Photo-induced electron transfer in supramolecular materials of titania nanostructures and cytochrome c†
Clemerson F. B.
Dias‡
a,
Juliana C.
Araújo-Chaves‡
ab,
Katia C. U.
Mugnol
ab,
Fabiane J.
Trindade
c,
Oswaldo L.
Alves
d,
Antonio C. F.
Caires§
a,
Sergio
Brochsztain
c,
Frank N.
Crespilho
e,
Jivaldo R.
Matos
f,
Otaciro R.
Nascimento
g and
Iseli L.
Nantes
*b
aCentro Interdisciplinar de Investigação Bioquímica CIIB Universidade de Mogi das Cruzes UMC, Mogi das Cruzes-SP, Brazil
bGrupo de Nanoestruturas para Biologia e Materiais Avançados (NanoBioMAv), Centro de Ciências Naturais e Humanas, Universidade Federal do ABC, Santo André-SP, Brazil. E-mail: ilnantes@ufabc.edu.br
cCentros de Engenharia, Modelagem e Ciências Sociais Aplicadas, Universidade Federal do ABC, Santo André-SP, Brazil
dLaboratório de Química do Estado Sólido, Universidade Estadual de Campinas, Campinas-SP, Brazil
eInstituto de Química de São Carlos (IQSC), Universidade de São Paulo (USP), Av. Trab. São-carlense, 400 CP 780, São Carlos, SP CEP 13560-970
fLaboratório de Análise Térmica Prof. Dr Ivo Giolito, Instituto de Química, Universidade de São Paulo, São Paulo-SP, Brazil
gGrupo de Biofísica “Sérgio Mascarenhas”, Instituto de Física, Universidade de São Paulo, Campus São Carlos, São Carlos-SP, Brazil
First published on 13th June 2012
Abstract
In the present paper, we report on the molecular interaction and photochemistry of TiO2 nanoparticles (NPs) and cytochrome c systems for understanding the effects of supramolecular organization and electron transfer by using two TiO2 structures: P25 TiO2 NPs and titanate nanotubes. The adsorption and reduction of cytochrome c heme iron promoted by photo-excited TiO2, arranged as P25 TiO2 NPs and as nanotubes, were characterized using electronic absorption spectroscopy, thermogravimetric analysis, and atomic force microscopy. In an aqueous buffered suspension (pH 8.0), the mass of cytochrome c adsorbed on the P25 TiO2 NP surface was 2.3 fold lower (0.75 μg m−2) than that adsorbed on the titanate nanotubes (1.75 μg m−2). Probably due to the high coverage of titanate nanotubes by adsorbed cytochrome c, the low amount of soluble remaining protein was not as efficiently photo-reduced by this nanostructure as it was by the P25 TiO2 NPs. Cytochrome c, which desorbed from both titanium materials, did not exhibit changes in its redox properties. In the presence of the TiO2 NPs, the photo-induced electron transfer from water to soluble cytochrome c heme iron was corroborated by the following findings: (i) identification by EPR of the hydroxyl radical production during the irradiation of an aqueous suspension of TiO2 NPs, (ii) impairment of a cytochrome c reduction by photo-excited TiO2 in the presence of dioxane, which affects the dielectric constant of the water, and (iii) change in the rate of TiO2-promoted cytochrome c reduction when water was replaced with D2O. The TiO2-promoted photo-reduction of cytochrome c was reverted by peroxides. Cytochrome c incorporated in the titanate nanotubes was also reversibly reduced under irradiation, as confirmed by EPR and UV-visible spectroscopy.
Introduction
N/p-doped semiconductors have been used in the harvesting of the abundantly available solar energy that is delivered to earth (3 × 1021 KJ yr−1).1,2 A typical apparatus for light harvesting consists of a semiconductor, usually titanium dioxide, deposited on a conductive substrate coated by a molecular antenna that absorbs a significant portion of visible light.2 When light strikes the doped material, an electric potential is created when the excited dye transfers one electron to the conductance band of the semiconductor, generating an electrical current.1–3 From the cathode, the electron is transferred back to the dye via a mediator agent present in the electrolyte solution. Different forms of TiO2 have been used in the photovoltaic apparatus, such as TiO2 NP, rutile, thin films, and nanotubes.4–13 A landmark study on the use of TiO2 structures to construct an apparatus for harvesting solar energy was conducted by O'Regan and Grätzel14 using a transparent film of titanium dioxide particles associated with a dye sensitizer.In parallel, the photo-induced redox process involving heme proteins has been described in the literature using dyes and aromatic compounds as sensitizers.15–20 These reactions exhibit interesting mechanisms and could also be feasible in practical applications. As an example, the aromatic imide, N,N′-bis(2-phosphonoethyl)-1,4,5,8-naphthalenetetracarboxylic diimide (PNDI), self-assembled with cytochrome c as a thin film on the surface of non-porous silica gel particles, was able to photo-reduce cytochrome c.
Considering the potential applicability, several studies have focused on different aspects of the interaction, electron transfer, and photochemistry of cytochrome c with semiconductors.21–23 Among the semiconductors, titanium dioxide (TiO2), an n-type semiconductor material, has attracted special interest because of factors such as good biocompatibility, stability, and environmental safety. TiO2 also has photovoltaic effects that make it a material with broader applications in electronic and chemical engineering, the energy industry, the military, etc.1–3
The electron transfer from TiO2 to ferric cytochrome c has been described over the last three decades.21–27 In the existing literature on the interaction of cytochrome c with TiO2, electrochemistry studies have been recurrent.11,21,23–30 On the other hand, the photochemistry of TiO2–cytochrome c systems has been explored predominantly using nanoporous TiO2 and TiO2 nanoparticles (NPs).11–13,21–30 Therefore, because of the immediate appeal for applications in biosensing, hydrogen generation, and light harvesting of the cytochrome c–TiO2 interactions, a more systematic and detailed approach to some basic aspects of these interactions and photochemistry needs to be developed. In the systems comprising cytochrome c–TiO2, details about the protein structure and functionality have been neglected. Not only studies on the development of new materials, but also systematic investigations of the mechanisms underlying their function and organization, are particularly relevant because they contribute to more efficient future innovative designs.
In the present study, we associated a more accurate view of the basic aspects governing the interactions and photochemistry of TiO2–cytochrome c systems with novel types of organization and electron transfer by using two TiO2 structures: TiO2 NPs and titanate nanotubes. Although both materials were able to promote the photo-reduction of the heme iron, different molecular arrangements were found.
Materials and methods
Chemicals
Samples of sodium titanate nanotubes (BET surface area = 128 m2 g−1) were prepared according to ref. 31 and 32. The characterization of this material is described in these references.The TiO2 anatase used was the standard material Degussa P25 (BET surface area = 53.2 m2 g−1), which herein will be referred to as TiO2 NPs (nanoparticles). All other chemicals were purchased from Sigma-Aldrich Co. All aqueous suspensions and solutions were prepared with deionized water (mixed bed of ion exchanger, Millipore®), and the pH was measured using a combined glass electrode (Orion Glass pH SURE-FLOWTM). The reference electrode (ROSSTM, model 8102) was filled with OrionFilling Solutions (ROSSTM). The pH meter was calibrated using METREPAK pHydrion standard buffer solutions (Brooklyn, NY).
Adsorption and desorption of cytochrome c associated with TiO2 NPs and titanate nanotubes
Cytochrome c–titanium nanostructures were obtained by the direct adsorption of a 20 μM (∼0.25 mg mL−1) protein solution incubated in the dark with 0.5 mg mL−1 TiO2 NPs or titanate nanotubes in a 5 mM phosphate buffer (pH 8.0). After incubation, the samples (30 mL) were sonicated for 30 s, stirred for 30 min and fractioned in three 10 mL aliquots. One aliquot was maintained in the dark. The second and third aliquots were gently stirred in ice and under irradiation with a 125 W high-pressure mercury vapour lamp with illumination intensity = 26.77 mW cm−2 on the sample area (3.14 cm2) for 3 h; in the dark, the former remained in air, and the latter remained in an argon atmosphere. Further, in the dark, all aliquots were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM for 10 min, and the supernatants were removed and stored for further cytochrome c dosage. The pellets were washed twice with the same quantity (10 mL) of phosphate buffer (5 mM, pH 8.0) under sonication in the dark and in an argon atmosphere. After the second wash, the cytochrome c present in the TiO2 NPs and in the nanotubes was desorbed with 2.5 mL of a 1 M NaCl solution in an argon atmosphere and in the dark.
000 RPM for 10 min, and the supernatants were removed and stored for further cytochrome c dosage. The pellets were washed twice with the same quantity (10 mL) of phosphate buffer (5 mM, pH 8.0) under sonication in the dark and in an argon atmosphere. After the second wash, the cytochrome c present in the TiO2 NPs and in the nanotubes was desorbed with 2.5 mL of a 1 M NaCl solution in an argon atmosphere and in the dark.
The kinetics of cytochrome c reduction by TiO2 NPs in aqueous and deutered aqueous media
Suspensions were obtained by the direct adsorption of 20 μM cytochrome c incubated with 0.5 mg mL−1 TiO2 NPs in a 5 mM phosphate buffer prepared in H2O (pH 8.0) or in D2O (pD 8.4). The adsorption occurred in the dark, with stirring and subsequent irradiation from a 125 W Hg UV lamp with illumination intensity = 26.77 mW cm−2 on the sample area (3.14 cm2) in an argon atmosphere. Aliquots (300 μL) were taken after 0, 1, 2, 3, 4, 5, 7, 9, 11, 13, and 15 min of irradiation, followed by centrifugation at 10000 RPM for 10 min. The spectra of the supernatants obtained at each irradiation interval were recorded using a Shimadzu Model 1501 MultiSpec (Tokyo, Japan). The optical path length was 0.1 cm at 409 nm for all measurements.
The obtained curves could be best fitted according to the first-order relationship:
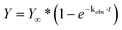 | (1) |
where Y and Y∞ are the absorbances at 549 nm at a given time and at infinite time, respectively, and kobs is the observed first-order rate of light-promoted cytochrome c reduction in the system containing the TiO2 NPs.
Thermogravimetric analysis (TGA) measurements
Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) were carried out using a Model TGA 2950 high-resolution thermogravimetric analyzer (HR–TGA) from TA Instruments (New Castle, DE), under a nitrogen atmosphere (60 mL min−1) using a sample mass between 4–6 mg at temperatures ranging from room temperature to 800 °C, with a maximum heating rate of 10 °C min−1.EPR measurements
The direct EPR measurements of samples were obtained using a Bruker ELEXSYS EPR System E-580 X-band spectrometer under the following conditions: gain: 45 dB; modulation amplitude: 1.0 mT; microwave power: 0.64 mW; microwave frequency: 9.47 GHz; centre magnetic field: 240.0 mT; scan range of magnetic field: 410.0 mT; temperature variation: between 4 and 50 K; time constant: 20.48 ms; and conversion time: 81.92 ms. After mixing, the solutions were quickly introduced into an EPR quartz tube that had previously been cooled in liquid nitrogen. After freezing, the sample was introduced into the microwave cavity at low temperature and the EPR measurements were performed.Vibrational spectroscopy experiments
Fourier transform infrared (FT-IR) spectroscopy measurements were recorded using a Perkin-Elmer FT-IR spectrophotometer mod. Spectrum 100 in the range 4000–400 cm−1 with a resolution of 4 cm−1 for 64 scans. The samples were analyzed as KBr pellets.Atomic force microscopy
The atomic force microscopy (AFM) images were obtained using a Shimadzu SPM 9600 scanning probe microscope by measuring the interaction forces between a triangular silicon tip and the sample surface in contact mode with a 30 μm scanner at room temperature (20 °C) and atmospheric pressure (760 mm Hg). The resonance frequencies of the cantilever were adjusted to 200 kHz. The samples were films prepared with titanate nanotube solutions in a 5 mM sodium phosphate buffer (pH 8.0) in the presence and in the absence of cytochrome c. A droplet of these suspensions (40 μL) was deposited onto a small silicon plate (0.5 cm2), previously subjected to 1 h of stringent cleaning,33 followed by the use of a paper filter to remove the excess solution. Drying was completed at ambient temperature in a Petri plate. The images were treated using the equipment software.Light source power measurements
The measurements of light source power used in the present study were determined using the equipment Laser Power Meter Model FieldMate with PowerMax thermal sensors, and the values obtained were: 116.14, 55.9 and 26.77 mW cm−2 for the three different distances of illumination (3, 7, 13 cm) and the samples had area under illumination = 3.14 cm2.Results and discussion
TiO2 NPs and titanate nanotubes exhibit specific characteristics towards cytochrome c adsorption
Native Fe3+ cytochrome c was incubated with TiO2 NPs and titanate nanotubes as described in Materials and Methods. Significant adsorption of cytochrome c on the titanate nanotubes in comparison with the TiO2 NPs was evident from the visual analysis of the supernatants and pellets obtained after the centrifugation of the respective samples containing the protein and the nanostructures (Fig. 1). | ||
| Fig. 1 From left to right: cytochrome c adsorbed in TiO2 NPs and re-suspended in water after the first washing and with the supernatant; cytochrome c solution; cytochrome c adsorbed in titanate nanotubes with supernatant and after the first washing. | ||
This result was expected because, as previously observed, the TiO2 nanotubes carry a stronger negative surface charge than the TiO2 NPs and, under neutral conditions, present negatively binding sites for electrostatic interactions with alkaline proteins such as cytochrome c (pI of horse heart cytochrome c used in the present study = 10.2).34
Therefore, a set of experiments was carried out to better characterize the adsorption of cytochrome c on the titanate nanotubes and the TiO2 NPs. The morphology of the titanate nanotubes before and after the adsorption of cytochrome c was studied using AFM.
In the absence and in the presence of cytochrome c, AFM images (Fig. 2) clearly show the appearance of the titanate nanotubes adsorbed on the surface of Si. Fig. 2A shows the top views of the AFM images of the titanate nanotubes in the absence of cytochrome c, and Fig. 2B shows the titanate nanotubes after the adsorption of cytochrome c. In the AFM images obtained in the absence of cytochrome c, only the isolated bundles of the nanotubes were discernible (Fig. 2A). The surface profile is compatible with bundles containing several nanotubes, and the distortions of the nanotube surface are due to the tip trajectory and indentation represented in the lowest right panel of Fig. 2A. However, the AFM images of the cytochrome c-containing titanate nanotubes show that, in the presence of the protein, the bundle-organized nanotubes dispersed on the Si surface were substituted by randomly positioned, closely packaged, superimposed nanotubes. In the latter situation, the height profile of the single nanotubes (diameter: ∼10 nm), according to the TEM images,35 is compatible with the partial disaggregation of the nanotubes from the original bundle organization, owing to surface recovery by cytochrome c (lowest right panel of Fig. 2B). However, the close packing of a large amount of cytochrome c-containing nanotubes is possibly related to a strong interaction between the cytochrome c molecules and the surface of the titanate nanotubes, and reflects the ability of cytochrome c molecules to build up supramolecular structures with this material.
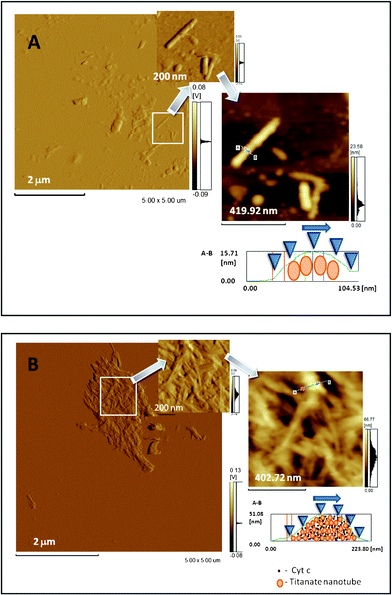 | ||
| Fig. 2 Atomic force microscopy images of titanate nanotubes. (A) AFM top view image of nanotubes in the absence of cytochrome c prepared in a 5 mM phosphate buffer (pH 8.0), and panels showing zoomed images of the isolated nanotubes under these conditions. The right panel is the image presented as trace height (nm) and the left and central panels are presented as deflection trace (volts). (B) AFM top-view image of nanotubes after adsorption of cytochrome c and panels showing zoomed images under these conditions. The right panel is the image presented as trace height (nm) and the left and central panels are presented as deflection trace (volts). In (A) and (B) the lowest right panels are the height profiles of the titanate nanotubes before and after the adsorption of cytochrome c, respectively. Atomic force microscopy images were obtained by measuring the interaction forces of the tip with the sample surface, in the contact mode, at room temperature (20 °C) and at atmospheric pressure (760 mm Hg). Triangular silicon tips were used for this analysis, and the resonant frequency of the cantilever was found to be 200 kHz. Based on the literature,31,32 the contours with ∼10 nm measured in the top view were considered as being from an individual titanate nanotube. The figures shown here are representative of a set of analyses showing similar characteristic patterns in the absence and in the presence of cytochrome c. | ||
In order to understand the AFM results, it is necessary to understand the chemistry involved in the supramolecular structure. It is well known that oxidized surfaces are good adsorbents for proteins and polypeptides.36 In the case of the titanate nanotubes, adsorption can be promoted by the interaction of the surface-terminating anionic groups of the titanate nanotubes and the positively charged amino groups (–NH3+) of alkaline protein cytochrome c.37–39 For the amino group moieties, both charge–charge interactions and ionic hydrogen bonds can be established between –NH3+ and any oxygen-containing TiO2 surface group. Despite knowing the precise origin of the interaction between the titanate nanotubes and cytochrome c, we cannot unequivocally distinguish between these possibilities. Further, as will be shown in the following FT-IR experiments, the water adsorbed on the TiO2 surfaces plays an important role in the molecular interaction of the TiO2 NPs and cytochrome c, as evidenced by the vibrational normal mode (ν O–H) of both cytochrome c and the TiO2 NPs. However, the typical binding energies for such ionic H bonds are in the range 10–30 kcal mol−1. For this case, cytochrome c, which consists of 19 lysines, is highly attractive to the titanate nanotubes.28,37–39 Another possible interaction between the titanate nanotubes and cytochrome c is based on the adsorption of cytochrome c and phosphate ions onto the TiO2 surface at a pH of 8. Just for the illustration of a similar study in the literature, Chusuei et al.,40 on the basis of spectroscopic data, have proposed that positively charged calcium forms an inner-sphere complex with a TiO2 surface and that phosphate is then bound in the TiO2-calcium layer. Further, Ronson and McQuillan41 have shown that phosphate forms a bidentate surface complex with TiO2, and at sufficiently high concentrations of phosphate, there is additional interfacial phosphate that is electrostatically adsorbed with Ca2+ providing the linkage. Similar results were obtained by Jézéquel and Chu, who reported the effects of pH and divalent cations on the adsorption of anions onto TiO2 NPs. The authors demonstrated that anion adsorption onto TiO2 decreased with increasing pH because of the decrease in the number of positively charged binding sites on the TiO2 surface.42
The adsorption of cytochrome c by TiO2 NPs and by titanate nanotubes was detected by infrared spectroscopy.
Fig. 3A shows the vibrational spectra of TiO2 NPs (light gray line) and the complex TiO2 NP–cytochrome c (black line) in comparison with the native Fe3+ cytochrome c FT-IR spectrum shown in the inset. The spectral band in the ∼3200 cm−1 region is assigned to water complexes, strongly bound with the TiO2 surface.43 The FT-IR spectrum of the TiO2 NPs exhibits the bands of the OH group stretching at 3200–3550 cm−1 (O–H asymmetrical and symmetrical stretching vibrations) and the bending vibrations (δ H–O–H) at 1639 cm−1, which are consistent with the presence of water on the surface of the TiO2 NPs. The peculiarities of water adsorption on a TiO2 surface result from the presence of polar hydroxyl groups, as well as from the existence of relatively strong electron-acceptor centres, namely coordinate non-saturated Ti4+ and Ti3+. More specifically, the FT-IR spectrum of the TiO2 NPs shown in Fig. 3A exhibits a band peaking at 3424 cm−1, which can be assigned to one type of surface OH group, located on the same crystallographic TiO2 planes. This result is suggestive of OH groups lying directly above the surface cations, with the axes oriented by the electrostatic field perpendicular to the surface. The peak at 3424 cm−1 is consistent with water molecules, which are bound by weak hydrogen bonds with each other and with OH groups on the TiO2 surface. In the complex TiO2 NP–cytochrome c, the shift of the particular vibrational normal mode (ν O–H) of both cytochrome c and the TiO2 NPs from 3424 cm−1 to 3419 cm−1 corroborates the occurrence of the molecular interaction of cytochrome c with the TiO2 NPs. The adsorption of cytochrome c is evident from the appearance of the band peaking at 2923 cm−1, which is assigned to the carboxylic acid of amino acids and the amide II band. The amide I band is hard to identify here because it is overlapped by the TiO2 NP band at around 1640–1650 cm−1.
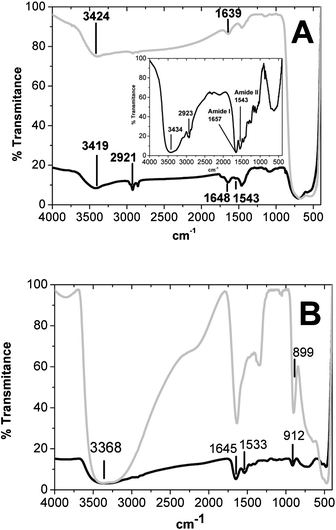 | ||
| Fig. 3 (A) Vibrational spectra of the TiO2 NPs (light gray line) and the TiO2 NP–cytochrome c complex (black line) in comparison with the native Fe3+ cytochrome c FT-IR spectrum shown in the inset. (B) Vibrational spectra of the titanate nanotubes (light gray line) and cytochrome c associated with nanotubes (black line). | ||
Fig. 3B shows the vibrational spectra of the titanate nanotubes (light gray line) and cytochrome c associated with the nanotubes (black line). Nanotube structures exhibit an FT-IR spectrum (Fig. 3B) with a broad and intense band peaking at 3368 cm−1 (O–H stretching vibrations), and the vibrational normal modes assigned to bending vibrations (δ H–O–H) is compatible with the presence of water molecules associated with the surface and the internal space of the titanate nanotubes.
The absorption band at 899 cm−1 can be assigned to the stretching vibration of short Ti–O bonds involving non-bridging oxygen coordinated with sodium ions. This band was shifted to 912 cm−1 in the complex with cytochrome c. The molecular interaction between cytochrome c and the titanate nanotubes is evident because of the presence of amide I and amide II bands of cytochrome c in the complex cytochrome c–titanate nanotubes. The amide I and amide II bands of cytochrome c are slightly shifted from 1657 and 1543 cm−144 to 1645 and 1533 cm−1, respectively, which remains compatible with a folded protein.
The cytochrome c-containing nanotubes exhibited a significant decrease in the spectral area in the frequency interval 2412 cm−1 to 3667 cm−1. Considering that the thermogravimetric analysis (TGA) did not reveal any change in the total water content (see below), this occurrence could be attributed to the changes in the symmetry of the O–H groups of the complex cytochrome c–titanate nanotubes.
The vibrational spectra of both the TiO2 NPs and the titanate nanotubes show that both titanium-containing structures could adsorb cytochrome c, but the differences in the amount of adsorbed protein were determined by TGA analysis and the electronic absorption spectra of the desorbed protein.
The thermal behaviour of the TiO2 NPs, cytochrome c-containing TiO2 NPs, titanate nanotubes, and the cytochrome c-containing titanate nanotubes was investigated using thermogravimetry, and the TGA–DTA–DTG data (where DTA is differential thermoanalysis and DTG is derivative of thermogravimetric analysis) obtained for these materials are shown in Table 1 and Fig. 1SA, B, C, D, and E of the ESI.†
| Analyzed material | Water | Organic material | ||
|---|---|---|---|---|
| Endothermic peaks (°C) | % Content | Endothermic peaks (°C) | % Content | |
| a TGA–DTG curves not shown in the Fig. 2S panels.† | ||||
| TiO2 NPsa | — | — | — | — |
| Titanate nanotubea | 84.07 | 14.45 | — | — |
| Cytochrome c | 68.94 | 14.65 | 344.52 and 442.37 | 41.05 and 44.61 |
| Cytochrome c–TiO2 NP (physical mixture) | 47.2 | 1.8 | 360.23 and 468.83 | 6.6 and 8.63 |
| Cytochrome c–TiO2 NP (dark)a | 47.23 and 177.2 | 0.25 and 0.14 | 354.27 and 451.13 | 1.61 and 0.83 |
| Cytochrome c–TiO2 NP (hν) | 55.29 and 176.65 | 0.52 and 0.31 | 353.46 and 457.35 | 2.94 and 1.43 |
| Cytochrome c–titanate nanotube (physical mixture) | 83.62 | 13.7 | 348.57 and 516.52 | 11.28 and 7.24 |
| Cytochrome c–titanate nanotube (dark)a | 74.8 | 11.6 | 346.7 | 17.25 |
| Cytochrome c–titanate nanotube (hν) | 73.51 | 13.55 | 344.85 | 18.57 |
The TGA–DTG curve for the TiO2 NPs (not shown) reveals that this material does not exhibit significant water content (Table 1). The TGA–DTG curves for the cytochrome c–TiO2 NP complex obtained in the dark (not shown) and under irradiation (Fig. 1SC†) are similar and show that weight loss occurs between 20 and 250 °C, which can be attributed to the loss of water molecules. The endothermic peaks at 68.9 °C (Fig. 1SA† and Table 1 data obtained from a sample of free cytochrome c), 47.2 °C and 170 °C (figure not shown and Table 1 data obtained from a sample of the cytochrome c–TiO2 NPs incubated in the dark) and 55.3 °C and 176 °C (Fig. 1SB† and Table 1 data obtained from a sample of the cytochrome c–TiO2 NPs incubated under irradiation) in the DTA curves confirm this attribution. In the complex cytochrome c–TiO2 NP, secondary mass losses between 250 and 600 °C correspond to the loss of two populations of cytochrome c molecules that are bound to the TiO2 NPs, as evidenced by the endothermic peaks at 344.5 and 442.4 °C (Fig. 1SA† and Table 1 data obtained from a cytochrome c solution), 354.3 °C and 451.1 °C (figure not shown and Table 1 data obtained from a sample of the cytochrome c–TiO2 NP incubated in the dark) and 356.4 and 457.3 °C (Fig. 1SC† and Table 1 data obtained from a sample of the cytochrome c–TiO2 NPs incubated under irradiation) in the DTA curves. Despite displaying similar thermal behaviour, a significant variation in the organic content was observed by comparing the TGA curves of the cytochrome c–TiO2 NP complexes obtained in the dark and under irradiation. Calculations revealed that around 10% and 20% of the total cytochrome c mass was adsorbed by the TiO2 NPs in the dark and under irradiation, respectively, and the effect of UV irradiation should be related to the reduction of heme iron. The water content of cytochrome c and the cytochrome c–TiO2 NP samples determined by TGA revealed that water contributed around 15% of the total cytochrome c mass before and after adsorption by the TiO2 NPs in the dark and under irradiation. However, in the complex of cytochrome c and the TiO2 NPs, there were two water populations: one population with a lower endothermic peak value and the other population with a higher value. These results suggest new interactions for the water molecules in the complex. The interaction of cytochrome c with the TiO2 NPs was also evident because of the increase in the cytochrome c population, with an endothermic peak at around 350 °C (Fig. 1SC† and Table 1), in contrast to free cytochrome c (Fig. 1SA† and Table 1) and cytochrome c physically mixed with the TiO2 NPs, whose highest cytochrome c population exhibited an endothermic peak around 450 °C (Fig. 1SB† and Table 1).
The TGA–DTG curve for the titanate nanotubes (figure not shown) revealed that this material was hydrated and water contributed approximately 14.5% of the total mass and had an endothermic peak at 84.07 °C (Table 1). The TGA–DTG curves for a cytochrome c titanate nanotube complex obtained in the dark (figure not shown) and under irradiation (Fig. 1SE†) were similar and showed that the weight loss occurred between 20 and 200 °C, which could be assigned to the loss of water molecules that were physically adsorbed on the surface of the titanate nanotubes (Table 1). The endothermic peak, around 74 °C in the DTA curves, confirmed this attribution (Table 1 and Fig. 1SE†). Mass losses were also observed between 250 and 600 °C, which showed the loss of one population of cytochrome c molecules that were bound to the titanate nanotubes, as evidenced by the endothermic peaks around 345 °C in the DTA curves (Table 1 and Fig. 1SE†). For the cytochrome c–titanate nanotube complex, the loss of weight revealed 90% adsorption from the total mass of protein present in the solution before the addition of the nanotubes, without a significant influence of the irradiation on the adsorption of cytochrome c by the nanotubes. Interestingly, the cytochrome c–titanate nanotube complex had one endothermic peak for water molecules, and the value of this peak is intermediate between that of the peak exhibited by free cytochrome c and by pure titanate nanotubes. In the cytochrome c–titanate nanotube complex, the total mass of water corresponded to the sum of the water content of the nanotubes and the protein. It is also important to note that the endothermic peaks around 350 °C were observed in the TGA–DTG curves of the cytochrome c physically mixed with the titanate nanotubes (Fig. 1SD† and Table 1), and, in this condition, an endothermic peak around 520 °C was also observed. This thermal behaviour suggests a high affinity between cytochrome c and the titanate nanotubes, which leads to the partial adsorption of the cytochrome c physically mixed with the nanotubes during the heating. In this condition, the titanate nanotubes probably protect cytochrome c from the heating, as evidenced by the increase in the temperature value of the highest endothermic peak of the protein.
The TGA data allow the calculation of the mass of cytochrome c adsorbed in the titania nanostructures. The mass of cytochrome c adsorbed on the surface of the titanate nanotubes (1.75 μg m−2) was 2.3 fold higher (0.75 μg m−2) than that adsorbed on the TiO2 NPs.
In Fig. 1 it was evident that the cytochrome c solution incubated with TiO2 NPs changed from a red-brown to a light red colour, which is typical of the reduced form of the heme protein. The colour change did not occur in samples maintained in the dark, and reduction of cytochrome c by TiO2 NPs subjected to illumination was corroborated by electronic absorption spectrum and EPR measurements.
Fig. 4A shows the UV-visible spectrum of 20 μM Fe3+ native cytochrome c before (gray line) and 3 h after incubation (gray line), with the 0.5 mg mL−1 TiO2 NPs in a 5 mM sodium phosphate buffer (pH 8.0) in air and under irradiation with a 125 W UV lamp. The spectrum shown as a black line refers to the supernatant obtained after the centrifugation of the TiO2 NP suspension in a cytochrome c solution.
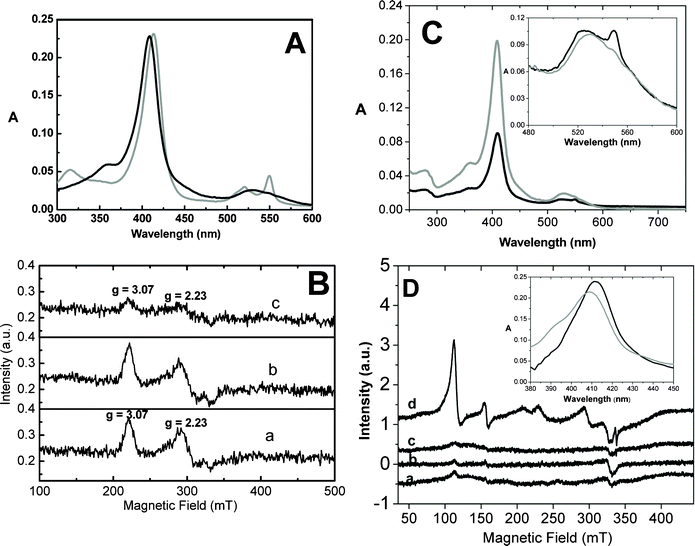 | ||
| Fig. 4 (A) Electronic absorption spectrum of 20 μM Fe3+ native cytochrome c before (black line) and 3 h after incubation (gray line) with 0.5 mg mL−1 TiO2 NPs in a 5 mM sodium phosphate buffer (pH 8.0) in air and under irradiation with a 125 W UV lamp. The optical path length was 0.1 cm for all measurements. (B) EPR spectra of 300 μM Fe3+ cytochrome c in water incubated for 30 min under irradiation with a 125 W UV lamp (line a), incubated for 30 min with 7.5 mg mL−1 TiO2 NPs in the dark (line b), and incubated for 30 min with TiO2 NPs under irradiation with a 125 W UV lamp (line c). (C) Electronic absorption spectrum of 20 μM Fe3+ native cytochrome c before (black line, optical length = 0.1 cm) and 3 h after incubation (gray line, optical length = 1 cm) with 0.5 mg mL−1 TiO2 nanotubes in 5 mM sodium phosphate buffer, pH 8.0, in air and under irradiation with a 125 W UV lamp. The inset corresponds to a zoom of the Q bands to show the slight increase of the 549 nm absorbance intensity indicative of a partial reduction of cytochrome c. (D) EPR spectra of 300 μM Fe3+ cytochrome c incubated with 7.5 mg mL−1 titanate nanotubes in water before irradiation (line a), after 5 min (line b), 15 min (line c), and 30 min (line d) of incubation under irradiation with a 125 W UV lamp. | ||
Cytochrome c was not reduced when incubated with the TiO2 NPs in acidic pH (6.0) and in the dark (not shown). The photo-reduction of cytochrome c by the TiO2 NPs was also observed by electron paramagnetic resonance (EPR) spectrometry. Fig. 4B, line a, shows the EPR spectrum of native cytochrome c before irradiation in the presence of the TiO2 NPs. The EPR spectrum is typical of the native low-spin-state cytochrome c with rhombic symmetry (spin ½, g1 = 3.07, and g2 = 2.23).45 A significant decrease in the cytochrome c EPR signal, indicative of the heme iron reduction, was not observed after the incubation of cytochrome c with the TiO2 NPs in the dark (line b), but only appeared when the protein was incubated with the TiO2 NPs under irradiation (line c).
The rate of photo-reduction of cytochrome c by the TiO2 NPs was dependent on the light source potency/area, because increasing the values from 116.14 mW cm−2 to 55.9 mW cm−2 decreased the kobs of the heme iron reduction from 12.0 to 6.0 × 10−3 s−1.
Native Fe3+ cytochrome c was also incubated for 3 h with 0.5 mg mL−1 titanate nanotubes in a 5 mM sodium phosphate buffer, pH 8.0, in air and under irradiation with a 125 W UV lamp. The electronic absorption spectrum of the supernatant obtained after centrifugation of the sample containing cytochrome c and titanate nanotubes revealed a significant decrease in cytochrome c content (Fig. 4C, gray line). This cytochrome c sample exhibited a relatively low concentration of the reduced form (see in the inset, the low increase in the absorbance intensity at 549 nm). This result suggests significant adsorption of the protein on the titanate nanotubes. The high coverage of titanate nanotubes by cytochrome c might impair the direct contact of non-adsorbed cytochrome c with the titanate nanotube surface, with consequent low yield of the reduced cytochrome c in solution.
However, cytochrome c adsorbed onto titanate nanotubes was reduced by irradiation, as evidenced by EPR and electronic absorption spectra (Fig. 4D and inset). Fig. 4D shows the EPR spectrum of cytochrome c adsorbed onto the titanate nanotubes after a 3 h incubation under irradiation. Cytochrome c-containing titanate nanotubes were centrifuged, thoroughly washed, and re-suspended in deionized water. The EPR spectrum, shown in Fig. 4D, line a, revealed the different populations of cytochrome c. Cytochrome c adsorbed on the titanate nanotubes remained predominantly in the low-spin state but had changes in the heme symmetry. A part of the low-spin-state protein exhibits an EPR spectrum of the alternative low-spin form (spin ½, g1 = 2.902, g2 = 2.225, and g3 = 1.510), a low-spin form that had been previously observed in the presence of cardiolipin, dodecylphosphate (DCP) vesicles, sodium dodecylsulphate (SDS), and TRIS-buffered sodium di-2-ethylhexylsulfosuccinate/hexane reverse micelles (pH = 8.5).45 A part of the low-spin-state cytochrome c population exhibited EPR signals suggestive of the heme group with a relatively high rhombic symmetry. Considering the high-intensity EPR signal inherent in the high-spin forms of cytochrome c, Fig. 4D, line a, shows that in the titanate nanotubes, only a low amount of the cytochrome c population was converted into the high-spin form. Fig. 4D, line a, shows that the high-spin state (spin 5/2, g1 = 6.0 and g2 = 2.0) and a small amount of the high-spin form with a rhombic symmetry (g = 4.3, magnetic field at 155 mT) were also detected. These high-spin cytochrome c forms were previously detected during the reaction of cytochrome c with t-butyl hydroperoxide (t-BuOOH), and in the presence of negatively charged liposomes.46,47 The irradiation of cytochrome c that was adsorbed on the titanate nanotubes also led to heme iron reduction, as indicated by the disappearance of the EPR signal when the sample was subjected to irradiation (Fig. 4D, lines b, c, and d) and a Soret band red shift detected by electronic absorption spectroscopy (inset of Fig. 4D).
The TiO2-promoted photo-reduction of cytochrome c was reverted by peroxides (not shown). Despite the production of singlet oxygen and a hydroxyl radical in the medium during TiO2 irradiation,48,49 the Fe3+-cytochrome c generated from TiO2-photo-reduced protein after treatment with peroxides (not shown) conserved the redox properties of the native protein, since the protein remained capable of reduction by diphenylacetaldehyde (DPAA) (not shown), a process dependent on the integrity of the native structure.15,50
Cytochrome c adsorbed on TiO2 NPs and titanate nanotubes retains the redox properties of native protein
Considering the adsorption of cytochrome c by the TiO2 NPs and the titanate nanotubes, the functionality of the protein desorbed from these materials was probed. Cytochrome c desorbed from the TiO2 NPs and the titanate nanotubes was reduced by DPAA, ascorbic acid, and β-mercaptoethanol (not shown). The capacity to be reduced by DPAA suggests the integrity of the tyrosine residues of the protein structure.50The pathway involved in photo-reduction of cytochrome c by TiO2 NPs and by titanate nanotubes
The possible mechanism of electron transfer from the TiO2 NPs to cytochrome c was also investigated. The occurrence of water photolysis was probed by the generation of the hydroxyl radical in the TiO2 samples illuminated in the presence and in the absence of molecular oxygen (Fig. 2SA†). The photo-induced electron transfer from water to cytochrome c promoted by the TiO2 NPs was also probed by changing the dielectric constant of the medium (Fig. 2SB†) and by replacing water with D2O. The presence of 20% dioxane in the medium was sufficient to completely prevent the reduction of cytochrome c by the TiO2 NPs (gray bars), but even 40% dioxane had no effect on the chemical reduction of cytochrome c by β-mercaptoethanol (light gray bars). The impairment of cytochrome c reduction by the TiO2 NPs promoted by dioxane was not due to the damage in the protein promoted by the TiO2 NPs in the presence of the solvent, since cytochrome c, not reduced by TiO2 NPs in the presence of dioxane, was promptly reduced after the addition of β-mercaptoethanol. The replacement of water by D2O increased by fivefold the rate of cytochrome c reduction by the TiO2 NPs, as compared to water as the reducing agent for cytochrome c (Fig. 2SC†). For more details about the pathway involved in the photo-reduction of cytochrome c by TiO2 NPs and by titanate nanotubes, see the supplementary material.† Therefore, the classical semiconductor mechanism seems to be involved in the photoreduction of cytochrome c by titania nanostructures.As mentioned before, in the last few decades, several research groups have contributed considerably to the characterization of the electro(photo)chemistry of semiconductor materials associated with heme proteins. In these studies involving heme proteins and semiconductors, cytochrome c and TiO2 is the predominant combination, and different architectures of these components have been constructed, especially by varying the TiO2 structural organization to forms such as nanoporous, mesoporous, nanoparticles, and nanotubes.4–13 Most of these high-quality studies have focused on the functionality of the system with respect to the generation of an electrical (photo) current and molecular hydrogen to be used as fuel. Furthermore, from our knowledge, only two studies have been concerned with the interaction of cytochrome c with titanate nanotubes, and these studies, along with all the existing literature in this area, have scarcely contributed to the elucidation of the structure, functionality, and reversibility of the TiO2-bound cytochrome c. Therefore, a biochemical approach to the systems composed by cytochrome c and titania nanostructures complements the characterization of these composites. Moreover, even explored themes such as cytochrome c photo-reduction and water splitting need to be revisited. The present study, for the first time, focused on the structure and function of the protein component by using advanced techniques such as EPR, FT-IR, and TGA. It was found that most of the cytochrome c molecules associated with the titanate nanotubes retain the spin state of the native protein (Fig. 4D), and the spin state changes observed for a fraction of the protein are reversible, since the cytochrome c removed from the titanate nanotubes exhibits the same spectral features as those of the native protein (Fig. 3S†). Further, the FT-IR analysis corroborated the preservation of cytochrome c folding when associated with the titanate nanotubes. The TGA analysis also provided additional relevant comparative information about the cytochrome c bound to the TiO2 NPs and the titanate nanotubes. The TGA analysis revealed a significant difference in the water content of the TiO2 NPs and the titanate nanotubes before association with cytochrome c. This analysis revealed that, although cytochrome c remained predominantly in a native-like structure, as indicated by the EPR and FT-IR analyses, the thermal stability of the protein was significantly affected after association with both the TiO2 NPs and the titanate nanotubes. In the case of the titanate nanotubes, because of the strong affinity for cytochrome c, the loss of thermal stability of the protein was observed even in the physical mixture. Cytochrome c bears the heme group covalently attached to the polypeptide chain, and the redox centre is unlikely to be lost, even when there are drastic changes in the protein structure. However, the redox properties of the heme group are finely modulated by the microenvironment of the protein. Further, these findings are particularly important for the proteins bearing the non-covalent heme, in which even moderate structural changes can result in drastic changes in the activity, and should be considered in the construction of other bio-composite materials.
In addition to a better comparative characterization of the cytochrome c associated with the TiO2 NPs and the titanate nanotubes, the present study contributes two more important findings: the formation of an unprecedented supramolecular structure after the association of cytochrome c with the titanate nanotubes, and a more accurate investigation of the photo-reduction mechanisms. With respect to the photo-reduction of cytochrome c by the titania nanostructures, for the first time the influence of the semiconductor structure on this photochemistry has been demonstrated. The data presented in Fig. 2SA, B, and C in the supplementary material† provide clear evidence that the TiO2 NP-containing system is analogous to some steps of the photo-phosphorylation process, and water acted as the reducing agent for the heme iron in an event made feasible by light absorption (Fig. 5). Unlike the photosynthetic apparatus, the oxidation of H2O to ˙OH occurs in the TiO2 NP-containing system because of the close potential values of the H2O/˙OH (+ 2.27 V vs. SHE) and TiO2vb/TiO2vb+ redox couples (+ 0.30 V vs. SHE). The redox potential for TiO2cb/TiO2cb− redox couples is −0.7 V vs. SHE, which is in principle sufficiently negative to promote the cytochrome c reduction in a thermodynamically favorable way.51,52 The latter is similar to the biological system diagram (left-hand side), in which light energy is used for creating a high-energy electron donor and a low-energy electron acceptor. In this case, the photo-reduction of cytochrome f in the photo-phosphorylation process also occurs in a thermodynamically favorable way. However, in a biological system, the electron transfer from the photo-system to cytochrome f via quinone intermediates allows the generation of the protomotive force, whose dissipation is coupled with ATP synthesis.
For electrochemical and bioelectrochemical formalism, it is worth noting that the potential values presented in Fig. 5 may vary slightly according to the experimental conditions.51–55 Nevertheless, they always follow the trend and the energy reaction pathway proposed in Fig. 5.
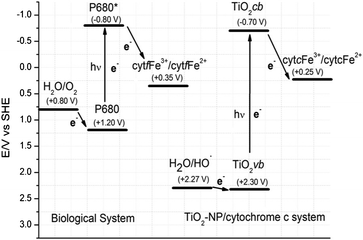 | ||
| Fig. 5 Comparative view of the photo-reduction of cytochrome f in the photo-phosphorylation process with cytochrome c reduction in the TiO2 NP–cytochrome c system. Redox potentials at pH 7.0 (vs. SHE) according to ref. 51–55. The intermediate steps of the photo-reduction of cytochrome f have been omitted for the sake of clarity. | ||
In the present study, the novel finding that the suspended TiO2 NPs can photochemically reduce cytochrome c in solution is particularly relevant because it allows the use of cytochrome c as a mobile electron carrier for an oxidant material added to the media. It is important to note that the present results show that, in a system containing TiO2 NPs, soluble photo-reduced cytochrome c can be recycled to the ferric form without any loss in structure or reactivity.
Conclusions
The most important findings that can be outlined from the presented results are as follows:- Different crystalline structures of titanium nanostructures exhibited significant differences in their affinity for cytochrome c; the cytochrome c mass that adsorbed on the titanate nanotubes (1.75 μg m−2) was 2.3 times higher than that adsorbed on the surface of the TiO2 NPs (0.75 μg m−2).
- The OH groups of water molecules adsorbed on the surfaces of the TiO2 NPs and the titanate nanotubes were involved in the molecular interaction of the TiO2 NPs and cytochrome c, as evidenced by the changes in the vibrational spectra of the titania structures after the cytochrome c adsorption.
- According to the EPR and TGA measurements, association with titanate nanotubes led to structural changes in cytochrome c.
- However, these structural changes were reversible, since the protein desorbed from the titanate nanotubes, as well as that from the TiO2 NP, retained the spectroscopic characteristics and redox properties of the native protein.
- Both the TiO2 NPs and the titanate nanotubes could promote the photo-reduction of cytochrome c.
- The photo-reduction of cytochrome c by TiO2 NPs probably involved net electron transfer from water to cytochrome c.
Acknowledgements
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) I. L. Nantes, Project Number 2008/04849-0 and F.N. Crespilho Project numbers: 2009/15558-1 and 2011/01541-0; Fundação de Amparo ao Ensino e Pesquisa da Universidade de Mogi das Cruzes (FAEP-UMC), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) I. L. Nantes Project Number 475235/2010-0 and F.N. Crespilho, CNPq Project number: 2011/01541-0; INEO and Rede NanoBioMed-Brasil, CAPES.The authors are grateful to Prof. Flavio L. de Souza and the MSc student Waldemir M. de Carvalho Jr for the measurements of light source powers used in the present study.
References
- M. Grätzel, Inorg. Chem., 2005, 44, 6841 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef.
- E. Coronad and E. Palomares, J. Mater. Chem., 2005, 15, 3593 RSC.
- N-G. Park, G. Schlichthörl, J. van de Lagemaat, H. M. Cheong, A. Mascarenhas and A. J. Frank, J. Phys. Chem. B, 1999, 103, 3308 CrossRef CAS.
- N-G. Park, J. van de Lagemaat and A. J. Frank, J. Phys. Chem. B, 2000, 104, 8989 CrossRef CAS.
- Q. Wang, W. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Gra1tzel, J. Phys. Chem. B, 2005, 109, 15397 CrossRef CAS.
- L. Kavan, B. O'Regan, A. Kay and M. Grätzel, J. Electroanal. Chem., 1993, 346, 291 CrossRef CAS.
- K. Hara, Y. Dan-Oh, C. Kasada, Y. Ohga, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Langmuir, 2004, 20, 4205 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef CAS.
- J. Y. Kim, S. H. Kim, H-H. Lee, K. Lee, W. Ma, X. Gong and A. Heeger, Adv. Mater., 2006, 18, 572 CrossRef CAS.
- E. Topoglidis, T. Lutz, J. R. Durrant and E. Palomares, Bioelectrochemistry, 2008, 74, 142 CrossRef CAS.
- J. H. Park, S. Kim and A. J. Bard, Nano Lett., 2006, 6, 24 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef CAS.
- M. L. Estevam, O. R. Nascimento, M. S. Baptista, P. D. Mascio, F. M. Prado, A. Faljoni-Alario, M. R. Zucchi and I. L. Nantes., J. Biol. Chem., 2004, 279, 39214 CrossRef CAS.
- V. A. Soares, D. Severino, H. C. Junqueira, I. L. S. Tersariol, C. S. Shida, M. S. Baptista, O. R. Nascimento and I. L. Nantes, Photochem. Photobiol., 2007, 83, 1254 CrossRef CAS.
- M. J. B. Hauser, A. Lunding and L. F. Olsen, Phys. Chem. Chem. Phys., 2000, 2, 1685 RSC.
- J. J. L. Carson and J. Walleczek, J. Phys. Chem. B, 2003, 107, 8637 CrossRef CAS.
- I. B. Campos, I. L. Nantes, F. A. Rodrigues and S. Brochsztain, J. Mater. Chem., 2004, 14, 54 RSC.
- K. M. Figueiredo, R. O. Marcon, I. B. Campos, I. L. Nantes and S. Brochsztain, J. Photochem. Photobiol., B, 2005, 79, 1 CrossRef CAS.
- E. Topoglidis, Y. Astuti, F. Duriaux, M. Grätzel and J. R. Durrant, Langmuir, 2003, 19, 6894 CrossRef CAS.
- Y. Astuti, E. Palomares, S. A. Haque and J. R. Durrant, J. Am. Chem. Soc., 2005, 127, 15120 CrossRef CAS.
- E. Topoglidis, C. J. Campbell, E. Palomares and J. R. Durrant, Chem. Commun., 2002, 1518 RSC.
- P. Cuendet and M. Grätzel, Bioelectrochem. Bioenerg., 1986, 16, 125 CrossRef CAS.
- E. Topoglidis, A. E. G. Cass, G. Gilardi, S. Sadeghi, N. Beaumont and J. R. Durrant, Anal. Chem., 1998, 70, 5111 CrossRef CAS.
- E. Topoglidis, T. Lutz, R. L. Willis, C. J. Barnett, A. E. G. Cass and J. R. Durrant, Faraday Discuss., 2000, 116, 35 RSC.
- L. Liu, N. Wang, X. Cao and L. Guo, Nano Res., 2010, 3, 369 CrossRef CAS.
- D. V. Bavykin, E. V. Milsom, F. Marken, D. H. Kim, D. H. Marsh, D. J. Riley, F. C. Walsh, K. H. El-Abiary and A. A. Lapkin, Electrochem. Commun., 2005, 7, 1050 CrossRef CAS.
- G. Zhao, Y. Lei, Y. Zhang, H. Li and M. Liu, J. Phys. Chem. C, 2008, 112, 14786 CAS.
- K. J. McKenzie and F. Marken, Langmuir, 2003, 19, 4327 CrossRef CAS.
- F. P. Daly, H. Ando, J. L. Schimitt and E. A. Sturm, J. Catal., 1987, 108, 401 CrossRef CAS.
- O. P. Ferreira, A. G. Souza Filho, J. M. Filho and O. L. Alves, J. Braz. Chem. Soc., 2006, 73, 393 CrossRef.
- J. Jass, T. Tjärnhage and G. Puu, Methods Enzymol., 2003, 367, 199 CAS.
- R. E. Dickerson and R. Timkovich, in The Enzymes, ed. P. D. Boyer, Academic Press, New York, 1975, vol. 11, pp. 398–547 Search PubMed.
- B. C. Viana, O. P. Ferreira, A. G. Souza-Filho, J. Mendes-Filho and O. L. Alves, J. Braz. Chem. Soc., 2009, 20, 167 CrossRef CAS.
- L-C. L. Huang and H.-C. Chang, Langmuir, 2004, 20, 5879 CrossRef CAS.
- M. Meot-Ner, J. Am. Chem. Soc., 1984, 106, 1257 CrossRef.
- M. Meot-Ner, L. W. Sieck, J. F. Liebman and S. Scheiner, J. Phys. Chem., 1996, 100, 6445 CrossRef CAS.
- G. R. Moore, G. W. Pettigrew, Cytochrome c, Springer-Verlag, Heidelberg, 1990 Search PubMed.
- C. C. Chusuei, D. W. Goodman, M. J. Stipdonk, D. R. Justes, K. H. Loh and E. A. Schweikert, Langmuir, 1999, 15, 7355 CrossRef CAS.
- T. K. Ronson and A. J. McQuillan, Langmuir, 2002, 18, 5019 CrossRef CAS.
- H. Jézéquel and K. H. Chu, Environ. Chem. Lett., 2005, 3, 132 CrossRef.
- C. E. Bamberger and G. M. Begun, J. Am. Ceram. Soc., 1987, 70, C48 CrossRef.
- I. B. Campos, I. L. Nantes, F. A. Rodrigues and S. Brochsztain, J. Mater. Chem., 2004, 14, 54 RSC.
- K. C. U. Mugnol, R. A. Ando, R. Y. Nagayasu, A. Faljoni-Alario, S. Brochsztain, P. S. Santos, O. R. Nascimento and I. L. Nantes, Biophys. J., 2008, 94, 4066 CrossRef CAS.
- I. L. Nantes, A. Faljoni-Alario, O. R. Nascimento, B. Bandy, R. Gatti and E. J. H. Bechara, Free Radical Biol. Med., 2000, 28, 786 CrossRef CAS.
- M. R. Zucchi, O. R. Nascimento, A. Faljoni-Alario, T. Prieto and I. L. Nantes, Biochem. J., 2003, 370, 671 CrossRef CAS.
- Y. Nosaka, T. Daimon, A. Y. Nosaka and Y. Murakami, Phys. Chem. Chem. Phys., 2004, 6, 2917 RSC.
- S. Wang, R. Gao, F. Zhou and M. Selke, J. Mater. Chem., 2004, 14, 487 RSC.
- T. A. Rinaldi, I. L. S. Tersariol, F. H. Dyszy, F. M. Prado, O. R. Nascimento, P. Di Mascio and I. L. Nantes, Free Radical Biol. Med., 2004, 36, 802 CrossRef CAS.
- M. R. Allen, A. Thibert, E. M. Sabio, N. D. Browning, D. S. Larsen and F. E. Osterloh, Chem. Mater., 2010, 22, 1220 CrossRef CAS.
- A. Ujishima, T. N. Rao and D. A. Tryk, J.Photochem. and Photobiol. C: Photochemistry Reviews 1, 2000, 1, 1–21 CrossRef.
- L. Krishtalik and I. Biochim, Biochim. Biophys. Acta, Bioenerg., 1986, 849, 162 CrossRef CAS.
- P. Wardman, J. Phys. Chem. Ref. Data, 1989, 18, 1631 CrossRef.
- S. E. Martinez, D. Huang, M. Ponomarev, W. A. Cramer and J. L. Smith, Protein Sci., 1996, 5, 1081 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20996a/ |
| ‡ C. F. B. D. and J. C. A. contributed equally to the manuscript. |
| § Deceased March 12th, 2012. |
| This journal is © The Royal Society of Chemistry 2012 |