A highly efficient synthesis of dithiocarbamates in green reaction media
Najmadin
Azizi
* and
Elham
Gholibeglo
Chemistry and Chemical Engineering Research Center of Iran, P.O. Box 14335-186, Tehran, Iran. E-mail: azizi@ccerci.ac.ir; Fax: +98 21 44580762.
First published on 11th July 2012
Abstract
A deep eutectic solvent (DES) and polyethylene glycol (PEG), promoted the environmentally friendly and fast synthesis of dithiocarbamate derivatives via a one-pot, three-component condensation of an amine, carbon disulfide, and a variety of electrophilic reagents in high yields and short reaction times without organic solvents and tedious work-up. These green solvents can be recovered and recycled for subsequent reactions.
Room temperature ionic liquids represent a class of alternative green solvents currently receiving serious consideration is not only important on the laboratory scale but also for industrial applications with the promise of both environmental and technological benefits. By virtue of their nonflammable, chemical and thermal stability, and outstanding solvation ability and negligible vapour pressure, ionic liquids have been proposed as new solvents. Unfortunately, cost and toxicity for some aquatic species and high purity are the main disadvantage of these green solvents that limits their use for chemistry and industry. On the other hand, deep eutectic solvents (DES), consisting of an ammonium salt and hydrogen-bond donor compounds such as urea, acids, amines, and carbohydrates are alternatives to ionic liquids for organic transformations. These green and stable eutectic based solvents are cheaper and easier to make; it is necessary to take two or three solids, mix them together and heat until melting. Furthermore, choline chloride ChCl and urea are both naturally occurring biocompatible compounds as they are easily biodegradable and are not hazardous if they are released back into nature, so processes that use this deep eutectic solvent are economically green.1
Dithiocarbamates are of significant importance as biologically active natural and synthetic products,2 potent anticancer agents3 and an important class of fungicides to protect crops from fungal diseases.4 The biological activity of the dithiocarbamates is increased when they are in the form of heavy metal salts as versatile classes of ligands with the ability to stabilize transition metals in a wide range of oxidation states and efficient ligands in surface science and nanomaterials chemistry.5 Furthermore, they are also useful building blocks for the synthesis of biologically active heterocyclic compounds and solid support grafting materials.6 Therefore, the syntheses of biologically important dithiocarbamates has received considerable attention, and there are some reports on the synthesis of dithiocarbamate derivatives in the literature.7 However, there are various limitations such as long reaction times, use of organic solvents, high temperatures, moderate yields and limited substrates. Given the widespread availability of amines, carbon disulfide, epoxides, alkyl halides and unsaturated enones, there is substantial interest in developing an efficient one-pot, three-component reaction from these simple starting materials. Thus, several efforts have been made to develop simple green methodologies for a one-pot reaction in novel reaction media such as water and ionic liquids.8
Our research has aimed at developing green chemistry by using water and deep eutectic solvents as a reaction media.9 Herein, we describe a simple, highly efficient and eco-friendly method for the fast synthesis of organic dithiocarbamates via a one-pot, three-component condensation of aliphatic amines, carbon disulfide, and an activated alkene–epoxide–alkyl halide to produce the corresponding dithiocarbamates in high yields at room temperature in choline chloride–urea and polyethylene glycol (PEG).
In an initial experiment, phenyl glycidyl ether 1 (1 mmol) was treated with CS2 (2 mmol) and diethyl amine 2 (1 mmol) in polyethylene glycol 200 (2 mL) and choline chloride–urea; (1 mL) in the absence of any catalyst at room temperature. After 60 min, the phenyl glycidyl ether was consumed, and the corresponding dithiocarbamate 3 was formed as the only detectable product and isolated in 95% and 97% yields respectively after simple a work-up (Scheme 1). To test the feasibility of a large-scale reaction, 1 (50 mmol) was treated with 2 (50 mmol) and CS2 (80 mmol) in PEG (20 mL) and DES (15 mL) at room temperature. The product was isolated in 80% and 84% yields after 80 min respectively.
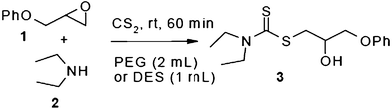 | ||
| Scheme 1 Optimization of reaction conditions. | ||
Under optimized reaction conditions, to explore the scope of this new three-component coupling, we investigated different amines and epoxides and the results of this investigation are shown in Table 1. This procedure is quite general, and a wide range of structurally varied aliphatic amines such as primary, allylic, benzylic and hindered and unhindered and secondary amines were used in this procedure with excellent results. Primary amines such as benzylamine and n-butylamine, sec-butylamine and hindered amines such as tert-butylamine undergo efficient ring opening reactions with epoxides to give the corresponding 2-hydroxy dithiocarbamate with excellent results (Table 1). Furthermore, secondary amines such as pyrrolidine, piperidine, dibutylamine and diethylamine show higher yields of products with short reaction times. With regard to epoxides, the reactions proceeded smoothly with the commercially available epoxides such as glycidyl phenyl ether, allyl glycidyl ether, isopropyl glycidyl ether, 1,2-epoxycyclohexane and 2-phenyloxirane. The results indicate the usefulness of this method. The substituents do not show any significant effects on the reaction times and the yields of the products. The cleavages were regio- and stereoselective and can be explained by the steric and electronic factors associated with the epoxide and the dithiocarbamate anion. The major isomer in all of these reactions with alkyl and aryl-substituted epoxides was the secondary alcohol, which resulted from the usual nucleophilic attack of bulky dithiocarbamate anions at the less substituted carbon atom of epoxides in high to quantitative yields.
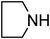
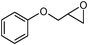
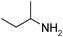
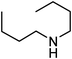
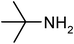
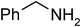
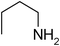
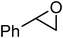
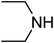
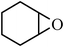
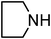
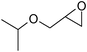
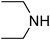
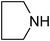
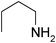

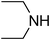
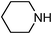
The high yield, short reaction time, and originality of this green process prompted us to explore the three-component coupling reactions of an aliphatic amine, carbon disulfide and alkyl halides under the same reaction conditions (Table 2). In fact, treatment of a wide variety of alkyl halides with aliphatic amines and carbon disulfides at room temperature allows satisfactory to good yields of a variety of dithiocarbamates 5, in PEG and DES, mainly in short reaction times. The results are summarized in Table 2. The in situ generated dithiocarbamate anions react with alkyl chlorides, bromides and iodides effectively and no marked difference was observed in terms of reaction time and yield. Primary, allylic, benzylic, and hindered and unhindered secondary and tertiary alkyl primary amines were used in this protocol with excellent results.
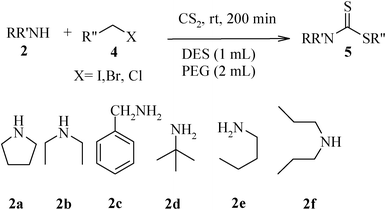

We further explored the potential of these green solvents for the synthesis of dithiocarbamate derivatives from α,β-unsaturated carbonyl compounds. Treatment of CS2 (1 mmol) and diethyl amine (1 mmol) with methyl acrylate (1 mmol) in urea–choline chloride eutectic salt (1 mL) and PEG (2 mL) was performed to give the corresponding dithiocarbamate 7. The reaction was carried out with a very simple procedure in DES and PEG at room temperature under mild reaction conditions and with excellent yields (Table 3).
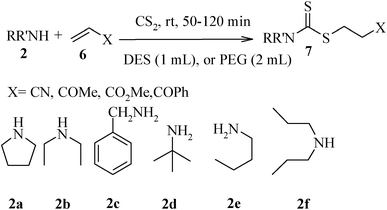
| Entry | Unsaturated enones | Amines | Yields (%)a | Ref. | |
|---|---|---|---|---|---|
| PEG | DES | ||||
| a NMR yields | |||||
| 1 |
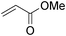
|
2a | 95 | 97 | 8e |
| 2 | 2b | 95 | 97 | 8e | |
| 3 | 2c | 90 | 88 | 8e | |
| 4 | 2d | 80 | 82 | 8e | |
| 5 | 2e | 84 | 80 | 8e | |
| 6 | 2f | 76 | 72 | 8e | |
| 7 |

|
2a | 97 | 97 | 8a |
| 8 | 2b | 97 | 95 | 8a | |
| 9 | 2c | 80 | 90 | 8e | |
| 10 | 2d | 84 | 78 | 8e | |
| 11 | 2e | 76 | 80 | 8e | |
| 12 |
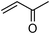
|
2a | 88 | 78 | 8e |
| 13 | 2b | 84 | 82 | 8e | |
| 14 | 2c | 72 | 80 | 8e | |
| 15 |
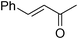
|
2a | 80 | 80 | 8a |
| 16 | 2b | 74 | 72 | 8a | |
| 17 |
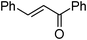
|
2b | 75 | 78 | 8e |
Recycling of DES and PE
The combination of atom economy and ease of recovery and reuse of these novel reaction media are expected to contribute to the development of a green protocol for the simple and fast preparation of dithiocarbamates at room temperature. The recycling of DES and PEG was examined using the reaction of phenyl glycidyl ether CS2 and diethyl amine as starting martials under optimized conditions. The recovered DES and PEG were then reused for three runs without obvious loss of activity. After completion, water (10 mL) was added to the reaction mixture, shaken vigorously, and solid or viscous liquid was separated by filtration. Deep eutectic solvent and polyethylene glycol were recovered from filtrate by evaporating the water phase at 80 °C under vacuum and recycled by reusing for the next batch (Table 4).| Entry | Cycle | Yields (%) | |
|---|---|---|---|
| DES | PEG | ||
| 1 | Fresh | 97 | 95 |
| 2 | First recycle | 95 | 95 |
| 3 | Second recycle | 92 | 92 |
| 4 | Third recycle | 82 | 85 |
Conclusions
In summary, we have developed a simple, highly efficient, and green method for the preparation of dithiocarbamate derivatives under fast and additive free conditions using low melting mixtures and polyethylene glycol as a novel and green reaction medium.10–12 The procedure offers simple experimental procedures, short reaction times, low cost, efficient yields, and is organic solvent-free, which makes this method a useful and attractive strategy in view of economic and environmental advantages. Further studies in our laboratory are under way to develop one-pot green multicomponent reactions in this reaction media.Acknowledgements
The financial support of this work provided by the Chemistry and Chemical Research Center of Iran and the Iran National Science Foundation (INSF) is gratefully appreciated.References
- (a) A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70 RSC; (b) C.-D. Gu and J.-P. Tu, RSC Adv., 2011, 1, 1220 RSC; (c) Z. Chen, B. Zhou, H. Cai, W. Zhu and X. Zou, Green Chem., 2009, 11, 275 RSC; (d) Z. Chen, W. Zhu, Z. Zheng and X. Zou, J. Fluorine Chem., 2010, 131, 340 CrossRef CAS; (e) K. Haerens, E. Matthijs, K. Binnemans and B. Van der Bruggen, Green Chem., 2009, 11, 1357 RSC; (f) J. T. Gorke, F. Srienc and R. J. Kazlauskas, Chem. Commun., 2008, 1235 RSC; (g) C. A. Angell, Y. Ansari and Z. F. Zhao, Faraday Discuss., 2012, 154, 9 RSC; (h) Z. Maugeri and P. Domínguez de María, RSC Adv., 2012, 2, 421 RSC; (i) A. P. Abbott, G. Frisch, H. Garrett and J. Hartley, Chem. Commun., 2011, 47, 11876 RSC.
- (a) E. D. Caldas, M. Conceicu, M. C. C. Miranda, L. Souza and J. F. Lima, J. Agric. Food Chem., 2001, 49, 4521 CrossRef CAS; (b) M. Beji, H. Sbihi, A. Baklouti and A. Cambon, J. Fluorine Chem., 1999, 99, 17 CrossRef CAS.
- (a) Y. Zhang and P. Talalay, Cancer Res., 1994, 54, 1976s CAS; (b) C. C. Conaway, Y. M. Yang and F. L. Chung, Curr. Drug Metab., 2003, 3, 233 CrossRef; (c) S. L. Cao, Y. P. Feng, Y. Y. Jiang, S. Y. Liu, G. Y. Ding and R. T. Li, Bioorg. Med. Chem. Lett., 2005, 15, 1915 CrossRef CAS; (d) R. T. Li, X. L. Hou and Z. M. Ge, Bioorg. Med. Chem. Lett., 2006, 16, 4214 CrossRef.
- (a) L. Ronconi, C. Marzano, P. Zanello, M. Corsini, G. Miolo, C. Macca, A. Trevisan and D. Fregona, J. Med. Chem., 2006, 49, 1648 CrossRef CAS; (b) W. Walter and K.-D. Bode, Angew. Chem., Int. Ed. Engl., 1967, 6, 281 CrossRef CAS; (c) G. H. Elgemeie and S. H. Sayed, Synthesis, 2001, 1747 CrossRef CAS.
- (a) Y. Zhao, W. Perez-segarra, Q. Shi and A. Wei, J. Am. Chem. Soc., 2005, 127, 7328 CrossRef CAS; (b) A. Ivachtchenko, S. Kovalenko, O. V. Tkachenko and O. Parkhomenko, J. Comb. Chem., 2004, 6, 573 CrossRef CAS.
- (a) S. Goubert-Renaudin, R. Schneider and A. Walcarius, Tetrahedron Lett., 2007, 48, 2113 CrossRef CAS; (b) T. S. Griffin, T. S. Woods and D. L. Klayman, in Advances in heterocyclic chemistry, ed. A. R. Katritzky and A. J. Boulton, Academic Press, New York, 1975, vol. 18, p 99 Search PubMed.
- (a) D. Chaturvedi and S. Ray, Tetrahedron Lett., 2006, 47, 1307 CrossRef CAS; (b) D. Chaturvedi and S. Ray, Monatsh. Chem., 2006, 137, 311 CrossRef CAS; (c) D. Chaturvedi, N. Mishra and V. Mishra, Synthesis, 2008, 355 CrossRef CAS; (d) D. Chaturvedi, N. Mishra and V. Mishra, J. Sulfur Chem., 2007, 28, 39 CrossRef CAS; (e) D. Chaturvedi and S. Ray, Tetrahedron Lett., 2007, 48, 149 CrossRef CAS; (f) J.-L. Cui, Z. Ge, T. Cheng and R. Li, Synth. Commun., 2003, 33, 1969 CrossRef CAS; (g) W.-D. Rudorf, J. Sulfur Chem., 2007, 28, 295 CrossRef CAS; (h) G. S. M. Sundaram, C. Venkatesh, H. Ila and H. Junjappa, Synlett, 2007, 251 CAS; (i) G. Shi-Feng, W. Jie-Ping, P. Yuan-Jiang and S. Cui-Rong, Synlett, 2010, 973 Search PubMed; (j) H. Fang-bin, G. Ze-mei, C. Tie-ming and L. Run-Tao, Synlett, 2009, 648 Search PubMed; (k) A. Ziyaei-Halimjani and M. R. Saidi, Can. J. Chem., 2006, 84, 1515 CrossRef CAS.
- (a) B. C. Ranu, A. Saha and S. Banerjee, Eur. J. Org. Chem., 2008, 519 CrossRef CAS; (b) T. Chatterjee, S. Bhadra and B. C. Ranu, Green Chem., 2011, 13, 1837 RSC; (c) N. Azizi, B. Pourhasan, F. Aryanasab and M. R. Saidi, Synlett, 2007, 1239 CAS; (d) A. Alizadeh, S. Rostamnia, N. Zohreh and R. Hosseinpour, Tetrahedron Lett., 2009, 50, 1533 CrossRef CAS; (e) N. Azizi, F. Aryanasab, L. Torkiyan, A. Ziyaei and M. R. Saidi, J. Org. Chem., 2006, 71, 3634 CrossRef CAS; (f) N. Azizi, F. Aryanasab and M. R. Saidi, Org. Lett., 2006, 8, 5275 CrossRef CAS.
- (a) N. Azizi and M. R. Saidi, Organometallics, 2004, 23, 1457 CrossRef CAS; (b) N. Azizi and M. R. Saidi, Org. Lett., 2005, 7, 3649 CrossRef CAS; (c) N. Azizi, L. Torkiyan and M. R. Saidi, Org. Lett., 2006, 8, 2079 CrossRef CAS; (d) N. Azizi, F. Aryanasab and M. R. Saidi, Org. Biomol. Chem., 2006, 4, 4275 RSC; (e) N. Azizi, E. Batebi, S. Bagherpour and H. Ghafuri, RSC Adv., 2012, 2, 2289 RSC; (f) N. Azizi, N. Dado and A. Khaje, Can. J. Chem., 2012, 90, 1 CrossRef.
- General procedure: a mixture of amines (1 mmol), carbon disulfide (2 mmol), in urea–choline chloride (2
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (1 mL)12 ionic liquid, or PEG (2 mL) epoxides/unsaturated enones (1 mmol) were added into a test tube with a magnetic stirring bar. The test tube was stirred at room temperature at 60–200 min. After completion of reaction, water (10 mL) was added. The DES and PEG being soluble in water come in the water layer. The solid or viscous liquid was separated by filtration. DES and PEG were reused for a second run without much loss of product yields after vaporization of water at reduced pressure at 80 °C.
:1) (1 mL)12 ionic liquid, or PEG (2 mL) epoxides/unsaturated enones (1 mmol) were added into a test tube with a magnetic stirring bar. The test tube was stirred at room temperature at 60–200 min. After completion of reaction, water (10 mL) was added. The DES and PEG being soluble in water come in the water layer. The solid or viscous liquid was separated by filtration. DES and PEG were reused for a second run without much loss of product yields after vaporization of water at reduced pressure at 80 °C. - General procedure: a mixture of amines (2 mmol), carbon disulfide (2 mmol), in urea–choline chloride (2:1) (1 mL)11 ionic liquid, or PEG (2 mL) alkyl halides (1 mmol) were added into a test tube with a magnetic stirring bar. The whole mixture was stirred vigorously for 200 min. After the reaction was completed, water (10 mL) and ethyl acetate (10 mL) were added, The organic solvent was removed under reduced pressure to give the desired product. All compounds were known and IR and 1H NMR spectra was found to be identical to those described in literature.
- The general route for the synthesis of the ionic liquids: choline chloride (100 mmol) was mixed with urea (200 mmol) and heated to ca. 100 °C in air with stirring until a clear colourless liquid was obtained1.
| This journal is © The Royal Society of Chemistry 2012 |