B. Incledon, C. M. Yip and M. R. DeFelippis, Biophys. J., 2006, 91, 1905–1914 CrossRef CAS.
- M. Dong, M. B. Hovgaard, S. Xu, D. E. Otzen and F. Besenbacher, Nanotechnology, 2006, 17, 4003–4009 CrossRef CAS.
- S. K. Maji, M. H. Perrin, M. R. Sawaya, S. Jessberger, K. Vadodaria, R. A. Rissman, P. S. Singru, K.P. Nilsson, R. Simon, D. Schubert, D. Eisenberg, J. Rivier, P. Sawchenko, W. Vale and R. Riek, Science, 2009, 325, 328–332 CrossRef CAS.
- S. Onoue, K. Ohshima, K. Debari, K. Koh, S. Shioda, S. Iwasa, K. Kashimoto and T. Yajima, Pharm. Res., 2004, 21, 1274–1283 CrossRef CAS.
- S. Campioni, B. Mannini, M. Zampagni, A. Pensalfini, C. Parrini, E. Evangelisti, A. Relini, M. Stefani, C. M. Dobson, C. Cecchi and F. Chiti, Nat. Chem. Biol., 2010, 6, 140–147 CrossRef CAS.
- C. G. Glabe, J. Biol. Chem., 2008, 283, 29639–29643 CrossRef CAS.
- J. S. Pedersen, J. M. Flink, D. Dikov and D. E. Otzen, Biophys. J., 2006, 90, 4181–4194 CrossRef CAS.
- X. Zhou, J. Liu, B. Li, S. Pillai, D. Lin, J. H. Liu and Y. Zhang, Nanoscale, 2011, 3, 3049–3051 RSC.
- A. A. Golriz, T. Kaule, J. Heller, M. B. Untch, P. Schattling, P. Theato, M. Toda, S. Yoshida, T. Ono, H. J. Butt, J. S. Gutmann and R. Berger, Nanoscale, 2011, 3, 5049–5058 RSC.
- X. Liu, Y. Zhang, D. K. Goswami, J. S. Okasinski, K. Salaita, P. Sun, M. J. Bedzyk and C. A. Mirkin, Science, 2005, 307, 1763–1766 CrossRef CAS.
- R. K. Pai and S. Pillai, J. Am. Chem. Soc., 2008, 130, 13074–13078 CrossRef CAS.
- H. Yagi, T. Ban, K. Morigaki, H. Naiki and Y. Goto, Biochemistry, 2007, 46, 15009–15017 CrossRef CAS.
- H. LeVine, Protein Sci., 1993, 2, 404–410 CrossRef CAS.
- J. S. Pedersen, D. Dikov, J. L. Flink, H. A. Hjuler, G. Christiansen and D. E. Otzen, J. Mol. Biol., 2006, 355, 501–523 CrossRef CAS.
- C. B. Andersen, D. E. Otzen, G. Christiansen and C. Rischel, Biochemistry, 2007, 46, 7314–7324 CrossRef CAS.
- S. Xu, J. Phys. Chem. B, 2009, 113, 12447–12455 CrossRef CAS.
- J. J. Dong, J. M. Canfield, A. M. Mehta, J. E. Shokes, B. Tian, W. S. Childers, J. A. Simmons, Z. Mao, R. A. Scott, K. Warncke and D. G. Lynn, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13313–13318 CrossRef CAS.
- J. R. Fransson, J. Pharm. Sci., 1997, 86, 1046–1050 CrossRef CAS.
- A. Barman, W. Taves and R. Prabhakar, J. Comput. Chem., 2009, 30, 1405–1413 CrossRef CAS.
- O. V. Bocharova, L. Breydo, V. V. Salnikov and I. V. Baskakov, Biochemistry, 2005, 44, 6776–6787 CrossRef CAS.
- J. R. Requena, M. N. Dimitrova, G. Legname, S. Teijeira, S. B. Prusiner and R. L. Levine, Arch. Biochem. Biophys., 2004, 432, 188–195 CrossRef CAS.
- M. R. Krebs, C. E. Macphee, A. F. Miller, I. E. Dunlop, C. M. Dobson and A. M. Donald, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14420–14424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c2ra20651j |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.