DOI:
10.1039/C2RA20633A
(Paper)
RSC Adv., 2012,
2, 6079-6086
Diffusion of organic dyes in bovine serum albumin solution studied by fluorescence correlation spectroscopy†
Received
6th March 2012
, Accepted 2nd May 2012
First published on 6th June 2012
Abstract
The understanding of the transport of drugs and naturally occurring molecules in living cells and tissues requires a thorough knowledge of the diffusion behaviour of the molecular systems in these media. In this work, we studied the translational diffusion of three fluorescent molecules, electrically neutral coumarin 102 (C102), cationic rhodamine 6G (R6G) and anionic fluorescein (FL) in phosphate-buffered (pH 7) aqueous solutions of bovine serum albumin (BSA) protein in the absence and presence of common salt and urea using fluorescence correlation spectroscopy (FCS) by monitoring the fluorescence intensity fluctuations in a small confocal observation volume. The diffusion due to both free and BSA-bound molecules is observed in the case of the C102-BSA system. While no exchange between the bound and free states of the molecule is observed in this case, a rapid exchange between the two states is observed in the case of electrically charged hydrophilic dyes R6G and FL. This molecular picture, which is the first of its kind, is a reflection of a weaker binding of R6G and FL compared to C102 with the protein molecule. The binding sites of the probe molecules in BSA were identified based on the urea-induced change of diffusion of the probes in BSA.
1. Introduction
Serum albumins, which are the most abundantly found proteins in blood plasma, serve as depot proteins and transport proteins for a variety of compounds, like fatty acids, amino acids, bile salts, metals, hormones, drugs and pharmaceuticals.1–7 The nature of binding between small molecular probes and the albumins is an important topic of investigation for many reasons.8–13 Unlike human serum albumin (HSA), the crystal structure of bovine serum albumin (BSA), which has a molecular mass of 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 Da with 583 amino acids in a single polypeptide chain,14 is unknown. A model structure of BSA, which can be obtained from its amino acid sequence and crystal structure of related homologous protein, HSA, indicates that it is made up of three homologous domains (I, II, and III) divided into nine loops (L1–L9) by 17 disulphide bridges. Each domain in turn is the product of two sub-domains, A and B.15 BSA has two tryptophan (Trp) residues that possess intrinsic fluorescence. Trp-212 locates within a hydrophobic binding pocket in subdomain IIA, and Trp-134 on the surface of the albumin molecule in domain I.16 The principal ligand binding sites of BSA are located in the subdomain IIA and IIIA,17,18 which are termed binding sites I and II (Fig. 1).17,18
200 Da with 583 amino acids in a single polypeptide chain,14 is unknown. A model structure of BSA, which can be obtained from its amino acid sequence and crystal structure of related homologous protein, HSA, indicates that it is made up of three homologous domains (I, II, and III) divided into nine loops (L1–L9) by 17 disulphide bridges. Each domain in turn is the product of two sub-domains, A and B.15 BSA has two tryptophan (Trp) residues that possess intrinsic fluorescence. Trp-212 locates within a hydrophobic binding pocket in subdomain IIA, and Trp-134 on the surface of the albumin molecule in domain I.16 The principal ligand binding sites of BSA are located in the subdomain IIA and IIIA,17,18 which are termed binding sites I and II (Fig. 1).17,18
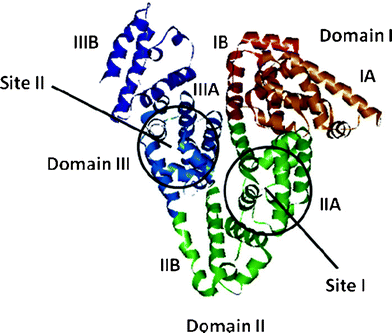 |
| | Fig. 1 A pictorial representation of the BSA model structure obtained from homology modelling indicating the domains and the binding sites. | |
In the present work, we explore the utility of FCS in studying the diffusion processes in phosphate-buffered (pH 7.0) BSA solution. FCS is a method in which spontaneous intensity fluctuation arising from a very small confocal volume (approx. 1 fL), defined by the tightly focused laser beam and a confocal pinhole, are correlated to quantify the temporal evolution of the system about its equilibrium state. Even though the basics of FCS were developed in the early 1970s,19,20 it became a popular technique after the development of confocal optics and highly sensitive detectors in the 1990s.21 Fluctuation of fluorescence arises due to diffusion of the fluorescent molecules in and out of the observation volume and changes in quantum yield due to different processes.21 Analysis of the correlation function provides information on the dynamic processes responsible for the fluctuation. The correlation function is fitted to a model, which contains information about the possible dynamic processes that may contribute to the fluctuation. Fitting of the data to these models gives dynamic parameters ranging from reaction times to diffusion coefficients. In the present study, we are particularly interested in the latter, which are related to the hydrodynamic radii of the diffusing molecules, thus offering a sensitive method for size determination.
FCS has been previously used to study the diffusion of fluorescent probes in many organized assemblies, which include vesicles and membranes,22–24 colloidal particles,25 ionic liquids,26–29 agarose gel,30,31 polymer matrices,32,33 micelles and 34–37 microemulsions,38 and to study protein aggregation39,40 and conformational changes of the protein.41–46 As BSA is a carrier protein, the mobility of the molecules in BSA solution is of fundamental importance in drug delivery and for the design of drug molecules. Considering the fact that a protein solution consists of both hydrophilic and hydrophobic domains, the diffusion of a probe may be location dependent with fast diffusion near the surface and slow diffusion in the core region. Andrade and coworkers studied the translational and rotational motion of BSA under physiological and acidic pH by FCS and time-resolved anisotropy measurements.47 Using anionic meso-tetrakis (p-sulfonatophenyl) porphyrin sodium salt (TSPP) as a probe molecule, they found a higher diffusion coefficient of TSPP in BSA at acidic pH. They attributed the result to a structural change of the protein at acidic pH and wobbling motion of the porphyrin at the binding site. Reyes and coworkers studied the effect of temperature and salt on the diffusion coefficient of BSA using a photon correlation spectroscopic technique.48 They found a decrease in the diffusion coefficient of BSA with ionic strength, which was rationalized in terms of conformational change of protein and protein aggregation at high ionic strength. Bhattacharyya and co-workers recently studied the interaction of C153 and R6G with immobilized human serum albumin (HSA) and determined the association sites of the probes in HSA by studying the effect of guanidinium hydrochloride on the binding kinetics of the probes with HSA.44 Salts, such as Na2SO4, NaCl, NaI, NaSCN, at high concentrations have significant effects on the conformational stability of biomacromolecules.49,50 Salt-induced association or dissociation of many biological macromolecules depends upon their “salting out” and “salting in” properties. The influence of an ion on the solubility and stability of protein follows the classical Hofmeister series.51 The anions appear to have a larger effect than the cations and are usually ordered CO32− > SO42− > S2O32− > H2PO4− > F− > Cl− > Br− > I− > ClO4− > SCN−.52 Early members of the series promote “salting out”, whereas the later members contribute to “salting in”. Earlier members provide stability to the protein structure, while the latter ones destabilize it. These salts can also affect the transport properties of BSA. Considering the fact that NaCl is found in nearly all biological systems, we investigated its effect on the diffusion properties of the dyes in BSA solution.
While the interaction between BSA and various fluorophores has been studied previously using conventional steady state and time-resolved techniques primarily to obtain information relating to the binding interaction, the present study focuses on the translational diffusion of electrically neutral and charged dye molecules (Chart 1) in aqueous solutions of BSA at neutral condition by FCS technique, an information that cannot be obtained by the conventional techniques. As is shown below, the results presented here provide deep insight into the nature of interaction of the molecular systems with BSA and on their diffusion behaviour.
 |
| | Chart 1 | |
2. Experimental section
2.1. Materials
Laser grade dye C102 was purchased from Exciton Inc. and used without further purification. R6G and Na2HPO4 (anhydrous) were obtained from Loba Chemie. FL and BSA (≥ 96%, fatty acid-free) were purchased from Sigma Aldrich and used as received. Sodium dihydrogen orthophosphate and urea were purchased from the local suppliers. Sodium chloride was purchased from Merck. MilliQ water was used for the present study.
2.2. Instrumentation
FCS measurements were carried out using a time-resolved confocal fluorescence microscope (MicroTime 200, PicoQuant), a modified Olympus IX 71, equipped with an Olympus UPlansApo NA 1.2 water immersion objective (60 ×), served as a microscope body. The output of the pulsed picosecond laser diodes (FWHM: 176 ps (405 nm), 144 ps (485 nm)) was coupled into the main optical unit using a polarization maintaining single mode optical fibre, guided through a dichroic mirror and then the collimated laser beam was directed into the entrance port of the inverted microscope. The sample was placed on a cover-slip and the directed laser beam was focused onto the sample using the water immersion objective. Fluorescence was collected by the same objective and dichroic mirror, passed through a 430-nm-long pass filter (for 405 nm) and 510 nm-long pass (for 485 nm) and then the signal was spatially filtered by focusing onto a 50 μm diameter pinhole to cut the out-of-focus signals, re-collimated and directed onto a (50/50) beam splitter prior to entering to two single photon avalanche photodiodes (SPADs). The fluorescence correlation traces were generated by cross-correlating signals from the two SPAD detectors. The data acquisition was performed with PicoHarp 300 TCSPC module in a Time-Tagged Time-Resolved (TTTR) mode, which stores all relevant information for each detected photon for further data analysis. Data analysis of the individual correlation curves was performed using the SymPhoTime software of PicoQuant. The correlation function of the fluorescence intensity is given by21| | 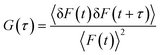 | (1) |
Here, <F(t)> is the average fluorescence intensity, δF(t) and δF(t + τ) are the deviations from the mean value at time t and (t + τ) and are given by
| | | δF(t) = F(t) − <F(t)>, δF(t + τ) = F(t + τ) − <F(t)> | (2) |
The correlation curves were fitted to the following 3D diffusion model, which includes the contribution of the triplet state as well:21
| | 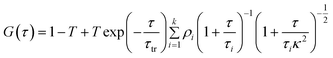 | (3) |
, where ρi is given by
| | 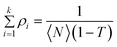 | (4) |
, which implies
| | 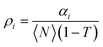 | (5) |
In the above expressions, the diffusion time, τi, denotes the average time that a dye molecule resides in the confocal volume, τtr is the lifetime of the triplet state of the molecule, τ is the delay or lag time, 〈N〉 is the average number of molecules in the observation volume, T is the fraction of the molecules in the triplet state, αi is the fraction of the molecules with diffusion time τi. κ is the structure parameter of the observation volume and is given by κ = ωz/ωxy, where, ωz and ωxy are the longitudinal and transverse radii, respectively, of the observation volume. We note in this context that eqn (3) is applicable when the fluorescence quantum yield of the free and the bound probes are very similar. That this condition is fulfilled in the present case is evident from the fact that C102 is a rigid probe, which shows very little fluorescence enhancement upon binding with BSA.9 The fluorescence efficiency of FL and R6G also is not sensitive to the polarity of the medium and, hence, no enhancement is expected upon binding.
The structure parameter of the excitation volume was calibrated using R6G in water of known diffusion coefficient (426 μm2 s−1).53,54 The estimated excitation volume was found to be 0.8 fL for 485 nm excitation and 0.4 fL for 405 nm excitation. At this excitation volume the obtained Dt value of R6G (440 μm2 s−1) is very close to the reported value. The translational diffusion coefficient (Dt) was calculated using the following equation employing the τi value obtained from the fit to eqn (3).
| | 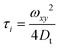 | (6) |
The concentrations of the samples were maintained at ∼10–20 nM throughout the experiment. Excitation power was 3.6 μW for 485 nm and 3.7 μW for 405 nm excitation. All the experiments were carried out in phosphate-buffered aqueous solution (pH 7, 0.02 M) at 25 °C. Each measurement was repeated several times (at least 15 times) to check the reproducibility of the data. The diffusion coefficients reported in this manuscript represent average values obtained from these measurements. The size of error is calculated from the deviation from the average value.
3. Results and discussion
3.1. Diffusion in bulk solvent
Fig. 2 shows the autocorrelation curves of the three dye molecules. The translational diffusion coefficient (Dt) estimated from these data are 640 and 420 μm2 s−1 for C102 and FL, respectively. These Dt values are in good agreement with the reported values shown in Table 1.37,53,54 The hydrodynamic radii of these dyes estimated from the Stokes Einstein equation are 0.34 ± 0.03, 0.51 ± 0.01 and 0.51 ± 0.01 nm for C102, FL and R6G, respectively.
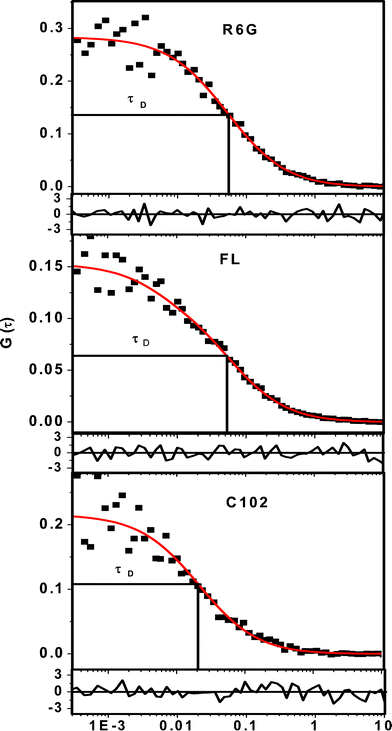 |
| | Fig. 2 Fluorescence correlation data points and best fits to those for R6G, FL and C102 in phosphate-buffered solution (pH = 7). λex for FL, R6G is 485 nm and 405 nm for C102. Residuals are shown at the bottom of each plot. | |
Table 1 Diffusion coefficients of the dyes in aqueous solution
| Medium |
|
D
t (μm2 s−1) |
|
| |
C102 |
R6G |
FL |
|
The quantity in brackets is the literature value of Dt from ref. 37.
The quantity in brackets is the literature value of Dt from ref. 53 .
The quantity in brackets is the literature value of Dt from ref. 54.
|
| Phosphate-buffered (pH = 7.0) |
640 ± 60 (600)a |
440 ± 15 (426)b |
420 ± 10 (425)c |
3.2. Diffusion in BSA solution
3.2.2. R6G and FL.
Unlike C102, the fluorescence correlation data could not be fit to a two-diffusion model for hydrophilic probes R6G and FL. Considering the hydrophilic nature of these systems and their relatively weak association with BSA,56 one expects exchange of the fluorescent probes between the free and bound state to be faster than the diffusion time through the observation volume. Hence, one obtains an average diffusion coefficient in these measurements with contributions from both the free and bound molecules. The treatment of the correlation data that takes into consideration the exchange dynamics of the system is as follows:57
Association of a fluorescent guest (A) with a non-fluorescent host (H) yielding a fluorescent complex (AH) can be represented as
| | 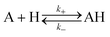 | (9) |
, where k+ and k− are the association and dissociation rate constants, respectively. If DA and DAH represent the diffusion coefficients of the free and bound molecules, respectively, the average diffusion coefficient of the system, ![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) t is given by
t is given by
| | | t = xADA + xAHDAH | (10) |
Where, xA and xAH are the mole fractions of the respective components. Under this condition, using eqn (6) and (10), one can express the average diffusion time, ![[small tau, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d4.gif) i, as:
i, as:
| | 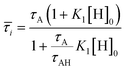 | (11) |
, where K1 is the binding constant of the system and is given by, K1 = k+/k−.
With increasing [H]0 value the observed diffusion time, i, shifts from τA to τAH. Fig. 5 shows the normalized correlation curves for FL as a function of the concentration of BSA. The K1 value is determined from the plot of ivs. [H]0 (Fig. 6). Thus the estimated K value for FL (1.50 ± 0.04 × 105 M−1) is 5 times higher than that obtained from the equilibrium dialysis method (2.8 × 104 M−1).56 However, a higher binding constant of ∼106 M−1 is also reported in the literature.58,59
![Normalized best fit correlation curves of FL for [BSA] of (i) 0 (ii) 8 (iii) 16 (iv) 25 (v) 40 (vi) 60 and (vii) 70 μM. The best fit lines along with the raw data points and fit parameters corresponding to each correlation curves are provided as supporting information.](/image/article/2012/RA/c2ra20633a/c2ra20633a-f5.gif) |
| | Fig. 5 Normalized best fit correlation curves of FL for [BSA] of (i) 0 (ii) 8 (iii) 16 (iv) 25 (v) 40 (vi) 60 and (vii) 70 μM. The best fit lines along with the raw data points and fit parameters corresponding to each correlation curves are provided as supporting information†. | |
![A plot of the mean diffusion times (i) of FL vs. [BSA]. The line represents the fit to the data according to eqn (11) from which K1 is estimated.](/image/article/2012/RA/c2ra20633a/c2ra20633a-f6.gif) |
| | Fig. 6 A plot of the mean diffusion times (i) of FL vs. [BSA]. The line represents the fit to the data according to eqn (11) from which K1 is estimated. | |
3.3 Effect of NaCl on diffusion
NaCl can have a significant effect on the diffusion behavior of the dyes in BSA as it affects the protein stability by altering the electrostatic interaction between the charged amino acid residues.60,61 The correlation curves for the systems in an aqueous solution of BSA (8 μM, pH 7) in the presence of various concentrations of NaCl are shown in Fig. 7 and the estimated values of the diffusion coefficients for the systems are collected in Table 2. Among all of the probes studied, C102 shows little effect upon addition of NaCl. The Dt value of C102 changes from 65 to 50 μm2 s−1 upon treatment with 1.5 M NaCl. For R6G, a steady decrease in the diffusion coefficient is observed with an increase in the concentration of NaCl.62
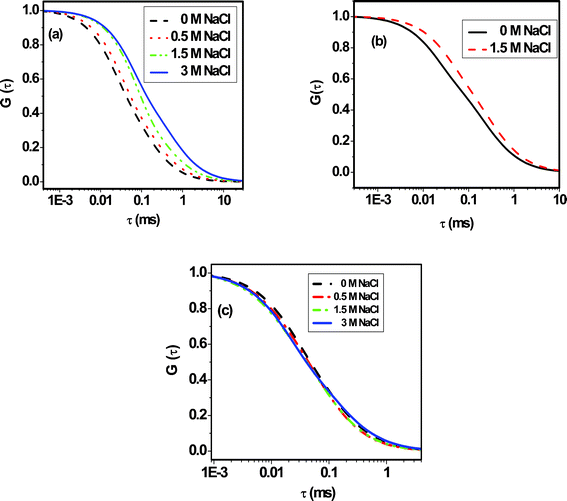 |
| | Fig. 7 The effect of NaCl on the correlation curves (normalized) of (a) R6G (b) C102 and (c) FL in the presence of a constant amount of (8 μM) of BSA. The raw data and best fit to them for each correlation curve are given in the supplementary information†. | |
Table 2 Diffusion coefficients of the dye molecules in BSA solution (8 μM) for different concentrations of NaCl
| Medium |
|
D
t (μm2 s−1) |
|
| |
C102 |
R6G |
FL |
| BSA (8 μM) |
65 ± 10 |
280 ± 40 |
270 ± 35 |
| BSA (8 μM) + 0.5 M NaCl |
|
170 ± 15 |
390 ± 15 |
| BSA (8 μM) + 1.5 M NaCl |
50 ± 5 |
90 ± 10 |
310 ± 15 |
| BSA (8 μM) + 3.0 M NaCl |
|
45 ± 5 |
220 ± 10 |
This observation can be rationalized as follows. Damodaran and Kinsella, while studying the binding of 2-nonanone with BSA, observed a decrease in the free ligand concentration in the solution upon addition of NaCl.49,50 This implies that with increasing NaCl concentration more dye molecules are transferred from the bulk to the protein phase and thus contribute to the decrease in the diffusion coefficient. It is not difficult to understand why the effect of NaCl is more pronounced in the case of R6G compared to C102. As the binding is relatively weaker in the case of R6G, more free molecules are present in the aqueous phase and their passage to the protein phase mainly leads to the significant change in the diffusion coefficient. Whereas in the case of C102, due to strong binding most molecules are in the bound state and hence little decrease in the diffusion coefficient is observed. It is difficult to suggest whether this small decrease is due to the structural change of BSA or not because there is small decrease in the diffusion coefficient of C102 in phosphate-buffered solution at pH 7.0 upon addition of 1.5 M NaCl (Fig. S8 and Table S1 of the supporting information†).
The case of FL is found to be not so straightforward as the diffusion coefficient passes through a maximum with an increase in the concentration of NaCl. Even though this implies that more than one factor – something that strengthens binding and something else that weakens it – contributes to this complex behavior, we are currently not in a position to comment on what these factors are. We have verified by performing several measurements that the trend is reproducible. We plan to address this point in more detail at a later stage.
3.4. Effect of urea on diffusion
The effect of urea on the correlation curves of the dyes in aqueous solution (pH 7) of BSA (8 μM) are shown in Fig. 8 and the estimated diffusion coefficients of the probes are listed in Table 3. Except R6G, the diffusion coefficients increase in all cases upon addition of urea.63 This increase is found to be most pronounced in the case of C102. The Dt value in this case increases by a factor of ∼4 in the presence of 4 M urea. As urea is a strong denaturing agent, it disrupts the native structure of the protein and affects the binding sites64 by exposing them to bulk water. As binding is the strongest in the case of C102, the effect of urea is most prominent in this case. In the case of FL, for a lower concentration of BSA (8 μM), when a large number of molecules are in the aqueous solution in the free state, urea-induced denaturation of protein does not have much impact on the diffusion of FL. But at higher BSA concentration, this change is more appreciable upon denaturation. Nearly a 2-fold enhancement in diffusion is observed in 60 μM BSA solution upon treatment with urea. This behavior is consistent with the fact that FL binds to the hydrophilic sites of BSA. The diffusion coefficient of R6G does not show any change in the presence of urea even at a higher concentration of BSA when most of the R6G molecules are in the bound state. This is perhaps a reflection of the fact that positively charged R6G molecules remain bound to the negatively charged amino acid residues (at pH 7) of the denatured protein. However, as denatured BSA has a larger hydrodynamic radius (4.3 nm) compared to native BSA (3.6 nm),65 one expects a 1.2-fold decrease of the Dt value of R6G upon addition of 4 M urea. Since this is not the case, it is evident that additional factor is involved, which nullifies the effect of the increasing hydrodynamic radius of denatured BSA on the Dt value. This factor is simply the change in binding strength between R6G and BSA on denaturation. Recently, Bhattacharyya and co-workers have shown a 1.2-fold decrease in the binding constant between R6G and HSA upon addition of 5 M guanidinium hydrochloride.44 Hence, it is not surprising to find a situation where the two factors counter-balance each other resulting in very little or negligible change in the Dt value of R6G in BSA.
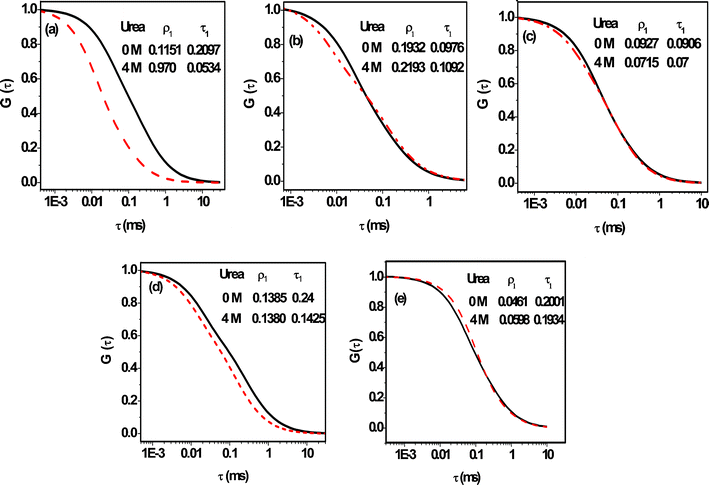 |
| | Fig. 8 The effect of urea (4 M) on the normalized correlation curves of (a) C102, (b) R6G and (c) FL in the presence of 8 μM BSA. Traces in (d) and (e) highlight the effect of the same amount of urea (4 M) on the correlation curves of FL and R6G in the presence of a larger quantity of BSA (60 μM). The curves with solid and dashed lines represent 0 and 4 M urea conditions, respectively. | |
Table 3 Diffusion coefficients (Dt, in μm2 s−1) of the dyes in 8 μM BSA and 60 μM BSA in the absence and presence of urea
| Fluorophores |
D
t in 8 μM BSA |
D
t in 60 μM BSA |
| 0 M urea |
4 M urea |
0 M urea |
4 M urea |
| C102 |
65 ± 10 |
250 ± 25 |
|
|
| R6G |
280 ± 40 |
250 ± 35 |
100 ± 10 |
105 ± 20 |
| FL |
270 ± 35 |
295 ± 45 |
90 ± 5 |
155 ± 20 |
The effect of urea on the diffusion of the fluorescent molecules allows identification of the binding sites of the probes in BSA, as the diffusion of the hydrophilic probes is not much affected by urea, but that of the hydrophobic probe is highly affected, implying that the hydrophobic domains are unfolded upon addition of urea without affecting the structure of hydrophilic domains. Bhattacharyya and co-workers also observed a similar behaviour recently in HSA upon addition of guanidinium hydrochloride, which is also a denaturing agent.44 One can perhaps obtain more definite information on the location of the probe molecules from the recent work of Leggio et al., who observed a multistep unfolding process of HSA induced by urea.65 They proposed two intermediates during the denaturation process. In the presence of 3 M urea domain I begins to open up, while domain II and III remain closed. At 4.35 M urea, domain II becomes unfolded. Domain III opens up only at high urea concentration (C > 8 M). In an earlier work, the binding site of C102 in HSA was assigned to subdomain IIA by a molecular docking study.9 Taking these literature reports into consideration along with careful consideration of our data we can conclude that C102 binds to the hydrophobic region IIA of BSA, which is almost completely denatured in the presence of 4 M urea showing a large increase in diffusion coefficient. R6G is expected to be associated with domain IIIA, which retains its structure even in the presence of 4 M urea. As FL shows small enhancement upon denaturation by urea, this probe is not entirely associated with domain III and may also be located in between subdomains IIA and IIIA.
4. Conclusion
The diffusion behaviour of electrically neutral and charged dye molecules in protein solution has been investigated using the FCS technique. The measured diffusion coefficients provide an insight into the nature and strength of the interaction between the guest and host, revealing the heterogeneity of the binding sites of the protein and fast exchange between the bound and free states of the molecules. The common salt and urea-induced changes of the diffusion behaviour allow identification of the association sites of the probes with the protein molecule.
Acknowledgements
This work is supported by the J. C. Bose Fellowship (to AS) and PURSE Grant (to the University of Hyderabad) of the Department of Science and Technology (DST). Thanks are also due to Council of Scientific and Industrial Research (CSIR) for the Fellowships (JRF/SRF) to S. P., K. S. and A. P.
References
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CAS.
- B. Sengupta and P. K. Sengupta, Biochem. Biophys. Res. Commun., 2002, 299, 400 CrossRef CAS.
- S. Makino, J. A. Reynolds and C. Tanford, J. Biol. Chem., 1973, 248, 4926 CAS.
- Y. Zhang and D. E. Wilcox, J. Biol. Inorg. Chem., 2002, 7, 327 CrossRef CAS.
- J. K. Choi, J. Ho, S. Curry, D. Qin, R. Bittman and J. A. Hamilton, J. Lipid Res., 2002, 43, 1000 Search PubMed.
- K. Kamikubo, S. Sakata, S. Nakamura, T. Komaki and K. Miura, J. Protein Chem., 1990, 9, 461 Search PubMed.
- X. Z. Feng, Z. Liu, L. J. Yang, C. Wang and C. L. Bai, Talanta, 1998, 47, 1223 CrossRef CAS.
- S. Chatterjee and T. S. Srivastav, J. Porphyrins Phthalocyanines, 2000, 4, 147 Search PubMed.
- B. Bhattacharya, S. Nakka, L. Guruprasad and A. Samanta, J. Phys. Chem. B, 2009, 113, 2143 CrossRef CAS.
- S. K. Pal, D. Mandal, D. Sukul, S. Sen and K. Bhattacharyya, J. Phys. Chem. B, 2001, 105, 1438 CrossRef CAS.
- A. Samanta, B. K. Paul and N. Gucchait, Biophys. Chem., 2011, 156, 128 CrossRef CAS.
- Y. J. Hu, Y. Liu and J. B. Wang, J. Pharm. Biomed. Anal., 2004, 36, 915 CrossRef CAS.
- Y. Q. wang, H. M. Zang and G. C. Zang, J. Pharm. Biomed. Anal., 2007, 43, 1869 CrossRef CAS.
-
J. F. Foster, Albumin Structure, Function and Uses, Pergamon Press, Oxford, UK, 1977 Search PubMed.
- A. Papadopoulou, R. J. Green and R. A. Frazier, J. Agric. Food Chem., 2005, 53, 158 CrossRef CAS.
- Y. Moriyama, D. Ohta, K. Hadiya, Y. Mitsui and K. Takeda, J. Protein Chem., 1996, 15, 265 CrossRef CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052 CAS.
- D. Magde, W. W. Webb and E. Elson, Phys. Rev. Lett., 1972, 29, 705 CrossRef CAS.
- D. Magde, E. L. Elson and W. W. Webb, Biopolymers, 1974, 13, 29 CrossRef CAS.
-
J. R. Lackowicz, Principle of Fluorescence Spectroscopy, 2006, Springer, New York, chapter 24 Search PubMed.
- J. Korlach, P. Scwille, W. W. Webb and G. W. Feigenson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 8461 CrossRef CAS.
- A. Benda, M. Benes, V. Marecek, A. Lhotsky, W. T. Hermens and M. Hof, Langmuir, 2003, 19, 4120 CrossRef CAS.
- J. Humpolickova, E. Gielen, A. Benda, J. Fagulova, J. Vercammen, M. VandeVen, M. Hof, M. Ameloot and Y. Engelborghs, Biophys. J., 2006, 91, L23 CrossRef.
- C. B. Muller, A. Loman, W. Richtering and J. Enderlein, J. Phys. Chem. B, 2008, 112, 8236 CrossRef CAS.
- J. H. Werner, S. N. Baker and G. A. Baker, Analyst, 2003, 128, 786 RSC.
- A. Sarkar, M. Ali, G. A. Baker, S. Y. Tetin, Q. Ruan and S. Pandey, J. Phys. Chem. B, 2009, 113, 3088 CrossRef CAS.
- J. Guo, G. A. Baker, P. Hillesheim, S. Dai, R. W. Shaw and S. M. Mahurin, Phys. Chem. Chem. Phys., 2011, 13, 12395 RSC.
- D. Sasmal, A. K. Mandal, T. Mondal and K. Bhattacharya, J. Phys. Chem. B, 2011, 115, 7781 Search PubMed.
- N. Fatin-Rouge, K. J. Wilkinson and J. Buffle, J. Phys. Chem. B, 2006, 110, 20133 CrossRef CAS.
- N. Fatin-Rouge, K. Starchev and J. Buffle, Biophys. J., 2004, 86, 2710 CrossRef CAS.
- A. Michelman-Ribeiro, H. Boukari, R. Nossal and F. Horkay, Macromolecules, 2004, 37, 10212 CrossRef CAS.
- T. Cherdhirankorn, A. Best, K. Koynov, K. Peneva, K. Muellen and G. Fytas, J. Phys. Chem. B, 2009, 113, 3355 CrossRef CAS.
- R. Erhardt, M. Zhang, A. Boker, H. Zettl, C. Abetz, P. Frederik, G. Krausch, V. Abetz and A. H. E. Muller, J. Am. Chem. Soc., 2003, 125, 3260 CrossRef CAS.
- R. Nörenberg, J. Klingler and D. Horn, Angew. Chem., Int. Ed., 1999, 38, 1626 CrossRef.
- H. Zettl, Y. Portnoy, M. Gottlieb and G. Krausch, J. Phys. Chem. B, 2005, 109, 13397 CrossRef CAS.
- S. Ghosh, U. Mandal, A. Adhikari and K. Bhattacharyya, Chem.–Asian J., 2009, 4, 948 Search PubMed.
- G. R. Burnett, G. D. Rees, D. C. Steytler and B. H. Robinson, Colloids Surf., A, 2004, 250, 171 Search PubMed.
- B. Sahoo, J. Balaji, S. Nag, S. K. Kaushalya and S. Maiti, J. Chem. Phys., 2008, 129, 075103 CrossRef.
- R. Ghosh, S. Sharma and K. Chattopadhyay, Biochemistry, 2009, 48, 1135 Search PubMed.
- K. Chattopadhyay, S. Saffarian, E. L. Elson and C. Frieden, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14171 CrossRef CAS.
- E. Sherman, A. Itkin, Y. Y. Kuttner, E. Rhoades, D. Amir, E. Haas and G. Haran, Biophys. J., 2009, 94, 4819 Search PubMed.
- U. Haupts, S. Maiti, P. Schwille and W. W. Webb, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13573 CrossRef CAS.
- D. K. Das, T. Mondal, A. K. Mandal and K. Bhattacharyya, Chem.–Asian J., 2011, 6, 3097 Search PubMed.
- D. K. Das, A. K. Das, A. K. Mandal, T. Mondal and K. Bhattacharyya, ChemPhysChem, 2012, 13, 1949 Search PubMed.
- D. K. Sasmal, T. Mondal, S. S. Mojumdar, A. Choudhury, R. Banerjee and K. Bhattacharyya, J. Phys. Chem. B, 2011, 115, 13075 Search PubMed.
- S. M. Andrade, S. M. B. Costa, J. W. Borst, A. V. Hoek and A. J. W. G. Viser, J. Fluoresc., 2008, 18, 601 CrossRef CAS.
- L. Reyes, J. Bert, J. Fornazero, R. Cohen and L. Heinrich, Colloids Surf., B, 2002, 25, 99 Search PubMed.
- S. Damodaran and J. E. Kinsella, J. Biol. Chem., 1981, 256, 3394.
- S. Damodaran and J. E. Kinsella, J. Biol. Chem., 1980, 255, 8503 Search PubMed.
- F. Hofmeister, Arch. Exp. Pathol. Pharmakol., 1888, 24, 247.
- Y. Zhang and P. S. Cremer, Curr. Opin. Chem. Biol., 2006, 10, 658 CrossRef CAS.
- Z. Petrasek and P. Schwille, Biophys. J., 2008, 94, 1437 CrossRef CAS.
- P. Kapusta, Technical Note, PicoQuant GmbH, July 2010, Rev1 Search PubMed.
- A. K. Gaigalas, J. B. Hubbard, M. McCurley and S. Woo, J. Phys. Chem., 1992, 96, 2355 CrossRef CAS.
- L. O. Andersson, A. Rehnstorm and D. L. Eaker, Eur. J. Biochem., 1971, 20, 371 Search PubMed.
- W. Al-Soufi, B. Reija, M. Novo, S. Felekyan, R. Kühnemuth and C. A. M. Seidel, J. Am. Chem. Soc., 2005, 127, 8775 CrossRef CAS.
- N. Barbero, E. Barni, C. Barolo, P. Quagliotto, G. Viscardi, L. Napione, S. Pavan and F. Bussolino, Dyes Pigm., 2009, 80, 307 CrossRef CAS.
- An important point to note here is that while measuring the diffusion behavior, BSA at higher concentrations (μM) contributes to the fluorescence signal and, hence, can influence the correlation curves. To avoid any error arising from this effect, the experiments were conducted with low excitation power (3.6 μW for 485 and 3.7 μW for 405 nm). The correlation function and count rate histogramm of 25 μM BSA and 60 μM is shown in the supplementary information (Figs S17 and S18†). The counts of BSA alone is negligible compared to the probes in BSA.
- S. Damodaran, Int. J. Biol. Macromol., 1989, 11, 2 Search PubMed.
- M. Yamasaki, H. Yano and K. Aoki, Int. J. Biol. Macromol., 1991, 13, 322 Search PubMed.
- That these changes are indeed due to NaCl-induced structural changes of BSA (and not due to any other effect arising out of salt addition) is evident from the results of control experiments (Figs S6–S8 and Table S1 of the supporting information†) performed on the probe molecules in aqueous buffered solution in the presence of NaCl without using any BSA.
- That this increase is not because of urea-induced change of the refractive index of the medium and consequent change of the confocal volume is verified by examining the effect of urea on the diffusion of the probes in the absence of BSA. The results of the control experiments are provided in the supplementary information. (Figs S3–S5 and Table S1†).
- K. Shikama, J. Biochem., 1968, 64, 55 Search PubMed.
- C. Leggio, L. Galantini, P. V. Konarev and N. V. Pavel, J. Phys. Chem. B, 2009, 113, 12590 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20633a/ |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![Normalized correlation curves of C102 for [BSA] of (i) 0 (ii) 1.0 (iii) 4.0 (iv) 8.0 (v) 16.0 μM. The curves shown are the best fits to eqn (3). The raw data points, best fits to them and the fit parameters are provided in the supporting materials (Fig. S1).](/image/article/2012/RA/c2ra20633a/c2ra20633a-f3.gif)
![A plot of the bound fraction (αb) of C102 vs. [BSA]. The line indicates the fit to the data according to eqn (7).](/image/article/2012/RA/c2ra20633a/c2ra20633a-f4.gif)