Mechanistic aspects of the horseradish peroxidase-catalysed polymerisation of aniline in the presence of AOT vesicles as templates†
Katja
Junker
a,
Giorgia
Zandomeneghi
b,
Zengwei
Guo
ac,
Reinhard
Kissner
b,
Takashi
Ishikawa
d,
Joachim
Kohlbrecher
e and
Peter
Walde
*a
aDepartment of Materials, ETH Zürich, Wolfgang-Pauli-Str. 10, CH-8093 Zürich, Switzerland. E-mail: peter.walde@mat.ethz.ch; Fax: +41 44 63 21265; Tel: +41 44 63 20473
bDepartment of Chemistry and Applied Biosciences, Wolfgang-Pauli-Str. 10, CH-8093 Zürich
cSwerea IVF, Argongatan 30, SE-43153 Mölndal, Sweden
dDepartment of Biology and Chemistry, Paul Scherrer Institute, CH-5231 Villigen PSI, Switzerland
eLaboratory of Neutron Scattering, ETH Zürich & Paul Scherrer Institute, CH-5232 Villigen PSI, Switzerland
First published on 14th May 2012
Abstract
The mechanism of the horseradish peroxidase (HRP)–H2O2-catalysed polymerisation of aniline in the presence of AOT vesicles was investigated. AOT (= bis-(2-ethylhexyl)sulfosuccinate) served as vesicle-forming surfactant and dopant for obtaining at pH = 4.3 and room temperature within 24 h under optimal reaction conditions the green emeraldine salt form of polyaniline in 90–95% yield. Based on UV/VIS/NIR and EPR measurements carried out during the polymerisation reaction, and based on changes in aniline and H2O2 concentrations and HRP activity, a mechanism is proposed. According to this “radical cation mechanism” chain growth occurs on the vesicle surface through addition of aniline radical cations to the growing polymer chain. H2O2 plays two essential roles, to oxidise the heme group of HRP, and to oxidise the growing polymer chain for allowing the stepwise addition of new aniline radical cations. The entire reaction can be divided into three kinetically distinct phases. In the first rapid phase (5–10 min), the actual polymer formation takes place to yield the emeraldine salt form of polyaniline in its bipolaron state. In the second and third slower phases (1–2 days) the bipolarons transform into polarons with unpaired electrons. During the reaction, the HRP activity is decreasing until the enzyme becomes inactive after polymer formation. Reactions carried out with partially deuterated anilines were analysed by 2H magic-angle spinning (MAS) NMR spectroscopy to demonstrate the regioselectivity of the chain growth: para-coupling of the aniline units clearly dominates. Association of the formed polyaniline with the vesicle membrane is evident from cryo-TEM and SANS measurements.
1 Introduction
The use of enzymes as catalysts for the in vitro synthesis of polymers currently is a promising field of research.1–4 In fact, enzymatic reactions usually are carried out under environmentally friendly, mild conditions, in water-rich reaction media. In addition, some enzyme-catalysed reactions are not regio- and stereoselective with narrow substrate specificity,5,6 thereby allowing enzymatic transformations of a variety of non-natural substrates.6,7 Furthermore, some of the enzymes tolerate high amounts of organic solvents, allowing the transformation of monomers and the formation of polymers which have low water solubilities.1–4 These characteristics make enzymatic reactions potentially useful for the development of energy efficient polymerisation processes.Among the various enzymes that have been studied as polymerisation catalysts, lipases and peroxidases belong to the most prominent ones.1–4 Both can transform non-natural or non-physiological monomers. However, lipases and peroxidases play a mechanistically different role in catalysing polymerisation reactions.8 Firstly, lipases catalyse condensation reactions that involve the controlled formation of covalent bonds via the formal transfer of electron pairs. In contrast, the application of peroxidases involves the (transient) formation of molecules with (delocalised) unpaired electrons (radicals) that may react in various ways with other molecules to form a variety of covalent bonds. Secondly, lipases take part directly in each reaction step that leads to the formation and elongation of oligomers and polymers, while peroxidases seem to play a role as catalysts only to activate (i.e. oxidise) the monomers.8 Therefore, the application of peroxidases1–4,9–12 to obtain polymeric products with a desired and defined chemical structure is more challenging. One example is the peroxidase-catalysed polymerisation of aromatic monomers, such as the polymerisation of aniline with horseradish peroxidase (HRP) and H2O2 as oxidant.13–17
In the HRP–H2O2-catalysed polymerisation of aniline, native HRP first is activated by H2O2 in a two-electron redox reaction to the oxidised form of HRP (so-called compound I) which then oxidises two molecules of aniline to yield two anilino radicals. This occurs in two one-electron redox reactions, via the formation of compound II. Finally, native HRP is recovered and two molecules of water are obtained (Scheme 1).18,19 This sequence of reactions is the peroxidase cycle of HRP.20 The actual formation of dimeric, oligomeric and polymeric forms of aniline then probably occurs away from the active site of HRP, in bulk solution or at appropriate interfaces. This latter possibility is of particular importance since a number of studies have shown that the presence of interfaces in the form of dissolved or dispersed structure-controlling agents, so-called templates (anionic polymers or surfactant assemblies), has a considerable influence on the chemical structure and properties of the polyaniline obtained with peroxidases.13–17,21,22 Under optimal reaction conditions, the templates promote the formation of the conductive, mainly linear emeraldine salt form of polyaniline (PANI, Scheme 2),23 with electronic transitions in the NIR region of the absorption spectrum, above about 900 nm, and at about 440 nm.23,24 Particularly efficient are templates containing the anionic form of a strong acid, for example sulfonated polystyrene.13a–c In the absence of templates, considerable amounts of ill-defined reaction products are formed, rich in unwanted ortho-coupled aniline units.9e In the presence of the templates the reaction becomes regioselective (mainly para N–C-coupling of the aniline units). Furthermore, although PANI itself is water insoluble, the templates prevent PANI precipitation from the aqueous solution in which the reaction is conducted.13–17 In addition, the templates also act simultaneously as dopants (counter ions), thereby promoting electron delocalisation within the PANI chain, i.e. the template dopants influence the extent of polaron formation, which is essential for intra-chain electron mobility. The characteristic polaron and bipolaron states of the emeraldine salt form of PANI are shown in Scheme 2. In the emeraldine salt form of PANI, polarons are favoured over bipolarons most likely due to a gain in resonance energy when bipolarons are converted into polarons.25,26
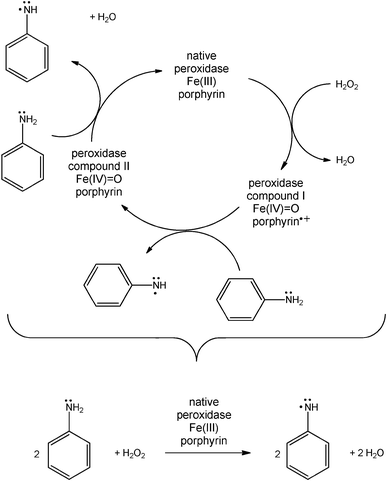 | ||
| Scheme 1 The peroxidase cycle of horseradish peroxidase isoenzyme C (HRP) with aniline as reducing substrate.18–20 The active site of HRP contains a heme group composed of Fe(III) and porphyrin. Native HRP is oxidised by hydrogen peroxide (H2O2) in a two-electron redox reaction to compound I, an oxyferryl species with a π-cation radical located on the heme group. Afterwards, two molecules of aniline are oxidised to anilino radicals in two one-electron redox reactions. Compound II is another oxyferryl species of HRP. Overall, in each peroxidase cycle, HRP catalyses the oxidation of two aniline molecules with one H2O2 to yield two anilino radicals and two water molecules. Compound II may also react with H2O2 to yield compound III containing Fe(III) with bound dioxygen, Fe(III)–O2•− (not shown); after the release of HO2• (hydroperoxyl radical), compound III turns back to native HRP.20 | ||
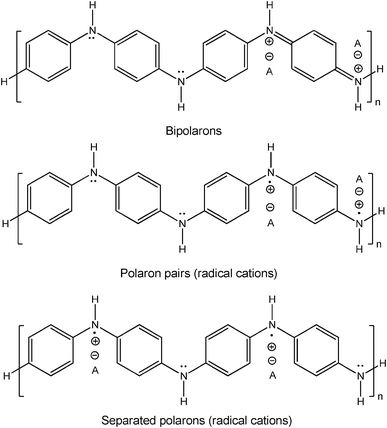 | ||
| Scheme 2 Emeraldine salt form of polyaniline (PANI), showing the chemical structures of the broadly accepted bipolaron and polaron states typical for conductive polymers.23 Bipolarons are diamagnetic and polarons are paramagnetic (electron spin S = ½). The presence of polarons with their unpaired electrons is responsible for the characteristic absorption in the NIR region of the absorption spectrum of the emeraldine salt form of PANI (“free-carrier tail” with absorption above about 900 nm due to the π → polaron band transition; in addition, there is also absorption around 440 nm due to polaron band → π* transition).24,35 Furthermore, the emeraldine salt form of PANI can be detected by EPR spectroscopy due to the presence of the paramagnetic polarons.26 A− stands for the counter ion (dopant). | ||
The aim of our investigation was to contribute to a better mechanistic understanding of the peroxidase–H2O2-catalysed polymerisation of aniline in the presence of bis-(2-ethylhexyl)sulfosuccinate (AOT) vesicles as chemical structure-controlling templates (Scheme 3). Recently, we have shown that AOT vesicles are excellent templates for this reaction,17 with improved properties as compared to the previously used SDBS–decanoic acid system.16 We now propose a mechanism for this complex reaction. Besides using conventional UV/VIS/NIR and EPR measurements to follow the progress of the reaction, 2H magic-angle spinning (MAS) NMR experiments with partially deuterated anilines were used for the first time to gain insight into (i) the general progress of the reaction, and (ii) the regioselectivity in the polymer chain growth (para versus ortho coupling of the monomer units). The determination of the changes of the aniline and H2O2 concentrations and the changes in HRP activity during polymerisation were particularly useful. Only with this type of analysis we could gain a deeper insight into the mechanism. Compared to our previous work,16,17 we have now a better understanding of the optimal reaction conditions that were determined empirically. We believe that our increased level of understanding is important for further developing this field of research.
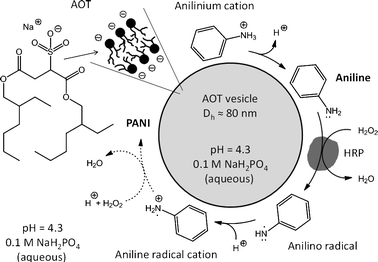 | ||
| Scheme 3 Highly schematic illustration of the reaction system investigated. The chemical structure-controlling templates used are anionic vesicles with an average diameter of about 80 nm, prepared at pH 4.3 in 0.1 M NaH2PO4 solution. The phosphate solution is present inside and outside of the vesicles. The vesicles are mainly unilamellar, formed from the negatively charged surfactant AOT, sodium bis-(2-ethyl-hexyl)sulfosuccinate. Horseradish peroxidase isoenzyme C (HRP) as catalyst and aniline as substrate (the pKa value of the anilinium cation is 4.6)16 are added to the vesicles, followed by addition of H2O2 which initiates the oxidation of aniline to the anilino radical, see Scheme 1. At pH = 4.3, the anilino radical must become protonated since the pKa value of the aniline radical cation is about 7.1.16 The formation of the aniline radical cation initiates the formation of aniline dimers, oligomers and finally polymeric forms of aniline (polyaniline, PANI). Under optimal reaction conditions, the PANI obtained is mainly the emeraldine salt form of polyaniline (Scheme 2) with at least some of the counter ions, A−, being AOT. Due to the positive charge of most of the aniline molecules and HRP (the pI of horseradish peroxidase isoenzyme C is 8.8),18b the reaction occurs in close proximity to the surface of the negatively charged vesicles.17 The formed PANI remains associated with the vesicles (no precipitation of reaction products). | ||
2 Materials and methods
2.1 Materials
Sodium bis(2-ethylhexyl)sulfosuccinate (AOT ≥ 99%), 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS2−(NH4+)2 ≥ 99%), ammonium peroxydisulfate (≥98%), 2,6-dibromoaniline (≥97%), and sodium deuteroxide (∼40% in D2O, 99.5 atom % D) were from Fluka. Aniline (99.8%) was purchased from Acros. Sodium phosphate monobasic (≥99%), polyaniline emeraldine salt (Aldrich, 42,832–9, Lot: #S24379–155, Mw > 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), polyaniline emeraldine base (Aldrich, 476706, Lot: #02903AD, Mw = 10000; and 530689, Lot: #14705TD, Mw = 65000), N-phenyl-1,4-phenylene-diamine (= p-aminodiphenylamine, PADPA, 98%), 4-bromoaniline (97%), sodium di-n-butylsulfosuccinate, benzene (99.5%), methanol-d1 (99.5 atom % D), deuterium oxide (99.9 atom % D), deuterium chloride (35 wt. % solution in D2O, 99 atom % D), chloroform (stabilised with ethanol, 99.8%), acetone (spectrophotometric grade ≥ 99.5%), and pinacyanol chloride (H2O content 1.9 mol mol−1) were from Sigma-Aldrich. Methanol-d3 (1,1,1-trideuteromethanol, 99.5 atom % D) was from Cambridge Isotope Laboratories. Hydrogen peroxide (30%), perchloric acid (60%), mercury (Pro Analysi), acetonitrile (uvasol 99.9%), and sodium di-n-hexylsulfosuccinate (for synthesis) were purchased from Merck. Sodium (sticks in paraffin oil 99%) was purchased from Chemie Brunschwig. Oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV), Ti-TPyp was from TCI Europe.
000), polyaniline emeraldine base (Aldrich, 476706, Lot: #02903AD, Mw = 10000; and 530689, Lot: #14705TD, Mw = 65000), N-phenyl-1,4-phenylene-diamine (= p-aminodiphenylamine, PADPA, 98%), 4-bromoaniline (97%), sodium di-n-butylsulfosuccinate, benzene (99.5%), methanol-d1 (99.5 atom % D), deuterium oxide (99.9 atom % D), deuterium chloride (35 wt. % solution in D2O, 99 atom % D), chloroform (stabilised with ethanol, 99.8%), acetone (spectrophotometric grade ≥ 99.5%), and pinacyanol chloride (H2O content 1.9 mol mol−1) were from Sigma-Aldrich. Methanol-d3 (1,1,1-trideuteromethanol, 99.5 atom % D) was from Cambridge Isotope Laboratories. Hydrogen peroxide (30%), perchloric acid (60%), mercury (Pro Analysi), acetonitrile (uvasol 99.9%), and sodium di-n-hexylsulfosuccinate (for synthesis) were purchased from Merck. Sodium (sticks in paraffin oil 99%) was purchased from Chemie Brunschwig. Oxo[5,10,15,20-tetra(4-pyridyl)porphyrinato]titanium(IV), Ti-TPyp was from TCI Europe.
Horseradish peroxidase isoenzyme C (HRPC Grade I, 280 U mg−1, RZ ≥ 3, Lot. number 0240160000) was purchased from Toyobo Enzymes. The peroxidase concentration was determined spectrophotometrically by using ε403 = 1.02 × 105 M−1 cm1 as molar absorbance.27
2,3,4,5,6-Pentadeuteroaniline (aniline-d5, 98 atom % D) was from Aldrich. 4-Deuteroaniline and 2,6-dideuteroaniline were synthesised by reductive debromination of 4-bromoaniline and 2,6-dibromoaniline, according to a published procedure.28 The products were purified by reductive electrolysis (to reduce trace amounts of mercury ions present) and by final distillation. The 1H NMR, 13C NMR and mass spectra of the purified products are given in Fig. S1 and Fig. S2, ESI.†
2.2 General analytical methods
Absorption measurements in the ultraviolet (UV), visible (VIS) and near infrared (NIR) region of the spectrum were recorded with either a Perkin Elmer Lambda 19, a Perkin Elmer Lambda 20, a Jasco V-670 or a Analytik Jena AG Specord S 600 diode array instrument at 25 °C, using quartz cuvettes from Hellma with path lengths of 0.1 cm, 0.5 cm or 1 cm. For UV/VIS absorption measurements of small volumes (below 0.1 mL) a NanoDrop ND1000 instrument from Thermo Scientific was used.Electron paramagnetic resonance (EPR) measurements were recorded with a Bruker EMX X-band spectrometer equipped with a TM cavity. The standard reaction solution was prepared as described below and transferred into a flat measuring cell immediately after hydrogen peroxide addition. The measurement itself usually was started 2 min after H2O2 addition. Spectra were measured at X-band microwave frequency with a modulation frequency of 100 kHz and modulation amplitudes of 1–4 G at room temperature. The line width (ΔHpp) represents the peak-to-peak distance in the EPR spectrum.
Dynamic light scattering (DLS) measurements were carried out with a ZetaSizer Nano from Malvern. Disposable polystyrene semi-micro cuvettes from BRAND with a path length of 1 cm were used.
Fourier transformed infrared (FTIR) spectra of PANI were recorded with an Alpha instrument from Bruker. About 1 mg PANI was finely ground with 100 mg dry KBr and pellets were pressed.
1H NMR (300 MHz) and 13C NMR (75.5 MHz) measurements were recorded on a Bruker AVANCE 300 instrument.
For the EI-MS analysis of the deuterated anilines synthesised, a Waters Micromass AutoSpec Ultima instrument with MassLynx 4.0 software was used.
The cryo transmission electron microscopy (Cryo-TEM) analysis of the AOT vesicles was performed as described previously.17
Small angle neutron scattering (SANS) measurements were performed at the SANS-I facility at the Swiss neutron spallation source SINQ, Paul Scherrer Institute Switzerland.29 Data were collected with a wavelength of 0.8 nm at three sample-to-detector distances, 2 m, 5 m and 18 m, to cover a wave vector transfer range of 0.0 to 3 nm−1 and normalised by standard procedures to absolute scale using H2O as a reference. The sample itself was prepared in D2O to get a large scattering contrast.
Three samples were measured (in 0.1 cm quartz cuvettes from Hellma at room temperature): a 3 mM AOT vesicle suspension, a reaction solution before the start of the reaction (sodium dihydrogen phosphate solution was added instead of H2O2 solution) and a reaction solution 16.5 h after H2O2 addition.
2.3 Vesicle preparation
AOT vesicles were prepared by the freezing–thawing extrusion method.17 First, a thin film of AOT was prepared inside a 100 mL round bottom glass flask by dissolution of 0.178 g (0.4 mmol) AOT in 5 ml chloroform and subsequent evaporation of the solvent. The film was dried overnight under high vacuum and then hydrated with 20 mL of a 0.1 M sodium dihydrogen phosphate solution (pH = 4.3). The obtained 20 mM AOT suspension was then frozen in liquid nitrogen and thawed in a water bath heated to 60 °C. After repeating this freezing–thawing procedure nine times, the suspension was extruded ten times through a 200 nm pore size Nucleopore polycarbonate membrane and ten times through a 100 nm pore size membrane, using The Extruder from Lipex Biomembranes (Vancouver, Canada). The size of the obtained vesicles was determined by DLS. The average hydrodynamic vesicle diameter measured immediately after preparation was about 80 nm with a polydispersity index of about 0.1.17 As shown previously,17 the size of the vesicles increased during storage.The vesicle suspension used for the SANS measurements were prepared as follows. A 0.1 M sodium dihydrogen phosphate solution was prepared by dissolving 239.9 mg sodium dihydrogen phosphate in 20 ml D2O. The “pH” was then adjusted to 4.22 (pH meter reading) using deuterium chloride and sodium deuteroxide.
2.4 Polymerisation of aniline by HRP–H2O2 in the presence of AOT vesicles
Polymerisation of aniline was carried out in a 0.1 M sodium dihydrogen phosphate solution at pH = 4.3 at room temperature. The typical reaction volume was 500.15 μL. The reactions were carried out in Eppendorf tubes. All components of the reaction system, except H2O2, were mixed first in the following sequence (the concentrations are given for the optimal reaction conditions): 357.2 μL sodium dihydrogen phosphate solution (0.1 M, pH = 4.3), 75 μL AOT vesicle suspension (20 mM), 49.9 μL aniline solution (40 mM in 0.1 M sodium dihydrogen phosphate solution, pH adjusted to 4.3 with HCl), and 6.8 μL HRP solution (3.4 mg HRP powder dissolved in 1 mL 0.1 M sodium dihydrogen phosphate solution, pH = 4.3, yielding 67.45 μM HRP, spectrophotometrically determined). After mixing, the reaction was triggered by adding 11.25 μL of a freshly prepared H2O2 solution (200 mM in water). The initial concentrations in the reaction system were: 3.0 mM AOT, 4.0 mM aniline, 0.92 μM HRP and 4.5 mM H2O2, pH = 4.3 (0.1 M NaH2PO4). The typical reaction time was 24 h.For the SANS measurements, the various solutions (AOT vesicle suspension, aniline solution and HRP solution) were made with a sodium dihydrogen phosphate solution prepared with D2O instead of H2O. Furthermore, the 200 mM H2O2 stock solution was prepared from 30% H2O2 by diluting with D2O.
2.5 Quantification of aniline, of H2O2 and of HRP activity during polymerisation
To determine the aniline concentration in the reaction mixture at different reaction times, 30 μL of the reaction solution were added to 1470 μL acetonitrile. After centrifugation to remove the reaction products, the UV/VIS spectrum of the supernatant solution was recorded. From the height of the signal at λ = 238 nm the aniline concentration was calculated by using ε238 = 10600 ± 300 M−1 cm1 as molar absorbance, as determined from a calibration curve prepared with known amounts of aniline, see Fig. S3, ESI.† The determined value agreed well with the value determined previously by Rajendiran and Swaminathan for aniline in pure acetonitrile (9550 M−1 cm−1).30 The reaction yield was defined through the amount of unreacted aniline. If no aniline remained in the system, the reaction yield was taken as 100%.
To follow the decrease of hydrogen peroxide in the reaction mixture the Ti-TPyp assay described by Takamura and collaborators was used.31 In a typical assay 125 μL of the reaction solution (undiluted or diluted in the case of samples with high amounts of H2O2) were added to 125 μL of a 4.8 M perchloric acid solution. Immediately afterwards, 125 μL of a 50 μM Ti-TPyp solution (prepared in 50 mM HCl) were added. After five min, 875 μL water were added and the sample was centrifuged. The absorption at λ = 432 nm (AS) of the supernatant was recorded with a 0.5 cm cell. The value was compared to the one of a blank sample (AB) that did not contain H2O2. The H2O2 content was then determined by measuring ΔA432 = AB − AS, and using a calibration curve obtained from measurements with known amounts of H2O2, see Fig. S4, ESI.†
The activity of HRP in the reaction mixture was measured by using ABTS2− as substrate. Oxidation of ABTS2− yields a stable radical, ABTS•−, which has an absorption maximum at λ = 414 nm.32 A typical assay solution (Vtot = 3 ml) contained 2692 μL 0.1 M sodium phosphate solution (pH = 6.0), 150 μL ABTS2− solution (5 mM in 0.1 M sodium phosphate solution, pH = 6.0), 8 μL of the reaction solution and 150 μL H2O2 solution (1 mM in water). The initial ABTS2− and H2O2 concentrations were 0.25 mM and 0.05 mM, respectively. The rate of ABTS•− formation was determined by measuring the increase in absorbance at λ = 414 nm every second during a period of t = 1 min at T = 25 °C using a 1 cm cell. ΔA414/Δt was taken as a measure for the activity of HRP (ε414(ABTS•−) = 3.6 × 104 M−1 cm1).32 See also ESI.†
2.6 2H MAS NMR measurements of the reaction system
2H magic-angle spinning (MAS) NMR measurements were carried out on a Bruker Biospin AVANCE spectrometer operating at 400 MHz 1H Larmor frequency, using a 4 mm double resonance Bruker MAS probe. Spectra were collected at 27 °C and MAS frequencies of about 2000 Hz. The single pulse experiments were measured with a 90° pulse of 16 μs, spectral width of 30 ppm, and acquisition time of 555.7 ms. The number of scans was 500, the recycle delay was 20 s, and a line broadening of 4 Hz was applied before Fourier transform.The usual reaction solution (see above) was prepared and divided into two samples before the addition of the H2O2 solution. For the measurements during the polymerisation reaction, the H2O2 solution was added and spectra were recorded, beginning about three hours after the start of the reaction. For measurements of solutions representing the situation before the start of the reaction, a sodium dihydrogen phosphate solution (0.1 M, pH = 4.3) was added instead of the H2O2 solution. All measurements were carried out in triplicates.
2.7 Investigation of aqueous solutions of sodium di-n-butylsulfosuccinate
The possible formation of vesicles or micelles in 0.1 M sodium dihydrogenphosphate solution (pH = 4.3) was investigated by using the dye pinacyanol chloride, as described previously.33 Aggregate formation leads to a change in the absorption spectrum of pinacyanol chloride due to interactions between the positively charged dye and negatively charged aggregates. Various sample solutions containing different amounts of sodium di-n-butylsulfosuccinate were prepared, up to a total concentration of 20 mM. The concentration of pinacyanol chloride in each solution was 2.9 μM: 5 μL 0.35 mM pinacyanol chloride (in methanol) were added to 0.6 mL sample solution and the absorbance was measured at 606 nm with a 1 cm cell and plotted against concentration of di-n-butylsulfosuccinate, see Fig. S5, ESI.† As a result, sodium di-n-butylsulfosuccinate up to at least 20 mM does not aggregate.34 For comparison, micelle-forming di-n-hexylsulfosuccinate was also used and was found to aggregate when its concentration was above about 5 mM.342.8 Isolation of reaction products
To isolate the formed PANI, acetone was poured into the reaction solution which led to an immediate product precipitation. After 10 to 20 min of stirring, the precipitate was collected by centrifugation and thoroughly washed with water. Finally, the precipitate was washed with 1 M HCl solution and then again with water. The solid, protonated product (containing Cl− as counter ions) was then dried in high vacuum for one or two days.3 Results
3.1 Optimisation of the reaction conditions
Due to the complexity of the vesicular reaction system investigated (Scheme 3), it is not surprising that the chemical structure and the yield of the reaction product(s) obtained (PANI) depend on many parameters.17 In particular, variations of the pH and of the AOT, HRP, H2O2, and aniline concentrations have an influence on the reaction.17 We aimed at optimising the system in order to obtain (i) the emeraldine salt form of PANI (presence of unpaired electrons as prerequisite for the electrical conductivity) as main reaction product; (ii) high yield of the reaction; and (iii) a stable and homogeneous suspension (no precipitation). This latter property is important for possible applications of “PANI ink” for the fabrication of conductive patterns with an inkjet printer (currently under investigation).To decide whether the emeraldine salt form of PANI with its polaron states preferentially formed, UV/VIS/NIR spectra of the reaction mixture were recorded during the reaction and after reaching equilibrium. The following criteria were considered to be relevant for obtaining a “good quality” product: (i) high absorption at wavelengths above about 900 nm (π → polaron band transition, see Scheme 2) and high absorption at about 440 nm (peak originating from the polaron band → π* transition, see Scheme 2);24,35 (ii) low absorption at about 500 nm (absence of extensive branching);23,24,36 (iii) no peak maximum at around 700 nm (absence of overoxidised polymeric products);16,24 and (iv) presence of a peak at around 300 nm since this peak is known to be typical too for the emeraldine salt form of PANI (π → π* transition).23,24
Based on these criteria, the following reaction conditions were found to be optimal: 3.0 mM AOT, 4.0 mM aniline, 4.5 mM H2O2, 0.92 μM HRP (isoenzyme C), pH = 4.3 (0.1 M NaH2PO4), room temperature (T = 23–25 °C). We refer to these conditions as “optimal conditions” throughout the text. It may be that other conditions work similarly well. However, we have chosen the concentration range such that the continuous monitoring of the reaction system with a conventional UV/VIS/NIR spectrophotometer and cells with a path length of 0.1–1.0 cm was possible. The AOT vesicles were prepared by polycarbonate membrane extrusion, yielding mainly unilamellar vesicles with an average diameter of about 80 nm (see Materials and methods).
Under the optimal conditions the reaction yield always was between 90 and 95% (see below). With the previous conditions,17 yields of only about 70% were reached since in that earlier study the concentration of H2O2 (1.0 mM) was too low for the amount of aniline used (1.33 mM),17 as becomes obvious based on the following considerations.
H2O2 plays two roles in the entire reaction. Firstly, H2O2 activates HRP according to the peroxidase cycle (Scheme 1); one molecule of H2O2 is required for the oxidation of two molecules of aniline. Secondly, H2O2 partially oxidises the oligomeric and polymeric products for obtaining the desired half-oxidised, emeraldine salt form of PANI (Scheme 2); without this latter non-enzymatic oxidation, the emeraldine salt form of polyaniline can not be obtained. The stoichiometric equation for the entire reaction is given in Scheme 4. For 4n aniline molecules 5n − 1 molecules H2O2 are required, n being the number of tetrameric emeraldine salt units in the polymeric reaction product, as shown in Scheme 2. According to this equation the molar ratio of H2O2 to aniline, R*, at the start of the reaction varies between 1 (for n = 1) and 1.25 (for n > 50). Based on the reaction yield and on the UV/VIS/NIR spectrum of the reaction system measured after reaching reaction equilibrium, it turned out that a slight excess of added H2O2 over aniline is optimal (4.5 mM H2O2 and 4.0 mM aniline, corresponding to an R*-value of 1.13, resulting in 90–95% yield;37 with 3.0 mM H2O2 and 4.0 mM aniline (R* = 0.75), the yield was ≈70%, with 4.0 mM H2O2 (R* = 1.0) it was ≈85%; with 5.0 mM H2O2, corresponding to an R*-value of 1.25, the yield was again 90–95%, but the formation of substantial amounts of unwanted overoxidised products was evident from the UV/VIS/NIR spectrum (shoulder at ≈750 nm, data not shown).38 Under the previous conditions,17 the molar ratio of H2O2 to aniline was 0.75 (= 1.0 mM:1.33 mM), which clearly was too low for obtaining high reaction yields.
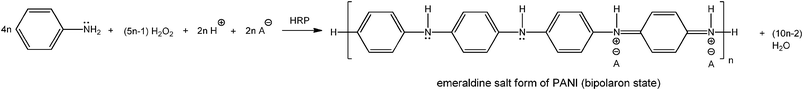 | ||
| Scheme 4 Stoichiometric equation for the HRP–H2O2-catalysed polymerisation of aniline into the emeraldine salt form of PANI; the bipolaron state is shown (see Scheme 2). A− represents the counter ion/dopant (probably mainly AOT). For each tetrameric repeating unit formed (n = 1), 3n H+ are released from the para-position of 3n aniline monomers | ||
Scheme 4 indicates that there should be no net release of protons for the HRP–H2O2-catalysed polymerisation of the neutral form of aniline (Ar–NH2) which is in clear contrast to the chemical polymerisation of aniline with ammonium peroxydisulfate, (NH4)2S2O8 as oxidant (release of sulfuric acid, i. e. H+),39,40 see Scheme S1a, ESI,† and compare with Scheme 4. For the HRP–H2O2-catalysed polymerisation of Ar–NH2, two protons (H+) actually are required for each tetrameric chain unit formed (Scheme 4). For the optimal reaction conditions, the situation is as follows: an amount of 2.0 mM H+ is needed for the polymerisation of 4.0 mM Ar–NH2. Since Ar–NH2 is in acid–base equilibrium with the anilinium cation (Ar–NH3+) and since the reaction is carried out at pH = 4.3, at which about 67% of all aniline molecules are present as Ar–NH3+ (pKa = 4.6),16 the consumption of Ar–NH2 during polymerisation results in a release of H+ from Ar–NH3+, until no more Ar–NH2 react and the level of unreacted Ar–NH2 stays constant. With a total concentration of aniline of 4.0 mM (= [Ar–NH2] + [Ar–NH3+]) at most 2.68 mM (= 0.67·4.0 mM) H+ are released from Ar–NH3+ during polymerisation due to this acid–base equilibrium. This is the case for 100% aniline consumption. Since 2.0 mM H+ are required for the polymerisation of 4.0 mM aniline (Scheme 4), a net excess release of 0.68 mM H+ from Ar–NH3+ (= 2.68 mM–2.0 mM) is expected if all initially present aniline is polymerised, which should result in a slight decrease in pH if the system is not buffered sufficiently.41 This is indeed what we observed. The measured pH of the reaction system changed from an initial value of 4.3 to 4.1 after completion of the reaction which agrees well with the calculated pH decrease based on the expected net release of H+, see ESI.†
3.2 Monitoring of changes occurring during the reaction
At the same time, the intensity at 400 nm and at 1000 nm first increased and then slightly decreased. During these changes in the NIR region of the spectrum a broad band with a maximum at about 1000 nm developed and slowly shifted with time to a slightly higher wavelength (small red shift). Another change occurred at about 310–320 nm (increase with time).
The time-dependent changes of A760, of A1000 and of the ratio of A1000:A500 are plotted in Fig. 1a (data for up to a reaction time of 24 h) and in Fig. 1b (data for the first 270 min only). The absorption spectrum of the reaction system obtained after 24 h is shown in Fig. 1c, illustrating the presence of those three bands that are characteristic for the emeraldine salt form of polyaniline, with highest absorption centred around 1020 nm, 410 nm, and 310 nm. Furthermore, the absorption around 500 nm was low.
![a, b: Time dependent changes of the VIS/NIR absorption spectrum during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles carried out at T = 25 °C. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), path length: 0.1 cm. Changes of the absorbance at λ = 1000 nm (A1000, filled square), at 760 nm (A760, empty circle) and changes in the ratio of A1000 to A500 (filled triangle) with reaction time. (a) For t = 0–1440 min (= 24 h); (b) for t = 0–270 min (= 4.5 h). Phase I, phase II and phase III of the reaction are indicated. c: UV/VIS/NIR absorption spectrum of the reaction system recorded after a reaction time of 24 h. Path length: 0.1 cm.](/image/article/2012/RA/c2ra20566a/c2ra20566a-f1.gif) | ||
| Fig. 1 a, b: Time dependent changes of the VIS/NIR absorption spectrum during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles carried out at T = 25 °C. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), path length: 0.1 cm. Changes of the absorbance at λ = 1000 nm (A1000, filled square), at 760 nm (A760, empty circle) and changes in the ratio of A1000 to A500 (filled triangle) with reaction time. (a) For t = 0–1440 min (= 24 h); (b) for t = 0–270 min (= 4.5 h). Phase I, phase II and phase III of the reaction are indicated. c: UV/VIS/NIR absorption spectrum of the reaction system recorded after a reaction time of 24 h. Path length: 0.1 cm. | ||
A1000:A500 was taken as one of the measures for the formation of the emeraldine salt form of PANI (see above). The higher the value of A1000:A500 was, the better we considered the “quality” of the reaction product. If, for example, the reactions were carried out with AOT vesicles at 2, 4, or 5 mM instead of 3 mM AOT, but otherwise identical conditions, A1000:A500 at final equilibrium was lower; at the same time the intensity of the peak at about 400 nm was also lower, see Fig. S8, ESI.† For this reason, 3 mM AOT was considered optimal in combination with 4 mM aniline, 4.5 mM H2O2 and 0.92 μM HRP (our optimal conditions).
The time-dependent changes in the UV/VIS/NIR spectrum of the reaction mixture for the optimal conditions can be divided into three phases: the first 5 min (phase I), from 5 min up to about 5 h (phase II), and above about 5 h (phase III). While phase I involved large changes in the absorption spectrum within a short time, the changes in phase II were moderate and slow, and in phase III they were small and even slower than in phase II (Fig. 1a, b).
![Time dependent changes of the intensity of the EPR spectrum during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles carried out at T = 25 °C. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−). a: Variation of the signal intensity with reaction time for t = 2–180 min. Phase I and phase II of the reaction are indicated. The relative intensity of a reaction system analysed after 22 h was 1.11 × 105. b: EPR spectrum of the reaction system recorded after t = 15, 17, 18, and 21.5 min (ΔHpp = 6.2–7.1 G, g-factor = 2.0071) and after 19.5 h (ΔHpp = 2.7 G, g-factor = 2.0070) . The arrows indicate the direction of the signal intensity changes with time.](/image/article/2012/RA/c2ra20566a/c2ra20566a-f2.gif) | ||
| Fig. 2 Time dependent changes of the intensity of the EPR spectrum during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles carried out at T = 25 °C. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−). a: Variation of the signal intensity with reaction time for t = 2–180 min. Phase I and phase II of the reaction are indicated. The relative intensity of a reaction system analysed after 22 h was 1.11 × 105. b: EPR spectrum of the reaction system recorded after t = 15, 17, 18, and 21.5 min (ΔHpp = 6.2–7.1 G, g-factor = 2.0071) and after 19.5 h (ΔHpp = 2.7 G, g-factor = 2.0070) . The arrows indicate the direction of the signal intensity changes with time. | ||
![Time dependent changes in the amounts of remaining aniline (filled circles) and H2O2 (empty triangles) and remaining HRP activity (empty squares) during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), T = 25 °C. 100% corresponds to the values measured before reaction start. Phase I and phase II of the reaction are indicated.](/image/article/2012/RA/c2ra20566a/c2ra20566a-f3.gif) | ||
| Fig. 3 Time dependent changes in the amounts of remaining aniline (filled circles) and H2O2 (empty triangles) and remaining HRP activity (empty squares) during the HRP-catalysed polymerisation of aniline in the presence of AOT vesicles. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), T = 25 °C. 100% corresponds to the values measured before reaction start. Phase I and phase II of the reaction are indicated. | ||
The activity of HRP (as measured with ABTS2−) was found to drop by about 70–80% during the first 5 min and then further dropped to reach after 30 min 10–15% of the initial value. After 24 h, the enzyme was completely inactive.
To better understand this undesired HRP inactivation, a series of control measurements were carried out. First, if HRP (0.92 μM) was incubated with 4.5 mM H2O2 ([H2O2]/[HRP] = 4.9 × 103) in the absence of aniline as reducing substrate, the HRP was inactivated with similar rate as during the aniline polymerisation; 64% of the initial activity was left after 4 min; after 20 min the enzyme activity reached 24% of its initial value, see Fig. S9, ESI.† These observations support previous findings that high concentrations of H2O2 lead to HRP inactivation, possibly due to hydroperoxyl (HO2•) formation.44,45
H2O2 is a known “suicide substrate”, the inactivation being particularly high at pH < 6.44
Second, if HRP (0.92 μM) was incubated together with AOT vesicles (3 mM AOT) in the absence of H2O2and in the absence of aniline, HRP remained fully active for at least 24 h, while without vesicles the activity dropped by about 25% during the same period of time, see Fig. S9, ESI.† This observation is in full agreement with our previous findings,17 demonstrating the stabilising effect of AOT vesicles on HRP.
Third, if new HRP (0.92 μM) was added to a reaction system obtained after reaching almost reaction equilibrium (t = 18 h), the activity of this newly added HRP rapidly dropped: after 4 h, the HRP was completely inactive, see Fig. S10, ESI.† This indicates that HRP inactivation is also caused by reaction products (or side products) formed during polymerisation.
HRP inactivation during the polymerisation of aniline was also observed if the HRP concentration was 0.092 μM instead of 0.92 μM. The percentage loss in activity after 5 min in both cases was very similar. With 0.092 μM HRP, however, the reaction yield reached only about 40%, i. e. 60% of the initially present aniline did not polymerise, see Fig. S14, ESI.† With 0.92 μM HRP, the yield always was between 90 and 95%, independent on whether the AOT concentration was 2, 3, 4, or 5 mM. The data for 3 mM AOT (optimal conditions) are given in Fig. 3.
:1.57 (HDO and D2O):0.25 (4-deuteroaniline). After 3 h reaction time, the peak at 7.33 ppm due to 4-deuteroaniline could not be detected anymore, while the intensity of the peak of deuterated water at 4.75 ppm clearly increased (Fig. 4b) with an integral relative to the integral of the CD3OH signal of 1.85. This increase in peak integral of deuterated water (Δwater = 1.85–1.57 = 0.28) was about the same as the decrease of the area of the 4-deuteroaniline signal (Δaniline = 0.25–0.00 = 0.25). The spectra recorded at later times (after a reaction time of up to18 h) showed that the positions and relative intensities of the peaks did not change significantly, indicating that D+ release from 4-deuteroaniline was complete after 3 h. The NMR analysis was repeated with two independently prepared reaction systems, obtaining very similar results. In fact, the average difference (Δwater − Δaniline) was 0.02 with maximum deviation of 0.04, thus close to zero. This indicates that D+ is released from the para-position of aniline during the reaction.
![2H MAS NMR spectra of the polymerisation reaction system before (left panel) and about three hours after starting the reaction, i.e. after H2O2 addition (right panel). [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), T = 27 °C. The peak at 3.27 ppm is assigned to CD3OH which was added at a concentration of 4 mM as an internal standard. The peak at 4.75 ppm is due to HDO and D2O, while the lines at lower field are assigned to deuterated aniline. (a) (b) Reaction mixture including 4-deuteroaniline. The peak integral ratios in (a) were: 1.00 (CD3OH) : 1.57 (HDO and D2O) : 0.25 (4-deuteroaniline). The integral ratios in (b) were: 1.00 (CD3OH) : 1.85 (HDO and D2O) : 0.00 (4-deuteroaniline). (c) (d) Reaction mixture with 2,6-dideuteroaniline. The integral ratios in (c) were: 1.00 (CD3OH) : 1.48 (HDO and D2O) : 0.62 (2,6-dideuteroaniline). In (d): 1.00 (CD3OH) : 1.42 (HDO and D2O) : 0.05 (2,6-dideuteroaniline). (e) (f) Reaction mixture with 2,3,4,5,6-pentadeuteroaniline. Integral ratios in (e): 1 (CD3OH) : 1.48 (HDO and D2O) : 1.62 (2,3,4,5,6-pentadeuteroaniline); in (f): 1.00 (CD3OH) : 1.68 (HDO and D2O) : 0.16 (2,3,4,5,6-pentadeuteroaniline).](/image/article/2012/RA/c2ra20566a/c2ra20566a-f4.gif) | ||
| Fig. 4
2H MAS NMR spectra of the polymerisation reaction system before (left panel) and about three hours after starting the reaction, i.e. after H2O2 addition (right panel). [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM; pH = 4.3 (0.1 M H2PO4−), T = 27 °C. The peak at 3.27 ppm is assigned to CD3OH which was added at a concentration of 4 mM as an internal standard. The peak at 4.75 ppm is due to HDO and D2O, while the lines at lower field are assigned to deuterated aniline. (a) (b) Reaction mixture including 4-deuteroaniline. The peak integral ratios in (a) were: 1.00 (CD3OH):1.57 (HDO and D2O):0.25 (4-deuteroaniline). The integral ratios in (b) were: 1.00 (CD3OH):1.85 (HDO and D2O):0.00 (4-deuteroaniline). (c) (d) Reaction mixture with 2,6-dideuteroaniline. The integral ratios in (c) were: 1.00 (CD3OH):1.48 (HDO and D2O):0.62 (2,6-dideuteroaniline). In (d): 1.00 (CD3OH):1.42 (HDO and D2O):0.05 (2,6-dideuteroaniline). (e) (f) Reaction mixture with 2,3,4,5,6-pentadeuteroaniline. Integral ratios in (e): 1 (CD3OH):1.48 (HDO and D2O):1.62 (2,3,4,5,6-pentadeuteroaniline); in (f): 1.00 (CD3OH):1.68 (HDO and D2O):0.16 (2,3,4,5,6-pentadeuteroaniline). | ||
If 2,6-dideuteroaniline was used instead of aniline (Fig. 4c,d), the intensity of the peak of deuterated water did not increase significantly during the polymerisation reaction (in Fig. 4c,d, Δwater = 0.06), although the peak at 7.26 ppm (originating from the two deuterium atoms in ortho-position of 2,6-dideuteroaniline) almost completely disappeared (Δaniline = 0.57). The average Δwater from three experiments on different samples was 0.07 ± 0.07. This indicates absence of significant ortho-couplings, clearly supporting prevalence of para-coupling of the monomers in the growing polymer chain.
If 2,3,4,5,6-pentadeuteraoaniline was used instead of aniline, the intensity of the peak of deuterated water again increased during polymerisation by about the same amount as when 4-deuteroaniline was used (Fig. 4e,f), i.e. for each aniline present about one D+ was released during polymerisation. The average difference between Δwater and Δaniline/5 was −0.02 ± 0.11, in agreement with predominance of para-coupling of the aniline monomers deduced from the previous experiments.
Please note that due to the presence of unpaired electrons in the emeraldine salt form of PANI and due to the fact that the formed polymer is bound to the vesicles, signals from PANI containing aniline repeating units with deuterium atoms bound to the aromatic rings (polymerisation of 2,6-dideuteroaniline and 2,4,5,6-pentadeuteraoaniline) could not be detected by these NMR measurements, as discussed previously.17
3.3 Cryo-TEM and SANS measurements
The reaction system was analysed by cryo-TEM. Fig. 5a–c shows images taken just before starting the reaction, i.e. before adding H2O2 (Fig. 5a), five minutes after initiating the reaction (Fig. 5b) and after 24 h (Fig. 5c). The electron micrographs show that the initially spherical vesicles underwent morphological changes during the polymerisation. Before addition of H2O2, the vesicles were rather uniform in size and mainly unilamellar with vesicle diameters in the range of 60–100 nm, see Fig. 5a. This is in agreement with the DLS analysis, see Materials and methods, and confirms previous data.17 In the electron microscopy images taken from the reaction system during the reaction, the presence of non-spherical vesicles with different sizes and an increase in polydispersity are obvious (Fig. 5b and Fig. 5c), in agreement with previous observations made in reactions carried out with vesicles formed from a 1:1 mixture of sodium dodecylbenzenesulfonate and decanoic acid16 or AOT.17 DLS measurements carried out during the polymerisation reaction also indicate that the reaction system became more heterogeneous with time (data not shown). Despite these changes in vesicle size and shape, the suspension remained stable (no visible precipitation).
![Cryo-TEM (a–c) and SANS (d) analysis of the reaction system. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM, T = 25 °C, pH = 4.3 (0.1 M H2PO4−). The cryo-TEM images were taken before starting the reaction by adding H2O2 (a); 5 min (b) and 24 h (c) after starting the reaction. The dark objects with high contrast seen in (b) and (c) are ice. SANS measurements were carried out of the vesicles, one day after vesicle formation, immediately before starting the reaction, and 16.5 h after starting the reaction, respectively. Length of the bar always 100 nm.](/image/article/2012/RA/c2ra20566a/c2ra20566a-f5.gif) | ||
| Fig. 5 Cryo-TEM (a–c) and SANS (d) analysis of the reaction system. [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM, T = 25 °C, pH = 4.3 (0.1 M H2PO4−). The cryo-TEM images were taken before starting the reaction by adding H2O2 (a); 5 min (b) and 24 h (c) after starting the reaction. The dark objects with high contrast seen in (b) and (c) are ice. SANS measurements were carried out of the vesicles, one day after vesicle formation, immediately before starting the reaction, and 16.5 h after starting the reaction, respectively. Length of the bar always 100 nm. | ||
SANS measurements showed that the bilayer thickness probably slightly increased from 1.85 (±0.08) nm (determined for AOT vesicles)46 to 1.92 (±0.08) nm (AOT vesicles after addition of aniline and HRP) to about 4.4 nm (after 16.5 h), see Fig. 5d, demonstrating the localisation of the formed reaction product in the region of the bilayer. A closer inspection of the SANS pattern indicates that the bilayer became inhomogeneous in the sense that it contained a lot of clusters with dimensions of about 4.4 nm. SANS measurements further indicated that the reaction system clearly became more polydisperse during the reaction (Fig. 5d), in agreement with DLS and cryo-TEM measurements. The size of the vesicles before starting the reaction was found to be about 80 nm, again in good agreement with the value determined by DLS.
3.4 Product analysis by FTIR spectroscopy
The FTIR spectrum of the reaction product isolated from the reaction mixture 48 h after starting the reaction showed the characteristic peaks due to the presence of the emeraldine salt form of PANI,17,23 see Fig. S11, ESI.† The product was isolated by acetone precipitation, followed by washing with water and then HCl (see Materials and methods), leading to almost complete removal of AOT. The recorded FTIR spectrum did not allow a detailed analysis of the molecular homogeneity of the product obtained, since the peaks are broad and their assignment is often ambiguous. Furthermore, and more importantly, the product obtained from a reaction carried out in the absence of AOT (Fig. S11, ESI†) had a similar, although not identical, FTIR spectrum, despite the fact that the two products clearly had rather different molecular properties, as discussed below. Wudl et al.25 pointed out that the presence of a band at 750 cm−1 is diagnostic for the formation of “ortho–para polymers”. The FTIR spectrum of PANI obtained in the absence of the vesicles had a clear band at 751 cm−1, while the FTIR spectrum of PANI obtained in the presence of the vesicles had a much smaller band at this wavelength (Fig. S11, ESI†). Based on this consideration, the PANI obtained in the presence of vesicles had less ortho-coupled aniline units than the PANI obtained without vesicles, which is in agreement with the expected template effect of the vesicles.3.5 Control measurements
Several control measurements were carried out in order to better understand the entire reaction system . In general, if one of the essential components of the reaction system was missing, the vesicle templates, HRP or H2O2, the reaction either did not occur or did not yield the desired product(s).Although the NMR measurements did not show significant differences between the reaction carried out in presence of AOT vesicles and without vesicles, taken the UV/VIS/NIR and the EPR measurements together, there is a clear confirmation of previous findings that AOT vesicles play their role as essential templates for obtaining—to at least some extent—the desired emeraldine salt form of PANI;17 the vesicles not only play a role as product “solubiliser”, see the Introduction.
If H2O2 was not present in the system containing AOT vesicles, aniline and HRP, no reaction occurred; the UV/VIS/NIR spectrum remained unchanged (Fig. S15, ESI†). This showed that H2O2 was required for the reaction and that no significant spontaneous oxidation of aniline by the dissolved oxygen occurred.
In the absence of HRP but otherwise under the optimal reaction conditions, no reaction occurred (Fig. S16, ESI†). This experiment demonstrated that aniline could not be oxidised with H2O2 alone under the conditions used.
If the polymerisation of aniline was carried out in presence of 3 mM sodium di-n-butylsulfosuccinate instead of 3 mM AOT, but otherwise under the optimal conditions, the reaction proceeded like in the surfactant free system, i.e. formation of a brown product and product precipitation with time (Fig. S17 and Fig. S18, ESI†). The reaction yield was again 90–95% (Fig. S14, ESI†).
These experiments convincingly demonstrate the importance of the templates to being an assembly of sulfonate group-bearing molecules. The presence of non-associated dialkylsulfonate counter ions does not support the formation of the emeraldine salt form of PANI, similarly to what was shown previously when studying the effect of polymeric or micellar templates formed from molecules with a benzenesulfonate group.13b
The following reaction conditions were used on the basis of the assumption that the arbitrarily chosen amount of PADPA present before H2O2 addition was obtained with HRP–H2O2 from aniline. Therefore, the required amount of H2O2 for obtaining the PADPA was subtracted from the amount of H2O2 used in experiments run under the optimal conditions. Two reactions were carried out (pH = 4.3, 0.1 M H2PO4−): (a) 3 mM AOT, 0.08 mM PADPA, 3.84 mM aniline, 4.42 mM H2O2; (b) 3 mM AOT, 0.20 mM PADPA, 3.60 mM aniline, 4.30 mM H2O2. These reactions mimic situations in which polymer chains can be obtained with a number average degree of polymerisation of 50 (for a) or 20 (for b), if each PADPA would grow through the addition of the aniline molecules present in the absence of HRP (4 mM/0.08 mM = 50; 4.0 mM/0.20 mM = 20). The UV/VIS/NIR spectra were recorded before and 48 h after H2O2 addition, see Fig. S19, ESI.† In both cases reactions occurred to a very small extent. Before H2O2 addition, there was no absorption above 350 nm; after 48 h, the spectrum of the reaction mixtures had weak broad peaks centred around 420 nm, 580 nm and 1050 nm, with a minimum at about 840 nm, very different from the spectrum of the emeraldine salt form of polyaniline obtained from aniline with HRP at reaction equilibrium (see Fig. 1c and Fig. S6, ESI†). Fig. 6 is a comparison of the UV/VIS/NIR absorption spectrum of the polyaniline obtained after t = 48 h from aniline with HRP in the presence of AOT vesicles under optimal reaction conditions (curve 1) with the absorption spectrum obtained from a reaction mixture containing PADPA, aniline and H2O2 in the absence of HRP, recorded after t = 48 h (curve 2). The two spectra not only differ with respect to the band positions but also with respect to the intensities of the bands.
![Comparison of the UV/VIS/NIR absorption spectrum of the polyaniline product obtained from aniline, HRP and H2O2 in the presence of AOT vesicles under optimal reaction conditions after t = 48 h (curve 1) with the UV/VIS/NIR absorption spectrum obtained from a mixture of PADPA, aniline and H2O2 in the presence of AOT vesicles without HRP after t = 48 h (curve 2) at T = 25 °C and pH = 4.3 (0.1 M H2PO4−). The initial reaction conditions for the spectrum 1 were: [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM. The initial reaction conditions for the spectrum 2 were: [AOT] = 3.0 mM; [PADPA] = 0.2 mM; [aniline] = 3.60 mM; [H2O2] = 4.30 mM. Please note that not only the peak positions are different but also the peak intensities. Curve 1 is taken from Fig. S6, ESI, curve 2 from Fig. S19, ESI.](/image/article/2012/RA/c2ra20566a/c2ra20566a-f6.gif) | ||
| Fig. 6 Comparison of the UV/VIS/NIR absorption spectrum of the polyaniline product obtained from aniline, HRP and H2O2 in the presence of AOT vesicles under optimal reaction conditions after t = 48 h (curve 1) with the UV/VIS/NIR absorption spectrum obtained from a mixture of PADPA, aniline and H2O2 in the presence of AOT vesicles without HRP after t = 48 h (curve 2) at T = 25 °C and pH = 4.3 (0.1 M H2PO4−). The initial reaction conditions for the spectrum 1 were: [AOT] = 3.0 mM; [aniline] = 4.0 mM; [HRP] = 0.92 μM; [H2O2] = 4.5 mM. The initial reaction conditions for the spectrum 2 were: [AOT] = 3.0 mM; [PADPA] = 0.2 mM; [aniline] = 3.60 mM; [H2O2] = 4.30 mM. Please note that not only the peak positions are different but also the peak intensities. Curve 1 is taken from Fig. S6, ESI,† curve 2 from Fig. S19, ESI.† | ||
These experiments clearly show that the presence of active HRP is required for the formation of PANI oligomers or polymers in their emeraldine salt form. HRP with H2O2 “produces” anilino radicals which become protonated to aniline radical cations (Scheme 1 and Scheme 3), which then add to the growing chain. Aniline molecules which are not oxidised do not contribute to chain growth. This finding is in agreement with the “radical cation mechanism” (Scheme S3, ESI†). The “nonclassical or reactivation chain polymerisation mechanism” (Scheme S2, ESI†) is not compatible with our experimental data.
These experiments show that the peroxidase–H2O2 oxidant system can not be replaced by (NH4)2S2O8 for obtaining the same reaction product, if all other reactions conditions are kept the same.
4 Discussion
After a quite laborious optimisation of the reaction system, the following initial conditions turned out to be ideal to obtain stable vesicular suspensions containing in high yield the emeraldine salt form of polyaniline that remained bound to the vesicles: 3.0 mM AOT, 4.0 mM aniline, 4.5 mM H2O2, 0.92 μM HRP, pH = 4.3 (0.1 M NaH2PO4), room temperature (T = 23–25 °C). The AOT vesicles served as templates and dopants, HRP as catalyst and H2O2 as oxidant. Changes of the concentration of one of the components led to changes of the reaction kinetics, product characteristics and yield. Under the optimal conditions above, the reaction was highly reproducible, and it was carried out dozens of times. In the following, we will rationalise these empirically found optimal conditions, and we will propose a reaction mechanism consistent with all experimental data presented.The molar ratio of reacted H2O2 to reacted aniline was 1.25,37 as stoichiometrically required for obtaining polymer chains in their emeraldine salt form with a high degree of polymerisation, see Scheme 4. The AOT concentration (3 mM) was high enough in order to: (i) efficiently bind aniline monomers before the reaction was initiated, (ii) keep the formed PANI dispersed (no product precipitation), and (iii) act as efficient counter ion (dopant) to promote polaron formation. Reactions with higher AOT concentrations (4 or 5 mM) resulted in lower A1000:A500 values, indicating a lower PANI “quality”, with similar reaction yield. The reason for this observation is not clear. It may, however, be related to a dilution of HRP and aniline monomers on the vesicle surface. If more vesicle surface is available for aniline binding, the surface concentrations decrease and may drop below the optimal ones. Simple calculations showed that on average about 20 HRP molecules would be associated on the surface of each vesicle, if all HRP molecules were bound to the vesicles (no free enzymes), see ESI.†
With 2H MAS NMR spectroscopy and three types of deuterium labelled anilines, it could be shown that para-coupling of the aniline monomers predominates (Fig. 4), a prerequisite for the formation of the emeraldine salt form of PANI. UV/VIS/NIR absorption and EPR measurements (Fig. 1 and Fig. 2) indicate that the emeraldine salt form of PANI indeed is obtained in the presence of the vesicles, similar to what has been demonstrated previously for the same reaction carried out with polymeric or micellar templates.8,13–15 Without templates, the reaction products are different (UV/VIS/NIR data), although the 2H MAS NMR and FTIR experiments were ambiguous about the effect of the vesicles on the molecular structure of the PANI.
A clear effect of the vesicles is their role as a solubiliser. While in the presence of the vesicles a stable PANI suspension is obtained, in absence of the vesicles the products precipitate during the course of the reaction.
UV/VIS/NIR measurements at the very early stage of the reaction, i.e. during the first 25–45 s, indicated that early reaction intermediates are very different if the reaction is carried out in the presence of vesicles (Fig. S7, ESI†) or in a template-free system (Fig. S13, ESI†). Although we do not have a direct proof, it is likely that the vesicles suppress the formation of π-dimers50 of early reaction intermediates, thereby preventing formation of the unwanted ortho-coupled, branched products. Formation of π-dimers23,50,51 as well as of molecules with phenazinium units48 lead to characteristic absorption peaks at 500–540 nm which we observed when the reaction was carried out without vesicles (Fig. 2 and Fig. S13, ESI†). In contrast, in the presence of the vesicles, the absorption at about 500 nm was low (Fig. 1 and Fig. S7, ESI†).
The AOT molecules in the vesicular aggregates acted as dopants,52,53 as counter ions of the formed polymeric products. In the emeraldine salt form of PANI, every second aniline repeating unit carries a positive charge with a corresponding dopant/counter ion (A− in Scheme 4). The “optimal” concentrations (4.0 mM aniline, 3.0 mM AOT) provided in principle enough AOT molecules to act as dopant/counter ion. In fact, 1.8–1.9 mM AOT is needed for a 90–95% polymerisation of 4.0 mM aniline. From the 3.0 mM AOT about 0.4 mM were present in bulk solution,17 while the remaining 2.6 mM formed the bilayers of the vesicles. Since the vesicles were mainly unilamellar with an average diameter of ≈80 nm, about 53% of these 2.6 mM AOT molecules in the bilayer (= 1.4 mM AOT) were in the outer layer (see ESI†), directly exposed to the added aniline and HRP molecules. This amount of exposed AOT was lower than the amount of AOT required for acting as dopant/counter ion (1.8–1.9 mM would be needed) to counter balance all positive charges in the PANI formed. It therefore seems that (i) either other anions partially acted as counter ions (H2PO4−), or (ii) the growing polymer chain led to deformations of the vesicles so that AOT molecules originally present in the inner monolayer became in contact with the PANI through molecular reorganisation within the bilayers. This second possibility finds support in the observation of changes in the vesicle size and morphology during the polymerisation (Fig. 5a–c).17,54
HRP is another important component of the entire reaction system, being responsible for triggering the reaction and promoting chain growth through formation of anilino radicals. Therefore, the observed HRP inactivation during the polymerisation (Fig. 3) is an important issue, since it seems a limiting factor and needs to be considered for further optimisation of the reaction system. The optimal HRP concentration (0.92 μM) was high enough to allow the oxidation of most of the aniline monomers (90–95%) before the complete HRP inactivation.55 If the HRP concentration was decreased to 0.092 μM, the reaction yield decreased to about 40% (Fig. S14, ESI†), since the enzyme was inactivated already after the oxidation of only 40% of the aniline molecules. The percentage decrease in HRP activity during polymerisation (0.92 μM and 0.092 μM) was about the same in both cases (Fig. S9, ESI†). This clearly demonstrates that high reaction yields require active HRP, being the anilino radicals that HRP “produces” not only needed for triggering the reaction but also for efficient chain growth. These results were a first indication that chain growth may occur via the radical cation mechanism (Scheme S3, ESI†) and not via the nonclassical or reactivation chain polymerisation (Scheme S2, ESI†). Experiments with PADPA and aniline supported further the radical cation mechanism (Fig. 6 and Fig. S19, ESI†).
The observed decrease in HRP activity during polymerisation is not surprising. HRP inactivation is known to occur if H2O2 is present in excess over a second electron donating (i.e. reducing) substrate–i.e. aniline–or at millimolar H2O2 concentrations in the absence of any reducing substrates, particularly at pH values below 6.5.44 Under these latter conditions, H2O2 may serve as oxidant as well as reductant to yield hydroperoxyl radicals, HO2•, which may be responsible for the HRP inactivation. It is also possible that some of the anilino radicals formed reacted with HRP and thus inhibited HRP, in a similar way as for phenoxyl radicals at pH = 7.0 in the HRP–H2O2-catzalysed oxidation of phenol.56 Loss in HRP activity during the polymerisation of phenol was also related to a direct interaction of HRP with the polymeric products,57 as shown in Fig. S10, ESI† the formed PANI products (or side products) indeed are inhibitory. Whatever the mechanism of HRP inactivation during the polymerisation, the enzyme certainly was not inactivated by the AOT vesicles.17 Furthermore, a stepwise addition of smaller portions of H2O2, as often applied to minimize HRP inactivation,13 did not lead to an improvement of the PANI quality. We therefore decided to initiate the reaction by adding H2O2 at once, and not stepwise. In any case, the observed inactivation of HRP during polymerisation affects the efficiency of the reaction. In fact, the polymerisation of 1 g aniline requires 0.11 g of HRP,58 a rather large amount for a catalyst. The molar ratio of aniline to HRP was 4348, i.e. one HRP molecule on average only made 2174 turnovers, or even less (reaction yield: 90–95%).
The phosphate salt stabilises AOT bilayers and in the absence of salt, no vesicles are formed.17 The H3O+ concentration, i.e. the pH, influences the degree of protonation of aniline and of the oligomeric and polymeric products—and as a consequence their interaction with the negatively charged vesicles. The pH also affects the rate of aniline oxidation by HRP since only the neutral form of aniline is oxidised.18 A reaction pH of 4.3 was previously found to be optimal, independent of whether vesicles or other templates were used.16,17
As already mentioned, the entire reaction occurs in three kinetically different phases. In the first phase which lasts for 5–8 min, almost all aniline and H2O2 molecules are consumed with the formation of products that have an absorption maximum at 750–770 nm (Fig. 1) and low intensity EPR signal (Fig. 2). This indicates that the formation of polymeric products occurs in this first phase already, and that the emeraldine salt form of PANI is obtained in its bipolaron state (Scheme 2) in this first phase. In the second phase which lasts up to about 250–300 min, the bipolarons are converted into polarons, as typically observed in the electrochemical polymerisation of aniline.23
This symproportionation is evident from the changes in the absorption spectrum (decrease in intensity at λ = 750–770 nm, Fig. 1), as well as from the increase in EPR signal (Fig. 2). In the third phase which is the time period beyond 250–300 min, the intensity at λ = 750–770 nm still decreases (Fig. 1) with further small increase in the EPR signal intensity (Fig. 2). A stable state seems to be reached after about 22 h.
All experimental data presented in this paper are consistent with the “radical cation mechanism” described by Genies and Tsintavis59 and Ding et al.,60 originally proposed for the electrochemical polymerisation of aniline. The tentative reaction steps are outlined in Schemes 5–8. Scheme 5 shows that at pH 4.3, anilinium cations (1) are in equilibrium with aniline (2); HRP is activated with H2O2 and then oxidises aniline monomers (2) to anilino radicals (3); the anilino radicals (3) become protonated to yield the aniline radical cation (4, pKa = 7.1).16Scheme 6 points up that two aniline radical cations (4a and 4b) combine to form a dimer (preferentially through a N–C para-coupling); the aniline dimer cation obtained (5) formally transforms into 6 through a release of a proton from the aromatic ring; after deprotonation of the dimer 6 to 7 (PADPA), 7 is oxidised by H2O2 to yield 8a (PBQ) on the vesicle surface and finally the diradical dication dimer 8b. This dimer combines with another aniline radical cation (4a) to yield a trimer (9) which after proton migration to 10 and deprotonation to 11 is oxidised by H2O2 to the trimer radical trication (12a and 12b), Scheme 7. After combination of 12b with another aniline radical cation (4b), the tetramer 13 is obtained. Proton migration to yield 14 and deprotonation yield the half oxidised tetramer dication 15, one repeating unit of the emeraldine salt form of polyaniline in its bipolaronic state, Scheme 8. In a similar way, the chain continues to grow. A key feature of this mechanism is that chain growth occurs through addition of aniline radical cations to the growing chain (“radical cation mechanism”59,60) and not through the addition of the neutral form of aniline, as shown to be the case for the chemical polymerisation of aniline by Wei et al.61
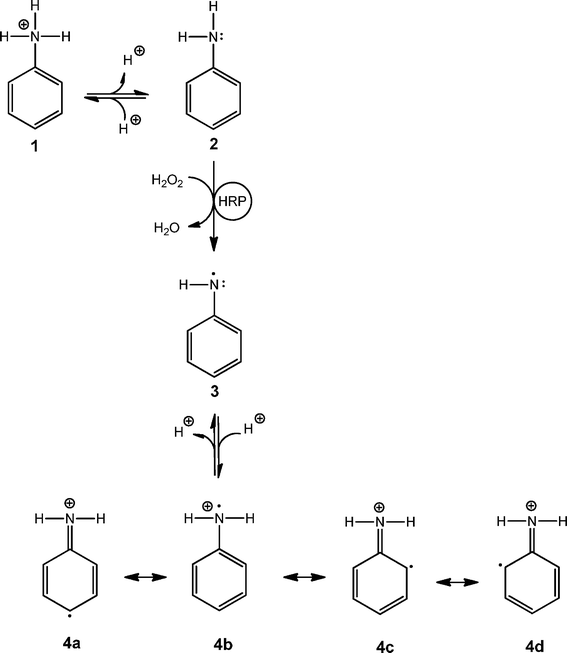 | ||
| Scheme 5 Formation of the anilinium cation radical. In aqueous solution, the anilinium cation (1, pKa = 4.6)16 is in equilibrium with the neutral form of aniline (2). The HRP–H2O2-catalysed oxidation of 2 leads to the formation of the anilino radical (3), see Scheme 1, which then becomes protonated to yield the aniline radical cation 4 (pKa = 7.1),16 in which the unpaired electron can be localised either on the carbon atom in para-position (4a), on the nitrogen atom (4b) or on one of the two carbon atoms in ortho-position (4c and 4d). | ||
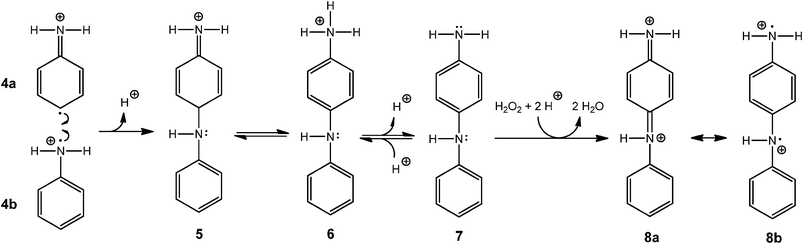 | ||
| Scheme 6 Plausible steps for the formation of the oxidised aniline dimer diradical dication. Two aniline radical cations (4a and 4b)—obtained through the HRP–H2O2-catalysed oxidation of aniline (Scheme 5) —react to form an aniline dimer dication (not shown, the reported pKa value of the secondary ammonium group is about −0.1),60 which after subsequent deprotonation yields 5 and after tautomerisation 6 (pKa = 4.70),62 which is in equilibrium with 7 (PADPA, p-aminodiphenylamine). During the conversion of 5 to 6 (tautomerism), the proton originally present in the para-position of the upper aniline is released and formally moves to the terminal nitrogen and then exchanges with the water molecules. Oxidation of 7 with H2O2 yields the dication 8a (PBQ, N-phenyl-1,4-benzequinonediimine). The mesomeric structure 8b is the aniline dimer diradical dication form. | ||
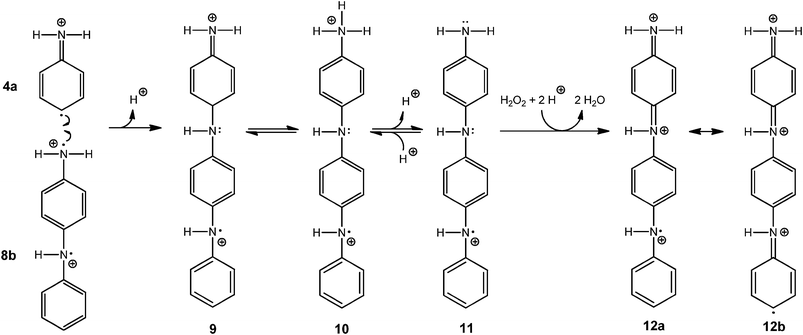 | ||
| Scheme 7 Plausible steps for the formation of the oxidised aniline trimer radical trication. An aniline dimer diradical dication (8b) reacts with an aniline radical cation (4a) to form after deprotonation the aniline trimer radical dication 9 which tautomerises to 10 and after deprotonation yields 11. Oxidation of 11 and protonation gives the aniline trimer 12, shown as trication radical with one unpaired electron either localised on the nitrogen atom of the aniline unit at the bottom (12a) or on the carbon atom in para-position of the aniline unit at the bottom (12b). | ||
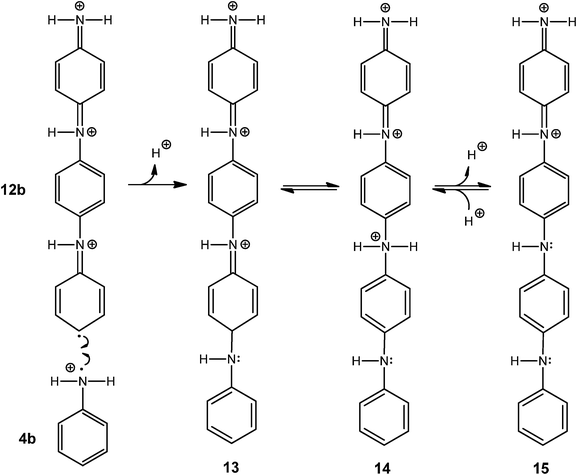 | ||
| Scheme 8 Plausible steps for the formation of the half-oxidised aniline tetramer dication. 12b reacts with 4b to form after deprotonation 13 which tauomerises to 14 and yields the aniline tetramer dication 15, the repeating unit of the emeraldine salt form of PANI in its bipolaron state, see Scheme 2 and Scheme 4. | ||
5 Concluding remarks
The work presented sheds some light onto the mechanism of the horseradish peroxidase-catalysed polymerisation of aniline in the presence of AOT vesicles as templates. The results obtained may be of relevance for similar reaction systems. With the newly developed 2H MAS NMR methodology it could be shown that preferentially para-coupling of the aniline monomers occurs. It is likely that the 2H MAS NMR method described can serve also for other comparable reaction systems, thereby contributing to a better understanding of similar polymerisation reactions.The vesicles have an influence on the regioselectivity of the polymer chain growth, possibly through a disturbance of the formation of π-dimers23,50 between aniline monomers and/or reaction intermediates. The localisation of the reaction on the surface of the ionic vesicles may hinder the formation of π-dimers.
The reaction system was optimised with respect to the initial concentrations used and to the order of adding the various components. These conditions allowed direct investigations by UV/VIS/NIR and EPR spectroscopy, cryo-TEM, SANS and 2H MAS NMR spectroscopy, in the latter case by using deuterated monomers. The reaction system is sensitive to changes of the concentration of one of the components, as demonstrated repeatedly throughout the work; the reaction kinetics, reaction yield and chemical structure of the products obtained strongly depend on the experimental conditions.
There still remain several open questions about the entire reaction, such as: what is the precise localisation of the aniline monomers, the reaction intermediates and the final reaction products on the surface of the vesicles? Is it possible to prove the suggested influence of the template on the formation of π-dimers? Are there defects in the polymer chain, e.g. caused by the presence of small amounts of phenazine or benzidine units? What is the molar mass of the product, how polydisperse are the polymers obtained? One point of interest is, of course, whether the stable green polyaniline suspension prepared in the way described can be applied. Preliminary experiments showed that the polyaniline obtained with HRP and H2O2 in the presence of vesicles as templates can be used as ink in a conventional ink jet printer (work in progress). The vesicular particles containing the emeraldine salt form of PANI on their surface are small enough not to block the nozzles of the printer.
The cost of the peroxidase certainly is a critical factor for the preparation of large volumes of PANI-vesicle suspensions. The amount of HRP used is high—and must be high—since the HRP is becoming inactivated during the polymerisation reaction, a clear disadvantage. Whether HRP could be replaced by more stable peroxidases—or other oxidoreductases—needs to be seen. Interestingly, HRP inactivation during polymerisation does not occur if the peroxidase is used as catalyst for atom transfer radical polymerisation (ATRP) reactions.12 Therefore, enzyme inactivation seems to be related to the details of the reaction catalysed.
Acknowledgements
We acknowledge EMEZ (Electron Microscopy Center, ETH Zürich), especially Peter Tittmann and Dr Roger Wepf, for technical support. The EI-MS analysis was carried out by the mass spectrometry service of the Laboratory of Organic Chemistry at ETH Zürich. The financial support by the Swiss National Science Foundation (200021–111696 and 200020–124690) is highly appreciated. Discussions with Dr Thomas Nauser (Department of Chemistry and Applied Biosciences, ETH Zürich) and Prof. Dr Beat Meier about the work are highly appreciated.References
- (a) S. Kobayashi, H. Uyama and S. Kimura, Chem. Rev., 2001, 101, 3793–3818 CrossRef CAS; (b) S. Kobayashi and A. Makino, Chem. Rev., 2009, 109, 5288–5353 CrossRef CAS; (c) S. Kobayashi, in Encyclopedia of polymer science and technology, John Wiley & Sons, New York, 2011. Search PubMed; (d) T. Oguchi, S. Tawaki, H. Uyama and S. Kobayashi, Macromol. Rapid Commun., 1999, 20, 401–403 Search PubMed.
- (a) R. A. Gross, A. Kumar and B. Kalra, Chem. Rev., 2001, 101, 2097–2124 CrossRef CAS; (b) R. A. Gross, M. Ganesh and W. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS.
- M. Reihmann and H. Ritter, Adv. Polym. Sci., 2006, 194, 1–49 CAS.
- K. Loos, Biocatalysis in polymer chemistry, Wiley-VCH, Weinheim, 2011. Search PubMed.
- (a) N. C. Price, L. Stevens, Fundamentals of enzymology, Oxford University Press, Oxford, 3rd edn 1999. Search PubMed; (b) A. Fersht, Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding, W. H. Freeman, New York, 1999. Search PubMed.
- L. Hedstrom, in Encyclopedia of life sciences (ELS), John Wiley & Sons, Chichester, 2010 Search PubMed.
- (a) O. Khersonsky, C. Roodveldt and D. S. Tawfik, Curr. Opin. Chem. Biol., 2006, 10, 498–508 CrossRef CAS; (b) K. Hult and P. Berglund, Trends Biotechnol., 2007, 25, 231–238 CrossRef CAS; (c) M. Svedendahl Humble and P. Berglund, Eur. J. Org. Chem., 2011, 3391–3401 Search PubMed.
- P. Walde and Z. Guo, Soft Matter, 2011, 7, 316–331 RSC.
- (a) P. Xu, A. Singh and D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 69–94 CAS; (b) A. Singh and D. L. Kaplan, Adv. Polym. Sci., 2006, 194, 211–224 CAS.
- R. Bouldin, A. Kokil, S. Ravichandran, S. Nagarajan, J. Kumar, L. A. Samuelson, F. F. Bruno and R. Nagarajan, ACS Symp. Ser., 2010, 1043, 315–341 Search PubMed.
- E. Ochoteco and D. Mecerreyes, Adv. Polym. Sci., 2010, 237, 1–19 Search PubMed.
- S. J. Sigg, F. Seidi, K. Renggli, T. B. Silva, G. Kali and N. Bruns, Macromol. Rapid Commun., 2011, 32, 1710–1715 CrossRef CAS.
- (a) L. A. Samuelson, A. Anagnostopoulos, K. S. Alva, J. Kumar and S. K. Tripathy, Macromolecules, 1998, 31, 4376–4378 CrossRef CAS; (b) W. Liu, A. L. Cholli, R. Nagarajan, J. Kumar, S. Tripathy, F. F. Bruno and L. A. Samuelson, J. Am. Chem. Soc., 1999, 121, 11345–11355 CrossRef CAS; (c) W. Liu, J. Kumar, S. Tripathy, K. J. Senecal and L. Samuelson, J. Am. Chem. Soc., 1999, 121, 71–78 CrossRef CAS; (d) W. Liu, J. Kumar, S. Tripathy and L. A. Samuelson, Langmuir, 2002, 18, 9696–9704 CrossRef CAS; (e) S. K. Sahoo, R. Nagarajan, S. Chakroborty, L. A. Samuelson, J. Kumar and A. L. Cholli, J. Macromol. Sci., Part A: Pure Appl. Chem., 2002, 39, 1223–1240 Search PubMed.
- V. Rumbau, J. A. Pomposo, J. A. Alduncin, H. Grande, D. Mecerreyes and E. Ochoteco, Enzyme Microb. Technol., 2007, 40, 1412–1421 CrossRef CAS.
- Y. Gu, C.-C. Chen and Z.-W. Ruan, Synth. Met., 2009, 159, 2091–2096 CrossRef CAS.
- Z. Guo, H. Rüegger, R. Kissner, T. Ishikawa, M. Willeke and P. Walde, Langmuir, 2009, 25, 11390–11405 CrossRef CAS , and 2010, 26, 7650, correction.
- Z. Guo, N. Hauser, A. Moreno, T. Ishikawa and P. Walde, Soft Matter, 2011, 7, 180–19 RSC.
- (a) D. Job and H. B. Dunford, Eur. J. Biochem., 1976, 66, 607–614 CAS; (b) H. B. Dunford, Peroxidases & catalases, John Wiley & Sons, Hoboken, 2nd edn, 2010. Search PubMed; (c) N. C. Veitch and A. T. Smith, Adv. Iorg. Chem., 2001, 51, 107–162 Search PubMed.
- M. A. Gilabert, A. N. P. Hiner, P. A. García-Ruiz, J. Tudela, F. García-Molina, M. Acosta, F. García-Cánovas and J. N. Rodríguez-López, Biochim. Biophys. Acta, 2004, 1699, 235–243 CAS.
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS.
- A. V. Caramyshev, E. G. Evtushenko, V. F. Ivanov, A. R. Barceló, M. G. Roig, V. L. Shnyrov, R. B. van Huystee, I. N. Kurochkin, A. Kh. Vorobiev and I. Yu. Sakharov, Biomacromolecules, 2005, 6, 1360–1366 CrossRef CAS.
- N. R. Curvetto, D. Figlas, A. Brandolin, S. B. Saidman, E. H. Rueda and M. L. Ferreira, Biochem. Eng. J., 2006, 29, 191–203 Search PubMed.
- E. Dmitrieva and L. Dunsch, J. Phys. Chem. B, 2011, 115, 6401–6411 Search PubMed.
- (a) W. S. Huang and A. G. MacDiarmid, Polymer, 1993, 34, 1833–1845 CrossRef CAS; (b) Y. Xia, J. M. Wiesinger, A. G. MacDiarmid and A. J. Epstein, Chem. Mater., 1995, 7, 443–445 CrossRef CAS.
- F. Wudl, R. O. Angus Jr., F. L. Lu, P. M. Allemand, D. J. Vachon, M. Nowak, Z. X. Liu and A. J. Heeger, J. Am. Chem. Soc., 1987, 109, 3677–3684 CrossRef CAS.
- (a) A. V. Kulikov, A. S. Komissarova, A. G. Ryabenko, L. S. Fokeeva, I. G. Shunina and O. V. Belonogova, Russ. Chem. Bull., 2005, 12, 2794–2804 Search PubMed; (b) V. I. Krinichnyi, H.-K. Roth, M. Schröder and B. Wessling, Polymer, 2006, 47, 7460–7468 Search PubMed.
- H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187–251 CrossRef.
- Y. Miura, H. Oka, E. Yamano and M. Morita, J. Org. Chem., 1997, 62, 1188–1190 Search PubMed.
- J. Kohlbrecher and W. Wagner, J. Appl. Crystallogr., 2000, 33, 804–806 CrossRef.
- N. Rajendiran and N. Swaminathan, Spectrochim. Acta, Part A, 1996, 52, 1785–1792 Search PubMed.
- (a) C. Matsubara, N. Kawamoto and K. Takamura, Analyst, 1992, 117, 1781–1784 RSC; (b) K. Takamura and C. Matsubara, Bull. Chem. Soc. Jpn., 2003, 76, 1873–1888 CrossRef CAS; (c) K. Takamura and T. Matsumoto, Appl. Spectrosc., 2009, 63, 579–584 CrossRef CAS.
- R. E. Childs and W. G. Bardsley, Biochem. J., 1975, 145, 93–103 CAS.
- T. Namani and P. Walde, Langmuir, 2005, 21, 6210–6219 CrossRef CAS.
- The critical concentration for micelle formation (cmc) of sodium di-n-butylsulfosuccinate in salt-free water at 25 °C is 187 ± 3 mM, of sodium di-n-hexylsulfosuccinate 12.5 ± 0.3 mM, and for AOT 2.56 ± 0.03 mM: S. Nave, J. Eastoe and J. Penfold, Langmuir, 2000, 16, 8733–8740 Search PubMed.
- G. M. do Nascimento and M. A. de Souza, in Nanostructured conductive polymers, ed. A. Eftekhari, John Wiley & Sons, Chichester, 2010, chapter 8, pp. 341–373 Search PubMed.
- High absorption at 500 nm for polymeric reactions products of aniline is typical for ill-defined, branched products with ortho-coupled aniline units 13 .
- A yield of 90% means that only 3.6 mM aniline reacted. Therefore, the molar ratio of reacted H2O2 to reacted aniline was 1.25 (= 4.5 mM:3.6 mM), which is the stoichiometric ratio required for obtaining long chains (n > 50).
- A yield of 90% means that 3.6 mM aniline reacted, leading to a molar ratio of reacted H2O2 to reacted aniline of 1.39 (= 5.0 mM:3.6 mM), which is higher than required for the emeraldine form of polyaniline (R* = 1.25, for n > 50).
- (a) I. Sapurina and J. Stejskal, Polym. Int., 2008, 57, 1295–1325 CrossRef CAS; (b) J. Stejskal, I. Sapurian and M. Trchová, Prog. Polym. Sci., 2010, 35, 1420–1481 CrossRef CAS.
- For the sake of simplicity, “H+” is used throughout the paper instead of the more appropriate notation “H+(aq)” or “H3O+(aq)”.
- (a) The dihydrogen phosphate solution used in all experiments (0.1 M H2PO4−, pH = 4.3) did not have efficient pH buffering capacity (pKa (H3PO4) = 2.2; pKa (H2PO4−) = 7.2). If “real” pH 4.3 buffer solutions were used instead, such as 0.1 M succinic acid–succinate (pKa1 = 4.2, pKa2 = 5.6), or 0.1 M citric acid–citrate (pKa1 = 3.1; pKa2 = 4.8; pKa3 = 6.4), or 0.1 M acetic acid–acetate (pKa = 4.7), there was no improvement in the “quality” of the PANI obtained.; (b) The use of an acetic acid–acetate buffer may lead to HRP inactivation in presence of H2O2 since it is known that acetic acid is oxidised by HRP to acetic acid radicals which then reacts with the heme group of HRP, the HRP inactivation being strongest at pH = 4.4; and linear alkanoic acids, like n-hexanoic acid, react similary: L. Huang, C. Colas and P. R. Ortiz de Montellano, J. Am. Chem. Soc., 2004, 126, 12865–12873. Search PubMed The possible oxidation of alkanoic acids by HRP in the presence of H2O2 is another reason for using AOT vesicles as templates instead of the SDBS–decanoic acid system which we investigated previously17.
- The UV spectrum of aniline dissolved in neutral aqueous solution has absorption peaks at 230 nm and at 278–280 nm; at pH 1.25 and lower, there is only one peak maximum at 254 nm (Ar–NH3+ (a) N. Rajendiran and M. Swaminathan, Spectrochim. Acta, Part A, 1996, 52, 1785–1792 Search PubMed; (b) S. Tajima, S. Shiobara, H. Shizuka and S. Tobita, Phys. Chem. Chem. Phys., 2002, 4, 3376–3382 RSC.
- L. Dennany, P. C. Innis, S. T. McGovern, G. G. Wallace and R. J. Forster, Phys. Chem. Chem. Phys., 2011, 13, 3303–3310 RSC.
- (a) R. Nakajima and I. Yamazaki, J. Biol. Chem., 1987, 262, 2576–2581 Search PubMed; (b) M. B. Arnao, M. Acosta, J. A. del Río, R. Varón and F. García-Cánovas, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1041, 43–47 Search PubMed; (c) M. B. Arnao, M. Acosta, J. A. del Río, R. Varón and F. García-Cánovas, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1041, 85–89 Search PubMed; (d) K. J. Baynton, J. K. Bewtra, N. Biswas and K. E. Taylor, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1994, 1206, 272–278 Search PubMed; (e) J. Hernández-Ruiz, M. B. Arnao, A. N. P. Hiner, F. García-Cánovas and M. Acosta, Biochem. J., 2001, 354, 107–114 CrossRef CAS.
- The reported pKa value of hydroperoxyl HO2• is 4.8: J. Rabani and S. O. Nielsen, J. Phys. Chem., 1969, 73, 3736–3744 Search PubMed.
- Literature data for the AOT bilayer thickness for different experimental systems varied between 1.9 nm and 2.1 nm (a) I. Grillo, P. Levitz and Th. Zemb, Langmuir, 2000, 16, 4830–4839 CrossRef CAS; (b) M. Skouri, J. Marignan and R. May, Colloid Polym. Sci., 1991, 269, 929–937 Search PubMed; (c) K. Fontell, J. Colloid Interface Sci., 1973, 44, 318–329 CAS; (d) F. Nallet, R. Laversanne and D. Roux, J. Phys. II, 1993, 3, 487–502 CrossRef CAS.
- N. Gospodinova and L. Terlemezyan, Prog. Polym. Sci., 1998, 23, 1443–1484 CrossRef CAS.
- Z. Ding, T. Sanchez, A. Labouriau, S. Iyer, T. Larson, R. Currier, Y. Zhao and D. Yang, J. Phys. Chem. B, 2010, 114, 10337–10346 Search PubMed.
- M. Trchová, I. Šeděnková, E. N. Konyushenko, J. Stejskal, P. Holler and G. Cirić-Marajanović, J. Phys. Chem. B, 2006, 110, 9461–9468 CrossRef CAS.
- It has been shown that π-dimers of the oxidised form of PADPA (i.e. face-to-face complexes of two aromatic radical cations interacting through their π-orbitals) form in acidified aqueous solutions, leading to absorption at about 500 nm: A. Petr, D. Wei, C. Kvarnström, A. Ivaska and L. Dunsch, J. Phys. Chem. B, 2007, 111, 12395–12398 Search PubMed.
- S. Yamabe, H. Nakata and S. Yamazaki, Org. Biomol. Chem., 2009, 7, 4631–4640 RSC.
- M. Zagorska, I. Kulszewicz-Bajer, O. Blet, P. Zawirska, B. Dufour, P. Rannou and A. Pron, Synth. Met., 2003, 138, 543–548 Search PubMed.
- K.-S. Lee, T. Hino and N. Kuramoto, Chem. Lett., 2007, 36, 340–341 Search PubMed.
- If 4.0 mM AOT were used instead of 3.0 mM AOT but otherwise identical conditions (4 mM aniline, 4.5 mM H2O2, 0.92 μM HRP, pH 4.3 (0.1 M H2PO4−)), the absorption at 1000 nm, at 440 nm and at 300 nm after reaching reaction equilibrium were lower, as well as the ratio A1000 nm:A500 nm, indicating a lower “quality” of the PANI obtained in the presence of 4.0 mM AOT as compared to the PANI obtained under the optimal conditions..
- The reaction yield never reached 100%, independent on whether vesicles were present or not. The reason for this is not clear. At least 5% of the initially added aniline remained..
- Q. Huang, Q. Huang, R. A. Pinto, K. Griebenow, R. Schweitzer-Stenner and W. Weber Jr., J. Am. Chem. Soc., 2005, 127, 1431–1437 CrossRef CAS.
- Y. Wu, K. E. Taylor, N. Biswas and J. K. Bewtra, Enzyme Microb. Technol., 1998, 22, 315–322 Search PubMed.
- Calculated for the optimal conditions with 4 mM aniline (M = 93.13 g mol−1) and 0.92 μM HRP (M = 44000 g mol−1).
- E. M. Genies and C. Tsintavis, J. Electroanal. Chem., 1985, 195, 109–128 Search PubMed.
- Y. Ding, A. B. Padias and H. K. Hall, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2569–2579 CrossRef CAS.
- (a) Y. Wei, G.-W. Jang, C.-C. Chan, K. F. Hsueh, R. Hariharan, S. A. Patel and C. K. Whitecar, J. Phys. Chem., 1990, 94, 7716–7721 CrossRef CAS; (b) Y. Wei, J. Chem. Educ., 2001, 78, 551–553 Search PubMed.
- J. P. Malval, J. P. Morand, R. Lapouyade, W. Rettig, G. Jonusauskas, A. Oberlé, C. Trieflinger and J. Daub, Photochem. Photobiol. Sci., 2004, 3, 939–948 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: additonal experimental data, reaction schemes and calculations. See DOI: 10.1039/c2ra20566a/ |
| This journal is © The Royal Society of Chemistry 2012 |