DOI:
10.1039/C2RA20598J
(Paper)
RSC Adv., 2012,
2, 5729-5737
The application of “backdoor induction” in bioinspired asymmetric catalysis†
Received
2nd April 2012
, Accepted 10th April 2012
First published on 13th April 2012
Abstract
Amino acid substituted monodentate triphenylphosphine ligands Lig-Aa1-Aa2-Aa3-Z 5 and 6, with a C-terminal ester or amide group, respectively, were prepared in a few simple synthetic steps. Supramolecular in situ formed complexes [Rh(COD)(Lig-Aa1-Aa2-Aa3-Z)2]BF43 and 4 with a prochiral coordination sphere of the metal are selective hydrogenation catalysts (up to 68% ee). The selectivity is induced by transmission of chirality via distant hydrogen-bonded amino acids with a Herrick-like secondary structure (“backdoor induction”).
Introduction
Rational design of new catalysts is often inspired by nature,1 combining transition metals with biooligomers composed of amino acid or nucleotide building blocks. In these bioinspired catalysts, the metal centre dominates the chemical activity, while the biooligomer has the potential to induce selectivity.
Gilbertson et al. provided pioneering examples of bioinspired catalysis, introducing various phosphine-containing amino acids into synthetic oligopeptides2 and applying them to transition metal mediated catalysis of “non-biological” reactions, e.g. rhodium-catalyzed hydrogenation or palladium-catalyzed alkylation. Several other authors have applied ligand-containing amino acids or oligopeptides to homogeneous or heterogeneous (resin-supported, “on bead”) transition metal catalysis,3 but often with only moderate selectivity. It was postulated that a well defined secondary structure of the biooligomer is essential for high selectivity in bioinspired catalysis.1
The secondary structure of 1,2′-disubstituted ferrocene peptides 1 is extensively studied and well understood.4–7 The preferred Herrick-conformation features (a) clockwise (P) helical chirality of the metallocene moiety induced by inter-strand hydrogen bonding of the attached L-amino acids, (b) both carbonyl oxygen atoms directly attached to ferrocene point outwards of the cleft formed by the two amino acid chains (E,E geometrical isomer), and (c) two 10-membered rings (β-turn) formed by inter-strand hydrogen bonding, Scheme 1.7
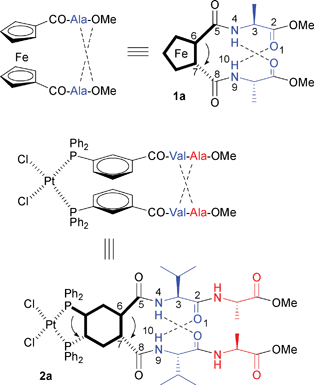 |
| | Scheme 1 Side view (top left) and top view (top right) of 1,2′-disubstituted ferrocene peptide 1a (Herrick-conformation).7Meta-Substituted SupraPhanePhos complex 2a: side view8 (middle) and stereochemical analysis7 (bottom). The upper central aromatic rings are indicated in bold; the arrows indicate the sign of helical chirality. | |
Recently, Breit reported supramolecular Rh(I) and Pt(II) PhanePhos-analogues 2 (named SupraPhanePhos),8 containing amino acid substituted monodentate phosphines9,10 crosslinked by inter-strand hydrogen bonding, Scheme 1. If we apply the nomenclature developed for 1,2′-disubstituted ferrocene peptides7 to complex 2a, a Herrick-conformation was found as well, with inter-strand hydrogen bonding inducing a β-turn (two 10-membered rings) and E,E-conformation of the two carbonyl groups bound directly to the central aromatic ring. In addition, the attached L-amino acids induce helical chirality of the stacked central aromatic rings; the clockwise (P) helical chirality of the chiral amino acid substituents is transferred to counterclockwise (M) helical chirality of the prochiral metal, Scheme 1. Breit used SupraPhanePhos ligands in rhodium-catalyzed asymmetric hydrogenation and obtained up to 51% ee.8,11
In this paper, we introduce several changes of Breit's ligands,8 based on the stereochemical analysis presented above, in order to improve the selectivity in catalysis. First, we used para-substitution of the central aromatic unit, Scheme 2, since it is insensitive to 180° rotation of one ligand (see ESI† for detailed stereochemical analysis). Second, in addition to the terminal ester derivatives, we introduced terminal amino acid amides in order to allow additional supramolecular interactions. And third, we used ligands substituted with three instead of two amino acids. According to our hydrogen bonding pattern analysis, Scheme 2,7,12 it is obvious that chains with one (or with two) amino acids form only two inter-strand hydrogen bonds, while chains with three amino acids are capable of forming four inter-strand hydrogen bonds, which increases the stability of the supramolecular complex and enables higher selectivity in asymmetric catalysis.
![Schematic representation (top view) of hydrogen bonding in [Rh(COD)(Lig-Aa1-Aa2-Aa3-Z)2]BF4 complexes 3 (Z = OMe or OEt) or 4 (Z = NH2); Lig = Ph2P-pC6H4C(O)-; the arrows indicate the sign of helical chirality.](/image/article/2012/RA/c2ra20598j/c2ra20598j-s2.gif) |
| | Scheme 2 Schematic representation (top view) of hydrogen bonding in [Rh(COD)(Lig-Aa1-Aa2-Aa3-Z)2]BF4 complexes 3 (Z = OMe or OEt) or 4 (Z = NH2); Lig = Ph2P-pC6H4C(O)-; the arrows indicate the sign of helical chirality. | |
Results and discussion
Synthesis and characterization
Synthesis of amino acid substituted phosphine ligands 5 and 6 was performed by standard peptide coupling/deprotection in solution, introducing the most expensive N-terminal phosphine building block Ph2P-pC6H4CO2H in the last step. The synthesis of tripeptides 5c–5j and 6d–6l is shown on Scheme 3, the analogous synthesis of amino acid derivatives Lig-Gly-OMe/NH25a/6a and dipeptides Lig-Gly-Phe-OMe/NH25b/6b is featured in the ESI.†
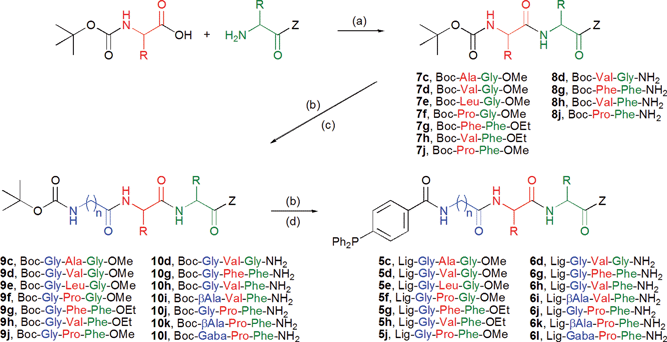 |
| | Scheme 3 Solution phase synthesis of ligands Lig-Aa1-Aa2-Aa3-Z 5 and 6. Reaction conditions: (a) TBTU/HOBt, DIPEA, DCM; (b) TFA/DCM (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); (c) Boc-Aa1-OH/TBTU/HOBt, DIPEA, DCM; (d) Ph2P-pC6H4-CO2H/TBTU/HOBt, DIPEA, DCM. :1); (c) Boc-Aa1-OH/TBTU/HOBt, DIPEA, DCM; (d) Ph2P-pC6H4-CO2H/TBTU/HOBt, DIPEA, DCM. | |
The ligands 5 and 6 were purified by column chromatography on silica and characterized by 1H–, 13C– and 31P–NMR and mass spectrometry. Interestingly, peptides containing N-alkylated amino acids like Pro often reveal a mixture of cis- and trans-isomers (Aa1-Pro tert-amide bond).13 However, 1H and 13C NMR (CDCl3) spectra of Pro-containing ligands 5f, 5j and 6j–6l show only the trans-isomer, assigned in accordance with the literature.13
Complexation of ligands 5 or 6 with [Rh(COD)(CH3CN)2]BF4 was studied in situ by NMR and CD spectroscopy. The folded Herrick-conformation of Rh(I)-complexes 3 or 4 with 1:2 metal to ligand stoichiometry is directed by the cis-bonded COD-ligand14 as well as by inter-strand hydrogen bonding, as previously described.8 The downfield shift of the amide proton (–C(O)–N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –CH2–) in Rh(I)-complexes 3a and 4a in 1H NMR spectra (CDCl3) strongly indicate hydrogen bonding, Fig. 1. The CD spectra of complexes 3 or 4 also support an ordered supramolecular structure in CH2Cl2 solution,15–17Fig. 2. The CD signal of the rhodium chromophore of 3 or 4 in the VIS region implies an asymmetric coordination sphere and supports the proposed “backdoor induction” of chirality. A correlation between the sign or amplitude of the CD signals and enantioselectivity in catalysis was not possible.
–CH2–) in Rh(I)-complexes 3a and 4a in 1H NMR spectra (CDCl3) strongly indicate hydrogen bonding, Fig. 1. The CD spectra of complexes 3 or 4 also support an ordered supramolecular structure in CH2Cl2 solution,15–17Fig. 2. The CD signal of the rhodium chromophore of 3 or 4 in the VIS region implies an asymmetric coordination sphere and supports the proposed “backdoor induction” of chirality. A correlation between the sign or amplitude of the CD signals and enantioselectivity in catalysis was not possible.
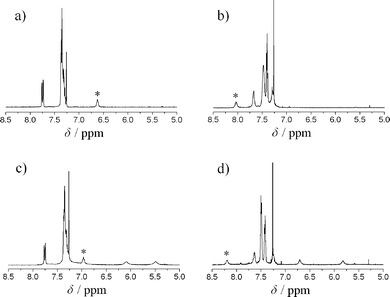 |
| | Fig. 1 Intercepts of 1H NMR (CDCl3) spectra of ligand Lig-Gly-OMe 5a (a) and its Rh(I) complex 3a (b), as well as ligand Lig-Gly-NH26a (c) and its Rh(I) complex 4a (d). The amide proton signals (–C(O)–N–CH2–) are starred. | |
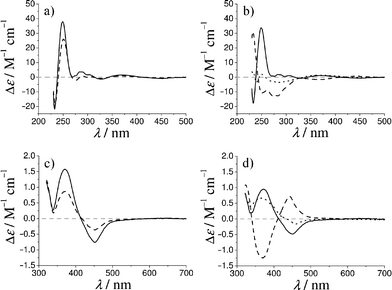 |
| | Fig. 2 CD (CH2Cl2) spectra of Rh(I) ester complexes 3d (left, full line) and 3j (left, dashed line), and Rh(I) amide complexes 4d (right, full line), 4j (right, dashed line) and 4k (right, dotted line). | |
|
|
| Run |
Ligand |
Time |
Temp. (°C) |
Conv.b (%) |
eec (%) R |
|
Reaction conditions: [Rh]:ligand:substrate = 1:2.4:100, p(H2) = 1 bar, DCM (0.1 M substrate).
Determined by 1H NMR.
Determined by GC (chiral column, Beta Dex 225).
|
| 1 |
5a, Lig-Gly-OMe |
2 h |
RT |
>98 |
0 |
| 2 |
5b, Lig-Gly-Phe-OMe |
2 h |
RT |
>98 |
1 (S) |
| 3 |
5c, Lig-Gly-Ala-Gly-OMe |
2 h |
RT |
>98 |
17 |
| 4 |
5c, Lig-Gly-Ala-Gly-OMe |
1 d |
–5 |
98 |
29 |
| 5 |
5d, Lig-Gly-Val-Gly-OMe |
2 h |
RT |
>98 |
32 |
| 6 |
5d, Lig-Gly-Val-Gly-OMe |
1 d |
–5 |
89 |
46 |
| 7 |
5e, Lig-Gly-Leu-Gly-OMe |
2 h |
RT |
>98 |
23 |
| 8 |
5e, Lig-Gly-Leu-Gly-OMe |
1 d |
–5 |
94 |
36 |
| 9 |
5f, Lig-Gly-Pro-Gly-OMe |
2 h |
RT |
>98 |
24 |
| 10 |
5f, Lig-Gly-Pro-Gly-OMe |
1 d |
–5 |
98 |
35 |
| 11 |
5g, Lig-Gly-Phe-Phe-OEt |
2 h |
RT |
>98 |
6 |
| 12 |
5g, Lig-Gly-Phe-Phe-OEt |
1 d |
–5 |
96 |
16 |
| 13 |
5h, Lig-Gly-Val-Phe-OEt |
1 d |
RT |
>98 |
19 |
| 14 |
5h, Lig-Gly-Val-Phe-OEt |
1 d |
–5 |
> 98 |
32 |
| 15 |
5j, Lig-Gly-Pro-Phe-OMe |
1 d |
RT |
>98 |
25 |
| 16 |
5j, Lig-Gly-Pro-Phe-OMe |
1 d |
–5 |
90 |
39 |
| 17 |
6a, Lig-Gly-NH2 |
2 h |
RT |
>98 |
0 |
| 18 |
6b, Lig-Gly-Phe-NH2 |
2 h |
RT |
>98 |
2 (S) |
| 19 |
6d, Lig-Gly-Val-Gly-NH2 |
2 h |
RT |
94 |
24 |
| 20 |
6g, Lig-Gly-Phe-Phe-NH2 |
2 h |
RT |
>98 |
49 |
| 21 |
6h, Lig-Gly-Val-Phe-NH2 |
2 h |
RT |
>98 |
61 |
| 22 |
6i, Lig-βAla-Val-Phe-NH2 |
2 h |
RT |
>98 |
7 |
| 23 |
6j, Lig-Gly-Pro-Phe-NH2 |
2 h |
RT |
97 |
57 |
| 24 |
6k, Lig-βAla-Pro-Phe-NH2 |
2 h |
RT |
>98 |
68 |
| 25 |
6l, Lig-Gaba-Pro-Phe-NH2 |
2 h |
RT |
85 |
12 (S) |
Achiral Lig-Gly-OMe 5a and Lig-Gly-NH26a gave a racemic product as expected (Table 1, runs 1 and 17, respectively); these ligands were used to optimize reaction conditions and product analysis. For chiral ligands substituted with one or two amino acids, only very low selectivity was obtained in catalysis (less than 10% ee), as exemplified by ligands Lig-Gly-Phe-OMe 5b or Lig-Gly-Phe-NH26b, runs 2 and 18, respectively. Ligands with long amino acid chains (five Aa or more) could form more inter-strand hydrogen bonds, but their preparation requires more reaction steps and gives lower overall yield. In addition, these ligands are more polar and less soluble in the reaction solvent (DCM). We therefore focused on ligands substituted with a sequence of three amino acids (Aa triplet). Gly is frequently found in β-turns,20 therefore it was used as the first amino acid (Aa1). βAla or GABA, γ-aminobutyric acid, were chosen at Aa1 as well, in order to test the efficiency of chirality transmission by increasing flexibility between the hydrogen-bonded amino acids and the coordination sphere of the prochiral metal. The second (Aa2) and third amino acids (Aa3) were varied for both 5 and 6, with the aim to study the influence of the Aa triplet sequence on the selectivity in catalytic hydrogenation.
Aa triplets that give more stable parallel β-sheet-like secondary structures21 could display higher selectivity in catalysis. However, a privileged Aa triplet that gives high selectivity for both esters 5 and amides 6 was not found. Instead, best results within series of esters 5 or amides 6 were obtained using different Aa triplets. Amides 6 revealed higher enantioselectivity in the studied hydrogenation, if compared to esters 5. At room temperature up to 68% ee was obtained for amides (run 24, Lig-βAla-Pro-Phe-NH26k) and up to 32% ee for esters (run 5, Lig-Gly-Val-Gly-OMe 5d).
Interestingly, if Lig-Gly-Val-Phe-NH26h is used, 61% ee was obtained (run 21). And if Gly at Aa1 in 6h is replaced with βAla in 6i, the formed supramolecular complex is more flexible and the transmission of chirality from the amino acid substituents to the coordination sphere of the prochiral metal is interrupted. As expected, the selectivity in catalysis is lost (7% ee, run 22, Lig-Gly-Val-Phe-NH26i). However, for Pro containing peptides, the more flexible βAla derivative 6k surprisingly shows slightly higher selectivity than the less flexible Gly derivative 6j, 68% ee and 57% ee (runs 24 and 25, respectively), while catalysis with the Gaba derivative 6l reveals only very low selectivity (12% ee, run 25).
In order to improve the selectivity, the catalysis was performed at a low temperature. At −5 °C the selectivity of ester ligands 5 could be enhanced for 10–14% ee; up to 46% ee was obtained (run 6, Lig-Gly-Val-Gly-OMe 5d). However, for amide ligands 6 the selectivity at −5 °C was not determined due to very low conversions even after prolonged reaction times. This finding can be explained by gel formation of most amides 6 in DCM at lower temperatures.
For most of the examined catalysts, (R)-configuration of the product was obtained, in contrast to Breit8 who obtained predominantly products with (S)-configuration. These results are in good agreement with our stereochemical analysis, see Scheme 1 and 2: L-amino acids induce (P) helical chirality of the prochiral catalytic metal in our complexes 3 or 4 (based on the para-substituted precursor) but (M) helical chirality of the catalytic metal in complexes 2 (based on the meta-substituted precursor).
Conclusions
Amino acid substituted monodentate phosphine ligands Lig-Aa1-Aa2-Aa3-R 5 and 6 were prepared by peptide coupling in solution. NMR and CD spectroscopic studies of in situ formed complexes [Rh(COD)(Lig-Aa1-Aa2-Aa3-R)2]BF43 and 4 indicate an ordered Herrick-like secondary structure with intra-molecular hydrogen bonds in solution. Ligands 5 and 6 were applied in rhodium-catalyzed asymmetric hydrogenation with selectivity up to 68% ee, the best result was obtained using Lig-βAla-Pro-Phe-NH26k.
In the presented supramolecular catalysts, an artificial pseudo C2-symmetrical peptide turn is formed that controls the chirality of the coordination sphere of the prochiral catalytic metal (“backdoor induction”), Scheme 4. Our results show a strong dependence of the distant amino acid substituents on the selectivity of the catalysis, indicating that the proposed transmission of chirality by “backdoor induction” might be an important tool in bioinspired asymmetric catalysis. This approach allows a more flexible design of the coordination sphere of the catalytic metal and opens the avenue for the development of new catalytic systems.
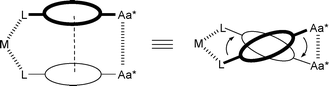 |
| | Scheme 4 “Backdoor induction” of chirality in a bioinspired catalyst. M = catalytic metal, L = ligand, oval = central (aromatic) unit, Aa* = chiral amino acid(s). The upper central unit is indicated in bold; the arrows indicate the sign of helical chirality. | |
Experimental section
Materials and instrumentation
Reactions were carried out in ordinary glassware and chemicals were used as purchased from commercial suppliers without further purification except where indicated. Pure L-amino acids were used. Reactions were monitored by TLC on Silica Gel 60 F254 plates (Merck); compounds were purified using automated flash chromatography (ISCO CombiFlash 75) equipped with a UV detector (254 nm) and prepacked columns (silica, RediSep).
Mass spectra were measured on a HPLC-MS system (Agilent Technologies 1200) coupled with 6410 Triple-Quadrupole mass spectrometer, operating in a positive ESI mode. High resolution mass spectra were obtained on a MALDI TOF-TOF instrument (Applied Biosystems 4800 plus) using CHCA matrix, except for Lig-Gly-OMe 5a where DHB matrix was used. CD spectra were recorded on a Jasco J-815 spectropolarimeter in 1 cm quartz Suprasil cells. NMR spectra were obtained on a Bruker Avance 300 or 600 spectrometers, operating at 300.13 or 600.13 MHz for 1H, 75.47 or 150.92 MHz for 13C and 242.93 MHz for 31P nuclei. Chemical shifts, δ (ppm), indicate a downfield shift from the internal standard, tetramethylsilane, TMS, for 1H-NMR and H3PO4 (85%) for 31P, or residual solvent signal for 13C-NMR (77.16 ppm CDCl3 or 39.52 ppm DMSO-d6). Coupling constants, J, are given in Hz.
For reactions at low temperature, an Intra-Cooler EK90/MT cryostat device (Thermo Fisher Scientific) was used. Enantiomeric excess was determined on gas chromatograph HP 5890-2 (FID detector) using chiral fused silica capillary column BetaDex 225 (Supelco, 30 m × 0.25 mm × 0.25 μm).
Synthesis, general procedures
Peptide coupling.
4-(Diphenylphosphino)-benzoic acid, Boc-protected amino acid or peptide, TBTU, HOBt and DIPEA were added to DCM and stirred at room temperature. After 1 h, N-unprotected amino acid or peptide was added to the reaction mixture and stirring was continued overnight (approximately 15 h). The reaction mixture was then washed with NaHCO3 (sat., aq.), citric acid (10%, aq.) and NaCl (sat., aq.), dried over Na2SO4, filtered, evaporated in vacuum and purified by automated flash chromatography on a prepacked silica column.
Boc-protecting group removal.
The corresponding Boc-protected peptide was dissolved in DCM–trifluoroacetic acid (1:1, 10 mL) and stirred for 2 h at room temperature. The volatiles were evaporated under reduced pressure and the viscous residue was dissolved in 15 mL of DCM. The residual trifluoroacetic acid was neutralized with excess of DIPEA (0.5 mL). This solution was used for further coupling.
The synthesis of precursors 7–10 is described in the Electronic Supplementary Information (ESI†).
NMR measurements.
(COD)Rh(CH3CN)2BF4 and ligand were each dissolved in 350 μL of CDCl3. Rhodium solution was added to the ligand solution and NMR spectra were recorded shortly after.
CD measurements15–17.
A dichloromethane solution of [(COD)Rh(CH3CN)2]BF4 (2 mL) was added to the solution of the ligand (2 mL), diluted to 5.0 mL and VIS-CD spectra of the intensive yellow solution were recorded. For UV-CD measurements, 300 to 500 μL of the solution was diluted to 5.0 mL and the spectra were recorded.
Synthesis of ligands 5 and 6
Lig-Gly-OMe, 5a.
4-(Diphenylphosphino)benzoic acid (312.8 mg, 1.02 mmol), HOBt (150.7 mg, 1.11 mmol), TBTU (321.5 mg, 1.00 mmol), DIPEA (0.660 mL, 3.99 mmol), H-Gly-OMe × HCl (135.7 mg, 1.08 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hexane gradient (TLC: Rf 0.42, EtOAc/Hexane = 1/1). Yield: 303.6 mg (79%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 3.80 (3H, s), 4.24 (d, 2H, J = 5 Hz), 6.63 (t, 1H, J = 4 Hz), 7.28–7.37 (m, 10H), 7.75 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz). 13C NMR (151 MHz, CDCl3): 41.87 (2), 52.60 (OMe), 127.07 (d, 3JCP = 6.5 Hz, 3), 128.81 (d, 3JCP = 6.5 Hz, 3′), 129.24 (4′), 133.69 (d, 2JCP = 19 Hz, 2), 133.78 (4), 134.03 (d, 2JCP = 20 Hz, 2′), 136.41 (d, 1JCP = 10.5 Hz, 1), 142.79 (d, 1JCP = 14 Hz, 1′), 167.27 (5), 170.56 (G1). ESI-MS (m/z): 378 (M+H+, 100%). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −4.98. ESI-MS (m/z): 378 (M+H+, 100%). MALDI-HRMS (m/z): calculated 378.1253 (C22H20NO3P + H+), found 378.1263.
Lig-Gly-Phe-OMe, 5b.
4-(Diphenylphosphino)benzoic acid (115.8 mg, 0.39 mmol), HOBt (57.2 mg, 0.42 mmol), TBTU (126.6 mg, 0.39 mmol), DIPEA (0.192 mL, 1.16 mmol), H-Gly-Phe-OMe (130 mg, 0.39 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hexane gradient (TLC: Rf 0.23, EtOAc/Hexane = 1/1). Yield: 136.5 mg (69%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 3.09 (dd, 1H, J1 = 14 Hz, J2 = 6 Hz), 3.16 (dd, 1H, J1 = 14 Hz, J2 = 6 Hz), 3.73 (s, 3H), 4.10 (d, 2H, J = 5 Hz), 4.89 (ddd, 1H, J1 = 8 Hz, J2 = J3 = 6 Hz), 6.41 (d, 1H, J = 8 Hz), 6.81 (t, 1H, J = 5 Hz), 7.06–7.09 (m, 2H), 7.19–7.24 (m, 3H), 7.29–7.38 (m, 12H), 7.70–7.74 (m, 2H). 13C NMR (151 MHz, CDCl3): 37.95 (2F3), 43.66 (1G2), 52.58 (OMe), 53.43 (2F2), 127.13 (d, 3JCP = 6.5 Hz, 3) ,127.33 (2F7), 128.77 (2F5), 128.82 (d, 3JCP = 7 Hz, 3′), 129.26 (4′), 129.32 (2F6), 133.51 (4), 133.63 (d, 2JCP = 18.5 Hz, 2), 134.03 (d, 2JCP = 20 Hz, 2′), 135.71 (2F4), 136.35 (d, 1JCP = 11 Hz, 1), 142.85 (d, 1JCP = 14 Hz, 1′), 167.48 (5), 168.70 (1G1), 171.76 (2F1). ESI-MS (m/z): 525 (M+H+, 100%). MALDI-HRMS (m/z): calculated 525.1938 (C31H29N2O4P + H+), found 525.1931.
Lig-Gly-Ala-Gly-OMe, 5c.
4-(Diphenylphosphino)benzoic acid (70.8 mg, 0.23 mmol), HOBt (33.3 mg, 0.25 mmol), TBTU (70.3 mg, 0.22 mmol), DIPEA (0.150 mL, 0.91 mmol), H-Gly-Ala-Gly-OMe (0.22 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hexane gradient (TLC: Rf 0.08, EtOAc/Hexane = 9/1). Yield: 56.2 mg (50%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.41 (d, J = 7 Hz, 3H), 3.70 (s, 3H), 3.97 (dd, 1H, J1 = 17 Hz, J2 = 5.5 Hz), 4.05 (dd, 1H, J1 = 17 Hz, J2 = 5.5 Hz), 4.11 (dd, 1H, J1 = 16.5 Hz, J2 = 5 Hz), 4.17 (dd, 1H, J1 = 16.5 Hz, J2 = 5 Hz), 4.57 (pseudo quintet, J = 7 Hz), 6.81 (t, J = 5 Hz, 1H), 6.87 (d, J = 7.5 Hz, 1H), 7.09 (t, J = 5 Hz, 1H), 7.29–7.36 (m, 12H), 7.75 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 18.12 (2A3), 41.30 (3G2), 43.81 (1G2), 49.11 (2A2), 52.47 (OMe), 127.21 (d, 3JCP = 5.5 Hz, 3), 128.81 (d, 3JCP = 7 Hz, 3′), 133.42 (4), 133.55 (d, 2JCP = 19 Hz, 2), 134.03 (d, 2JCP = 20 Hz, 2′), 136.32 (d, 1JCP = 10.5 Hz, 1), 142.88 (d, 1JCP = 14 Hz, 1′), 167.87 (5), 169.38 (3G1), 170.37 (1G1), 172.73 (2A1). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −4.89. ESI-MS (m/z): 506 (M+H+, 100%). MALDI-HRMS (m/z): calculated 506.1839 (C27H28N3O5P + H+), found 506.1827.
Lig-Gly-Val-Gly-OMe, 5d.
4-(Diphenylphosphino)benzoic acid (120.0 mg, 0.39 mmol), HOBt (52.4 mg, 0.39 mmol), TBTU (120.3 mg, 0.37 mmol), DIPEA (0.260 mL, 1.57 mmol), Boc-Gly-Val-Gly-OMe (0.39 mmol), DCM (50 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.18, DCM/MeOH = 95/5). Yield: 62.2 mg (30%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 0.95 (d, 3H, J = 7 Hz), 0.97 (d, 3H, J = 7 Hz), 2.15–2.20 (m, 1H), 3.70 (s, 3H), 3.97–4.05 (m, 2H), 4.14–4.21 (m, 2H), 4.37 (dd, 1H, J1 = 8.5 Hz, J2 = 6.5 Hz), 6.77 (t, 1H, J = 5 Hz), 6.94 (d, 1H, J = 8.5 Hz), 7.19 (t, 1H, J = 5 Hz), 7.29–7.37 (m, 12H), 7.75 (d, 2H, J = 8 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 18.20, 19.33 (2V4, 2V4′), 31.08 (2V3), 41.27 (1G2), 43.92 (3G2), 52.43 (OMe), 58.84 (2V2), 127.26 (d, 3JCP = 6 Hz, 3), 128.82 (d, 3JCP = 7 Hz, 3′), 129.25 (4′), 133.59 (4), 133.59 (d, 2JCP = 19 Hz, 2), 134.04 (d, 2JCP = 20 Hz, 2′), 136.41 (d, 1JCP = 11 Hz, 1), 142.79 (d, 1JCP = 14 Hz, 1′), 167.86 (5), 169.65 (3G1), 170.25 (1G1), 171.66 (2V1). ESI-MS (m/z): 534 (M+H+, 100%). MALDI-HRMS (m/z): calculated 532.2152 (C29H32N3O5P + H+), found 532.2150.
Lig-Gly-Leu-Gly-OMe, 5e.
4-(Diphenylphosphino)benzoic acid (98.7 mg, 0.32 mmol), HOBt (46.1 mg, 0.34 mmol), TBTU (100.8 mg, 0.31 mmol), DIPEA (0.210 mL, 1.27 mmol), H-Gly-Leu-Gly-OMe (0.32 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hex gradient (TLC: Rf 0.32, EtOAc/Hex = 9/1). Yield: 107.0 mg (62%). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 0.90 (d, 3H, J = 6.5 Hz), 0.92 (d, 3H, J = 6.5 Hz), 1.54–1.59 (m, 1H), 1.63–1.74 (m, 2H), 3.70 (s, 3H), 3.96 (dd, 1H, J1 = 18 Hz, J2 = 5 Hz), 4.02 (dd, 1H, J1 = 18 Hz, J2 = 5.5 Hz), 4.12 (dd, 1H, J1 = 16.5 Hz, J2 = 5 Hz), 4.18 (dd, 1H, J1 = 16.5 Hz, J2 = 5 Hz), 4.52–4.56 (m, 1H), 6.86 (d, 1H, J = 8 Hz), 6.87 (t, 1H, J = 5.5 Hz), 7.18 (t, 1H, J = 5 Hz), 4.29–4.35 (m, 12H), 7.75 (dd, 1H, J1 = 8 Hz, J2 = 1 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 22.07, 22.99 (2L5, 2L5′), 24.87 (2L4), 40.95, 41.29 (2L3, 3G2), 43.91 (1G2), 51.96 (2L2), 52.49 (OMe), 127.19 (d, 3JCP = 6.5 Hz, 3), 128.83 (d, 3JCP = 7 Hz, 3′), 129.27 (4′), 133.45 (4), 133.61 (d, 2JCP = 19 Hz, 2), 134.04 (d, 3JCP = 20 Hz, 2′), 136.34 (d, 1JCP = 11 Hz, 1), 142.94 (d, 1JCP = 14 Hz, 1′), 167.82 (5), 169.44, 170.30 (1G1, 3G1), 172.35 (2L1). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −4.90. ESI-MS (m/z): 548 (M+H+, 100%). MALDI-HRMS (m/z): calculated 548.2308 (C30H34N3O5P + H+), found 548.2318.
Lig-Gly-Pro-Gly-OMe, 5f.
4-(Diphenylphosphino)benzoic acid (122.2 mg, 0.40 mmol), HOBt (55.0 mg, 0.47 mmol), TBTU (128.9 mg, 0.40 mmol), DIPEA (0.270 mL, 1.63 mmol), H-Gly-Pro-Gly-OMe (0.39 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/EtOH gradient (TLC: Rf 0.08, EtOAc/Hexane = 9/1). Yield: 183.6 mg (86%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.89–2.26 (m, 3H), 2.38–2.45 (m, 1H), 3-45–3.54 (m, 1H), 3.61–3.68 (m, 1H), 3.73 (s, 1H), 4.01 (d, 2H, J = 5.5 Hz), 4.22 (dd, 1H, J1 = 18 Hz, J2 = 4 Hz), 4.29 (dd, 1H, J1 = 18 Hz, J2 = 4.5 Hz), 4.64 (dd, 1H, J1 = 8 Hz, J2 = 2 Hz), 7.18–7.21 (m, 2H), 7.28–7.37 (m ,12H), 7.77 (dd, 1H, J1 = 8 Hz, J2 = 1 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 41.37 (2P4), 27.86 (2P3), 41.36 (3G2), 42.68 (1G2), 46.63 (2P5), 52.47 (OMe), 60.23 (2P2), 127.12 (d, 3JCP = 6.5 Hz, 3), 128.80 (d, 3JCP = 7 Hz, 3′), 129.23 (4′), 133.64 (d, 2JCP = 19 Hz, 2), 133.83 (4), 134.02 (d, 3JCP = 20 Hz, 2′), 136.42 (d, 1JCP = 11 Hz, 1), 142.58 (d, 1JCP = 14 Hz, 1′), 167.22 (5), 168.53 (1G1), 170.29 (3G1), 171.14 (2P1). ESI-MS (m/z): 532 (M+H+, 100%). MALDI-HRMS (m/z): calculated 532.1995 (C29H30N3O5P + H+), found 532.2001.
Lig-Gly-Phe-Phe-OEt, 5g.
4-(Diphenylphosphino)benzoic acid (90.5 mg, 0.30 mmol), HOBt (40.8 mg, 0.30 mmol), TBTU (92.5 mg, 0.29 mmol), DIPEA (0.200 mL, 1.21 mmol), H-Gly-Phe-Phe-OEt (0.43 mmol), DCM (50 mL). Chromatography: silica (30 g), EtOAc/Hexane gradient (TLC: Rf 0.38, EtOAc/Hexane = 1/1). Yield: 100 mg (49%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.20 (t, 3H, J = 7 Hz), 2.95–3.11 (m, 4H), 4.02 (d, 2H, J = 5 Hz), 4.11 (q, 2H, J = 7 Hz), 4.62–4.69 (m, 1H), 4.71–4.77 (m, 1H), 6.34 (d, 1H, J = 7.5 Hz), 6.64 (d, 1H, J = 8 Hz), 6.86 (t, 1H, J = 5 Hz), 6.98–7.01 (m, 2H), 7.13–7.38 (m, 20H), 7.71 (d, 2H, J = 8 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 14.20 (CH3), 38.07, 38.22 (3,2F3), 43.71 (1G1), 53.56, 54.55 (3,2F2), 61.63 (OCH2), 127.12, 127.16 (3,2F7), 127.26 (d, 3JCP = 6.5 Hz, 3), 128.58, 128.73 (3,2F5), 128.83 (d, 3JCP = 7 Hz, 3), 129.27 (4′), 129.39, 129.44 (3,2F6), 133.51 (4), 133.61 (d, 2JCP = 19 Hz, 2), 134.05 (d, 2JCP = 20 Hz, 2′), 135.92, 136.40 (3,2F4), 136.41 (d, 1JCP = 11 Hz, 1), 142.88 (d, 1JCP = 14 Hz, 1′), 167.57 (5), 169.08 (1G1), 170.37 (2F1), 171.14 (3F1). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −5.30. ESI-MS (m/z): 686 (M+H+, 100%). MALDI-HRMS (m/z): calculated 686.2778 (C41H40N3O5P + H+), found 686.2797.
Lig-Gly-Val-Phe-OEt, 5h.
4-(Diphenylphosphino)benzoic acid (147.7 mg, 0.48 mmol), HOBt (65.4 mg, 0.48 mmol), TBTU (150.9 mg, 0.47 mmol), DIPEA (0.310 mL, 1.88 mmol), H-Gly-Val-Phe-OEt (0.59 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hexane gradient (TLC: Rf 0.17, EtOAc/Hexane = 1/1). Yield: 265.3 mg (86%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 0.87 (d, 3H, J = 7 Hz), 0.92 (d, 3H, J = 7 Hz), 1.21 (t, 3H, J = 7 Hz), 2.04–2.15 (m, 1H), 3.02–3.15 (m, 2H), 4.10–4.17 (m, 2H + 2H), 4.29 (dd, 1H, J1 = 8.5 Hz, J2 = 6.5 Hz), 4.84 (pseudo-q, 1H), 6.46 (d, 1H, J = 8 Hz), 6.70 (d, 1H, J = 8.5 Hz), 7.04–7.37 (m, 12H + 1H), 7.76 (d, 2H, J = 7.5 Hz). 13C NMR (75.48 MHz, CDCl3): 14.16 (3F9), 18.15, 19.23 (2V4), 31.29 (2V3), 38.01 (3F3), 43.73 (1G2), 53.33 (3F2), 58.61 (2V2), 61.61 (3F8), 127.15 (3F7), 127.29 (d, 3JCP = 6.5 Hz, 3), 128.59 (3F5), 128.79 (d, 3JCP = 7 Hz, 3′), 129.22 (4′), 129.38 (3F6), 133.54 (d, 2JCP = 19 Hz, 2), 133.68 (4), 134.00 (d, 2JCP = 20 Hz, 2′),135.96 (3F4), 136.40 (d, 1JCP = 10.5 Hz, 1), 142.61 (d, 1JCP = 14 Hz, 1′), 167.62 (5), 169.26 (1G1), 170.84 (2V1), 171.42 (3F1). ESI-MS (m/z): 445.2 (M–192), 638.3 (M+H+, 100%). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −6.05. ESI-MS (m/z): 638 (M+H+, 100%). MALDI-HRMS (m/z): calculated 638.2778 (C37H40N3O5P + H+), found 638.2797.
Lig-Gly-Pro-Phe-OMe, 5j.
4-(Diphenylphosphino)benzoic acid (64.2 mg, 0.21 mmol), HOBt (30.4 mg, 0.22 mmol), TBTU (61.8 mg, 0.19 mmol), DIPEA (0.140 mL, 0.85 mmol), H-Gly-Pro-Phe-OMe (0.59 mmol), DCM (50 mL). Chromatography: silica (12 g), EtOAc/Hexane gradient. Yield: 107.5 mg (83%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.78–1.88 (m, 1H), 1.94–2.03 (m, 2H), 2.34–2.41 (m, 1H), 2.99 (dd, 1H, J1 = 14 Hz, J2 = 7.5 Hz), 3.21 (dd, 1H, J1 = 14 Hz, J2 = 5.5 Hz), 3.37 (pseudo-t, 2H), 3.74 (s, 3H), 4.02 (dd, 1H, J1 = 18 Hz, J2 = 3.8 Hz), 4.18 (dd, 1H, J1 = 18 Hz, J2 = 4.2 Hz), 5.58 (d, 1H, J = 7 Hz), 4.87 (pseudo-q, 1H), 7.07–7.39 (m, 19H), 7.79 (d, 2H, J = 8 Hz). 13C NMR (75.48 MHz, CDCl3) δ (ppm): 24.80 (2P4), 27.19 (2P3), 37.93 (3F3), 42.64 (1G2), 46.37 (2P5), 52.50 (OMe), 53.12 (3F2), 60.08 (2P2), 127.14 (d, 3JCP = 6.5 Hz, 3), 127.16 (3F7), 128.47 (3F5), 128.83 (d, 3JCP = 7 Hz, 3′), 129.24 (4′), 129.33 (3F6), 133.72 (d, 2JCP = 19 Hz, 2), 133.98 (4), 134.06 (d, 2JCP = 19 Hz, 2′), 136.24 (3F4), 136.51 (d, 1JCP = 11 Hz, 1), 142.70 (d, 1JCP = 14 Hz, 1′), 167.09 (5), 168.27, 170.22 (1G1, 2P1), 171.99 (3F1). ESI-MS (m/z): 660 (M+K+, 100%). MALDI-HRMS (m/z): calculated 622.2465 (C36H36N3O5P + H+), found 622.2477.
Lig-Gly-NH2, 6a.
4-(Diphenylphosphino)benzoic acid (205.5 mg, 0.67 mmol), HOBt (90.6 mg, 0.67 mmol), TBTU (203.9 mg, 0.64 mmol), DIPEA (0.430 mL, 2.60 mmol), H-Gly-NH2 × HCl (84.2 mg, 0.76 mmol), DCM (50 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.3, DCM/MeOH = 10/0.6). Yield: 180.3 mg (78%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 4.17 (d, 2H, J = 5 Hz), 5.49 (s, 1H), 6.09 (s, 1H), 6.96 (t, 1H, J = 5 Hz), 7.28–7.38 (m, 12H), 7.75 (d, 2H, J = 8 Hz). 13C NMR (75,48 MHz, DMSO-d6) δ (ppm): 42.41 (1G2), 127.53 (d, 3JCP = 6.5 Hz, 3), 128.90 (d, 3JCP = 7 Hz, 3′), 129.25 (4′), 132.79 (d, 2JCP = 19 Hz, 2), 133.41 (d, 2JCP = 20 Hz, 2′), 134.40 (4), 136.05 (d, 1JCP = 11 Hz, 1), 140.75 (d, 1JCP = 13.5 Hz, 1′), 165.99 (5), 170.92 (1G1). ESI-MS (m/z): 363.3 (M+H+, 100%); MALDI-HRMS (m/z): calculated 363.1257 (C21H19N2O2P+H+), found 363.1258.
Lig-Gly-Phe-NH2, 6b.
4-(Diphenylphosphino)benzoic acid (112.2 mg, 0.37 mmol), HOBt (47.5 mg, 0.35 mmol), TBTU (113.2 mg, 0.35 mmol), DIPEA (0.235 mL, 1.42 mmol), H-Gly-Phe-NH2 (0.36 mmol) DCM (40 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.6, DCM/MeOH = 9/1). Yield 141.0 mg (79%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 2.81 (dd, 1H, J1 = 14 Hz, J2 = 9 Hz), 3.03 (dd, 1H, J1 = 14 Hz, J2 = 5 Hz), 3.77 (dd, 1H, J1 = 16 Hz, J2 = 6 Hz), 3.90 (dd, 1H, J1 = 16 Hz, J2 = 6 Hz), 4.45 (ddd, 1H, J1 = J2 = 9 Hz, J3 = 5 Hz), 7.09 (s, 1H), 7.14–7,17 (m, 1H), 7.20 (s, 2H), 7.21 (s, 2H), 7.26–7.32 (m, 6H), 7.38 (s, 1H), 7.41–7.43 (m, 6H), 7.84–7.86 (m, 2H), 8.02 (d, 1H, J = 8.5 Hz), 7.23 (t, 1H, J = 6 Hz). 13C NMR (75,48 MHz, DMSO-d6) δ (ppm): 37.46 (2F3), 42.70 (1G2), 53.76 (2F2), 126.21 (2F7), 127.51 (d, 3JCP = 6.5 Hz, 3), 128.04 (2F5), 128.91 (d, 3JCP = 7 Hz, 3′), 129.14 (2F6), 129.28 (4′), 132.84 (d, 2JCP = 19 Hz, 2), 133.43 (d, 2JCP = 20 Hz, 3′), 134.20 (4), 136.01 (d, 1JCP = 11 Hz, 1), 137.96 (2F4), 140.93 (d, 1JCP = 13.5 Hz, 1′), 166.17 (5), 168.66 (1G1), 172.84 (2F1). ESI-MS (m/z): 510.3 (M+H+, 100%); MALDI-HRMS (m/z): calculated 510.1941 (C30H28N3O3P+H+), found 510.1937.
Lig-Gly-Val-Gly-NH2, 6d.
4-(Diphenylphosphino)benzoic acid (189.1 mg, 0.62 mmol), HOBt (82.5 mg, 0.61 mmol), TBTU (169.9 mg, 0.53 mmol), and DIPEA (0.400 mL, 2.42 mmol), H-Gly-Val-Gly-NH2 (0.61 mmol), DCM (40 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.64, DCM/MeOH = 9/1), purified twice. Yield: 43.4 mg (16%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 0.98 (d, 3H, J = 6.5 Hz), 0.99 (d, 3H, J = 6.5 Hz), 2.09–2.20 (m, 1H), 4.14 (d, 2H, J = 5 Hz), 4.25–4.39 (m, 2H), 4.45 (dd, 1H, J1 = 8 Hz, J2 = 6 Hz), 7.17 (t, 1H, J = 5 Hz), 7.23 (s, 1H), 7.31–7.39 (m, 12H), 7.43 (t, 1H, J = 4.5 Hz), 4.75 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz), 7.91 (d, 1H, J = 8 Hz). 13C NMR (75,48 MHz, CDCl3) δ (ppm): 18.36, 19.32 (2V4, 2V4′), 31.37 (2V3), 42.89 (1G2), 44.03 (3G2), 59.14 (2V2), 133.39 (d, 3JCP = 6.5 Hz, 3), 133.58 (d, 3JCP = 7 Hz, 3′), 129.28 (4′), 133.39 (4), 133.58 (d, 2JCP = 19 Hz, 2), 134.04 (d, 2JCP = 20 Hz, 3′), 136.31 (d, 1JCP = 11 Hz, 1), 142.93 (d, 1JCP = 14 Hz, 1′), 167.93 (5), 169.75 (3G1), 171.72, 171.90 (1G1, 2V1). 31P NMR (242.93 MHz, CDCl3) δ (ppm): −5.25. ESI-MS (m/z): 519.2 (M+H+); MALDI-HRMS (m/z): calculated 519.2156 (C28H31N4O4P+H+), found 519.2151.
Lig-Gly-Phe-Phe-NH2, 6g.
4-(Diphenylphosphino)benzoic acid (78.1 mg, 0.26 mmol), HOBt (33.5 mg, 0.25 mmol), TBTU (84.4 mg, 0.26 mmol), DIPEA (0.165 mL, 1.00 mmol), H-Gly-Phe-Phe-NH2 (0.25 mmol), DCM (40 mL). Because the product gelates in DCM, the crude product was not extracted. Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.4, DCM/MeOH = 9/1). The fractions containing the product were evaporated; hexane (5 mL) was added and evaporated two times, to help evaporate residual DCM in the formed gel. Yield 95.6 mg (57%). 1H NMR (300.13 MHz, DMSO-d6) δ (ppm): 2.74–3.09 (m, 4H), 3.75 (dd, 1H, J1 = 16 Hz, J2 = 6 Hz), 3.91 (dd, 1H, J1 = 16 Hz, J2 = 6 Hz), 4.39–4.51 (m, 2H), 7.10 (s, 1H), 7.14–7.33 (m, 17H), 7.43–7.44 (m, 6H), 7.86 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz), 8.10 (d, 1H, J = 8 Hz), 8.13 (d, 1H, J = 7.5 Hz), 8.80 (t, 1H, J = 5.5 Hz). 13C NMR (75,48 MHz, DMSO-d6) δ (ppm): 37.25 (3F3), 37.41 (2F3), 42.69 (1G2), 53.93, 54.06 (3,2F2), 126.20 (2F7), 126.23 (3F7), 127.50 (d, 3JCP = 6.5 Hz, 3), 127.99 (2F5), 128.08 (3F5), 128.90 (d, 3JCP = 7 Hz, 3′), 129.11 (2F6), 129.16 (3F6), 129.26 (4′), 132.81 (d, 2JCP = 19 Hz, 2), 133.43 (d, 2JCP = 20 Hz, 3′), 136.00 (4), 136.01 (d, 1JCP = 11 Hz, 1), 137.64 (2F4), 137.90 (3F4), 137.96 (2F4), 140.96 (d, 1JCP = 13.5 Hz, 1′), 166.23 (5), 169.02 (1G1), 170.63 (2F1), 172.69 (3F1). ESI-MS (m/z): 657.4 (M+H+); MALDI-HRMS (m/z): calculated 657.2625 (C39H37N4O4P+H+), found 567.2621.
Lig-Gly-Val-Phe-NH2, 6h.
4-(Diphenylphosphino)benzoic acid (85.1 mg, 0.28 mmol), HOBt (43.5 mg, 0.32 mmol), TBTU (92.1 mg, 0.29 mmol), DIPEA (0.185 mL, 1.12 mmol), H-Gly-Val-Phe-NH2 (0.31 mmol), DCM (40 mL). Because the product gelates in DCM, no washing was performed. Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.4, DCM/MeOH = 9/1). The fractions containing the product were evaporated; hexane (5 mL) was added and evaporated two times, to evaporate residual DCM in the formed gel. The compound was dissolved in DCM/MeOH (20 mL) and washed with water (30 mL). The volatiles were evaporated and the precipitated compound was filtered off, washed with water and dried under vacuum. Yield: 35.8 mg (21%). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 0.70 (d, 3H, J = 7 Hz), 0.79 (d, 3H, J = 7 Hz), 1.92–1.97 (m, 1H), 2.84 (dd, 1H, J1 = 14 Hz, J2 = 9.5 Hz), 3.05 (dd, 1H, J1 = 14 Hz, J2 = 5 Hz), 3.87 (dd, 1H, J1 = 16 Hz, J2 = 6 Hz), 3.97 (dd, 1H, J1 = 16 Hz, J2 = 5.5 Hz), 4.09 (dd, 1H, J1 = 8 Hz, J2 = 8 Hz), 4.41 (ddd, 1H, J1 = 9.5 Hz, J2 = 8.5 Hz, J3 = 5 Hz), 7.02 (s, 1H), 7.14 (s, 1H), 7.14–7.18 (m, 1H), 7.226 (s, 2H), 7.233 (d, 2H, J = 1 Hz), 7.27–7.32 (m, 6H), 7.42–7.44 (m, 6H), 7.85–7.87 (m, 2H), 7.90 (d, 1H, J = 8 Hz), 7.92 (d, 1H, J = 8.5 Hz), 8.85 (t, 1H, J = 6 Hz). 13C NMR (75,48 MHz, DMSO-d6) δ (ppm): 17.60, 19.09 (2V4, 2V4′), 30.18 (2V3), 37.25 (3F3), 42.85 (1G2), 53.93 (3F2), 58.09 (2V2), 126.21 (3F7), 127.49 (d, 3JCP = 6.5 Hz, 3), 128.08 (3F5), 128.93 (d, 3JCP = 7 Hz, 3′), 129.06 (3F6), 129.29 (4′), 132.88 (d, 2JCP = 19 Hz, 2), 133.46 (d, 2JCP = 20 Hz, 3′), 134.172 (4),135.98 (d, 1JCP = 11 Hz, 1), 138.02 (3F4) 140.99 (d, 1JCP = 13 Hz, 1′), 166.47 (5), 169.53 (1G1), 170.56 (2V1), 172.87 (3F1). 31P NMR (242.93 MHz, DMSO-d6) δ (ppm): −6.77. ESI-MS (m/z): 609.3 (M+H+); MALDI-HRMS (m/z): calculated 609.2625 (C35H37N4O4P+H+), found 609.2631.
Lig-βAla-Val-Phe-NH2, 6i.
4-(Diphenylphosphino)benzoic acid (95.6 mg, 0.31 mmol), HOBt (42.7 mg, 0.32 mmol), TBTU (102.5 mg, 0.32 mmol), DIPEA (0.205 mL, 1.42 mmol), H-βAla-Val-Phe-NH2 (0.32 mmol), DCM (40 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.4, DCM/MeOH = 9/1). Yield: 111.5 mg (57%). 1H NMR (300.13 MHz, DMSO-d6) δ (ppm): 0.71 (d, 3H, J = 6.5 Hz), 0.72 (d, 3H, J = 6.5 Hz), 1.88 (m, 1H), 2.45 (m, 2H), 2.75–3.03 (m, 2H), 3.45 (m, 2H), 4.06 (dd, 1H, J1 = 8 Hz, J2 = 7 Hz), 4.41–4.49 (m, 1H), 7.07 (s, 1H), 7.14–7.30 (m, 12H), 7.40–7.43 (m, 5H), 7.81 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz), 7.88 (dd, 2H, J1 = 8 Hz, J2 = 3 Hz), 8.49 (t, 1H, J = 5.5 Hz). 13C NMR (75,48 MHz, DMSO-d6) δ (ppm): 18.02, 19.11 (2V4, 2V4′), 30.06 (2V3), 35.18 (1β2), 36.27 (1β3), 37.36 (3F3), 53.59 (3F2), 58.23 (2V2), 126.18 (3F7), 127.35 (d, 3JCP = 6.5 Hz, 3), 127.99 (3F5), 128.90 (d, 3JCP = 7 Hz, 3′), 129.09 (3F6), 129.26 (4′), 132.82 (d, 2JCP = 19 Hz, 2), 133.40 (d, 2JCP = 20 Hz, 3′), 134.84 (4), 136.07 (d, 1JCP = 11 Hz, 1), 137.91 (3F4), 137.96 (2F4), 140.51 (d, 1JCP = 13 Hz, 1′), 165.89 (5), 170.77, 170.90 (1β1, 2V1) 172.85 (3F1). ESI-MS (m/z): 623.4 (M+H+); MALDI-HRMS (m/z): calculated 623.2781 (C36H39N4O4P+H+), found 623.2782.
Lig-Gly-Pro-Phe-NH2, 6j.
4-(Diphenylphosphino)benzoic acid (122.6 mg, 0.40 mmol), HOBt (55 mg, 0.41 mmol), TBTU (125.4 mg, 0.39 mmol), DIPEA (0.270 mL, 1.63 mmol), H-Gly-Pro-Phe-NH2 (0.41 mmol), DCM (40 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.4, DCM/MeOH = 9/1). Yield: 105 mg (44%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.62–1.71 (m, 1H), 1.85–2.04 (m, 3H), 3.01 (dd, 1H, J1 = 14 Hz, J2 = 10 Hz), 3.30 (dd, 1H, J1 = 14 Hz, J2 = 5 Hz), 3.45–3.49 (m, 1H), 3.63–3.67 (m, 1H), 3.93 (dd, 1H, J1 = 16.5 Hz, J2 = 5 Hz), 4.10 (dd, 1H, J1 = 16.5 Hz, J2 = 4 Hz), 4.44 (dd, 1H, J1 = 8 Hz, J2 = 3 Hz), 4.61–4.65 (m, 1H), 5.54 (s, 1H), 6.44 (s, 1H), 6.99 (d, 1H, J = 8 Hz), 7.17–7.37 (m, 18H), 7.75 (dd, 2H, J1 = 8 Hz, J2 = 1 Hz). 13C NMR (75.48 MHz, CDCl3) δ (ppm): 24.41 (2P4), 28.83 (2P3), 36.72 (3F3), 42.89 (1G2), 47.45 (2P5), 54.57 (3F2), 61.22 (2P2), 126.91 (3F7), 127.37 (d, 3JCP = 6.5 Hz, 3), 128.58 (3F5), 128.82 (d, 3JCP = 7 Hz, 3′), 129.25 (3F6, 4′), 133.08 (4), 133.59 (d, 2JCP = 18.5 Hz, 2), 134.07 (d, 2JCP = 20 Hz, 2′), 136.25 (d, 1JCP = 11 Hz, 1), 137.74 (3F4), 143.14 (d, 1JCP = 14 Hz, 1′), 168.60 (5), 169.90, 171.15 (1G1, 2P1), 174.20 (3F1). ESI-MS (m/z): 607.4 (M+H+); MALDI-HRMS (m/z): calculated 607.2468 (C35H35N4O4P+H+), found 607.2476.
Lig-βAla-Pro-Phe-NH2, 6k.
4-(diphenylphosphino)benzoic acid (140.2 mg, 0.46 mmol), HOBt (66.6 mg, 0.49 mmol), TBTU (144.6 mg, 0.45 mmol), DIPEA (0.305 mL, 1.85 mmol), H-βAla-Pro-Phe-NH2 (0.46 mmol), DCM (40 mL). Chromatography: silica (12 g), DCM/MeOH gradient (TLC: Rf 0.46, DCM/MeOH = 9/1). Yield 191.5 mg (67%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.74–1.83 (m, 1H), 1.87–1.96 (m, 2H), 2.09–2.13 (m, 1H), 2.45 (ddd, 1H, J1 = 16.5 Hz, J2 = 7 Hz, J3 = 3.5 Hz), 2.54 (ddd, 1H, J1 = 16.5 Hz, J2 = 8 Hz, J3 = 4 Hz), 3.06 (dd, 1H, J1 = 14 Hz, J2 = 8 Hz), 3.14 (dd, 1H, J1 = 14 Hz, J2 = 6 Hz), 3.35–3.37 (m, 2H), 3.57–3.62 (m, 1H), 3.72–3.78 (m, 1H), 4.41 (dd, 1H, J1 = 8.5 Hz, J2 = 3 Hz), 4.66 (ddd, 1H, J1 = J2 = 8 Hz, J3 = 6 Hz), 5.30 (s, 1H), 6.26 (s, 1H), 6.93 (d, 1H, J = 8 Hz), 7.10 (t, 1H, J = 6 Hz), 7.12–7.14 (m, 2H), 7.18–7.24 (m, 3H), 7.28–7.36 (m, 12H), 7.70–7.72 (m, 2H). 13C NMR (75.48 MHz, CDCl3) δ (ppm): 24.75 (2P4), 28.56 (2P3), 34.66 (1β2), 35.83 (1β3), 37.06 (3F3), 47.74 (2P5), 53.78 (3F2), 60.84 (2P2), 127.01 (3F7), 127.09 (d, 3JCP = 6.5 Hz, 3), 128.63 (3F5), 128.77 (d, 3JCP = 7 Hz, 3′), 129.20 (3F6), 129.23 (4′), 133.61 (d, 2JCP = 19 Hz, 2), 133.99 (d, 2JCP = 20 Hz, 3′), 134.22 (4), 136.42 (d, 1JCP = 10.5 Hz, 1), 137.16 (3F4), 142.38 (d, 1JCP = 13.5 Hz, 1′), 167.48 (5), 171.44 (2P1), 172.68 (1β1), 173.65 (3F1). ESI-MS (m/z): 621.4 (M+H+); MALDI-HRMS (m/z): calculated 621.2625 (C36H37N4O4P+H+), found 621.2625.
Lig-Gaba-Pro-Phe-NH2, 6l.
4-(Diphenylphosphino)benzoic acid (131.4 mg, 0.43 mmol), HOBt (57.5 mg, 0.43 mmol), TBTU (142.9 mg, 0.45 mmol), DIPEA (0.280 mL, 1.69 mmol), H-Gaba-Pro-Phe-NH2 (0.43 mmol), DCM (40 mL). Chromatography: silica (12 g), EtOAc/DCM/MeOH gradient (TLC: Rf 0.3, EtOAc/DCM/MeOH = 8/2/1). Yield 89.1 mg (33%). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 1.59–1.70 (m, 2H), 1.73–1.81 (m, 2H), 2.00–2.06 (m, 1H), 2.17–2.24 (m, 2H), 2.41–2.46 (m, 1H), 2.95 (dd, 1H, J1 = 14.5 Hz, J2 = 11.5 Hz), 3.11–3.15 (m, 1H), 3.31–3.35 (m ,1H), 3.43–3.46 (m, 1H), 3.44 (dd, 1H, J1 = 14.5 Hz, J2 = 4 Hz), 3.89–3.95 (m, 1H), 4.36 (dd, 1H, J1 = 8.5 Hz, J2 = 4 Hz), 4.84 (ddd, 1H, J1 = 11.5 Hz, J2 = 9 Hz, J3 = 4 Hz), 5.30 (s, 1H), 6.49 (dd, J1 = 8 Hz, J2 = 4.5 Hz), 7.14–7.166 (m, 1H), 7.172 (s, 1H), 7.19–7.22 (m, 2H), 7.27–7.28 (m, 2H), 7.31–7.38 (m, 12H), 7.61–7.69 (m, 2H), 7.88 (d, 1H, J = 9 Hz); 13C NMR (75,48 MHz, CDCl3) δ (ppm): 24.57 (2P4), 25.36 (1γ3), 29.59 (2P3), 30.32 (1γ2), 34.66 (1β2), 36.18 (3F3), 38.42 (1γ4), 47.90 (2P5), 53.91 (3F2), 61.44 (2P2), 126.51 (3F7), 126.89 (d, 3JCP = 6.5 Hz, 3), 128.34 (3F5), 128.87 (d, 3JCP = 7.5 Hz, 3′), 129.24 (3F6), 129.34 (4′), 133.77 (d, 2JCP = 19 Hz, 2), 134.04 (4), 134.06 (d, 2JCP = 20 Hz, 3′), 136.34 (d, 1JCP = 10.5 Hz, 1), 138.55 (3F4), 143.03 (d, 1JCP = 14 Hz, 1′), 168.12 (5), 172.04 (2P1), 172.98 (1β1), 174.52 (3F1). ESI-MS (m/z): 635.4 (M+H+); MALDI-HRMS (m/z): calculated 635.2781 (C37H39N4O4P+H+), found 635.2788.
Metal complexes 3 and 4
[Rh(COD)(Lig-Gly-OMe)2]BF4, 3a.
[(COD)Rh(CH3CN)2]BF4 (1.20 mg, 3 μmol, 1 eq.), Lig-Gly-OMe5a (2.50 mg, 6.63 μmol, 2.2 eq.). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 2.25 (m, 4H), 2.55 (m, 4H), 3.76 (s, 6H), 4.25 (d, 4H, J = 6 Hz), 4.58 (ws, 4H), 7.29 (ws, 4H), 7.39–7.41 (m, 8H), 7.8 (ws, 12 H), 7.67 (ws, 4H), 8.03 (ws, 2H). 31P NMR (242.93 MHz, CDCl3) δ (ppm): 26.37 (d, 1JRh-P = 145 Hz).
[Rh(COD)(Lig-Gly-Val-Gly-OMe)2]BF4, 3d.
[(COD)Rh(CH3CN)2]BF4 (1.20 mg, 3 μmol, 1 eq.), Lig-Gly-Val-Gly-OMe3d (4.20 mg, 6.63 μmol, 2.2 eq.). 1H NMR (300.13 MHz, CDCl3) δ (ppm): 0.90 (d, 6H, J = 7 Hz), 0.96 (d, 6H, J = 7 Hz), 2.20–2.32 (m, 6H), 2.41–2.48 (m, 2H), 2.58–2.64 (m, 2H), 3.70 (s, 6H), 3.94 (d, 2H, J = 16 Hz), 3.99 (dd, 2H, J1 = 17.5 Hz, J2 = 5 Hz), 4.11 (dd, 2H, J1 = 17.5 Hz, J2 = 4.5 Hz), 4.41 (s, 2H), 4.49–4.53 (m, 4H), 4.74 (s, 2H), 7.11 (s, 6H), 7.44–7.63 (m, 26H), 8.17 (s, 2H). CD measurement: cUV = 39 μM, cVIS = 0.66 mM, CD (CH2Cl2) λ/nm (Δε/M−1 cm−1): 233 (−21.78), 249 (37.88), 370 (1.57), 452 (−0.76).
[Rh(COD)(Lig-Gly-Pro-Phe-OMe)2]BF4, 3j.
[(COD)Rh(CHCN)2]BF4 (1.01 mg, 2.66 μmol, 1 eq.), Lig-Gly-Pro-Phe-OMe5j (3.58 mg, 5.85 μmol, 2.2 eq. in 0.350 mL CDCl3). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 1.76 (s, 2H), 1.89 (s, 2H), 1.98 (s, 2H), 2.22 (s, 2H), 2.30 (s, 2H), 2.49 (s, 2H), 2.61 (s, 2H), 3.01–3.05 (m, 2H), 3.13–3.16 (m, 2H), 3.52 (s, 2H), 3.61 (s, 2H), 4.01 (d, 2H, J = 16 Hz), 4.39 (d, 2H, J = 16 Hz), 4.44 (s, 2H), 4.47 (s, 2H), 4.73 (s, 2H), 4.84–4.89 (m, 2H), 6.92 (s, 2H), 7.05–7.18 (m, 12H), 7.43–7.66 (m ,24H), 8.56 (s, 2H). CD measurements cUV = 39 μM, cVIS = 0.65 mM, CD (CH2Cl2) λ/nm (Δε/M−1 cm−1): 233 (−18.17), 250 (26.00), 371 (0.87), 452 (−0.36).
[Rh(COD)(Lig-Gly-NH2)2]BF4, 4a.
[(COD)Rh(CH3CN)2]BF4 (1.20 mg, 3 μmol, 1 eq.), Lig-Gly-NH26a (2.40 mg, 6.63 μmol, 2.2 eq. in 0.350 mL CDCl3). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 2.24 (ws, 4H), 2.53 (ws, 4H), 4.12 (d, 4H, J = 6 Hz), 4.58 (ws, 4H), 5.83 (ws, 2H), 6.71 (ws, 2H), 7.25 (ws, 4H), 7.41–7.43 (m, 8H), 7.47–7.51 (m, 12 H), 7.64 (ws, 4H), 8.19 (ws, 2H). 31P NMR (242.93 MHz, CDCl3) δ (ppm): 26.75 (d, 1JRh-P = 147 Hz).
[Rh(COD)(Lig-Gly-Val-Gly-NH2)2]BF4, 4d.
[(COD)Rh(CH3CN)2]BF4 (0.90 mg, 2.4 μmol, 1 eq.), Lig-Gly-Val-Gly-NH24d (2.70 mg, 5.21 μmol, 2.2 eq. in 0.350 mL CDCl3. 1H NMR (300.13 MHz, CDCl3) δ (ppm): 0.93 (d, 6H, J = 6 Hz), 0.95 (d, 6H, J = 7 Hz), 2.19–2.28 (m, 6H), 2.46–2.58 (m, 4H), 3.86–4.01 (m, 6H), 4.22–4.30 (m, 4H), 4.49 (s, 2H), 4.65 (s, 2H), 6.00 (s, 2H), 6.97 (s, 2H), 7.19 (s, 4H), 7.41–7.61 (m, 26H), 7.23 (s, 2H), 8.35 (s, 2H). CD measurements cUV = 39 μM, cVIS = 0.64 mM, CD (CH2Cl2) λ/nm (Δε/M−1 cm−1): 233 (−17.75), 249 (33.76), 371 (0.95), 450 (−0.48).
[Rh(COD)(Lig-Gly-Pro-Phe-NH2)2]BF4, 4j.
[(COD)Rh(CH3CN)2]BF4 (1.09 mg, 2.87 μmol, 1 eq.), Lig-Gly-Pro-Phe-NH26j (3.80 mg, 6.31 μmol, 2.2 eq. in 0.350 mL CDCl3). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 1.46 (s, 2H), 1.72 (s, 2H), 1.82 (s, 2H), 2.04 (s, 2H), 2.21 (s, 2H), 2.29 (s, 2H), 2.48 (s, 2H),2.60 (s, 2H), 2.86–2.90 (m, 2H), 3.44 (dd, 2H, J1 = 14 Hz, J2 = 2 Hz), 3.56–3.60 (m, 2H), 3.91 (s, 2H), 4.06 (dd, 2H, J1 = 15.5 Hz, J2 = 5.5 Hz), 4.22 (dd, 2H, J1 = 15.5 Hz, J2 = 3.5 Hz), 4.40 (dd, 2H, J1 = 8.5 Hz, J2 = 2.5 Hz), 4.48 (s, 2H), 4.60–4.63 (m, 2H), 4.69 (s, 2H), 5.92 (s, 2H), 7.12 (s, 2H), 7.14–7.49 (m, 38H), 7.66 (s, 4H), 8.59 (s, 2H). CD measurements cUV = 39 μM, cVIS = 0.66 mM, CD (CH2Cl2) λ/nm (Δε/M−1 cm−1): 232 (30.87), 250 (−10.87), 274 (−13.39), 283 (−12.73), 379 (−1.25), 442 (0.72).
[Rh(COD)(Lig-βAla-Pro-Phe-NH2)2]BF4, 4k.
[(COD)Rh(CH3-CN)2]BF4 (1.13 mg, 2.97 μmol, 1 eq.), Lig-βAla-Pro-Phe-NH26j (3.92 mg, 6.54 μmol, 2.2 eq. in 0.350 mL CDCl3). 1H NMR (600.13 MHz, CDCl3) δ (ppm): 1.71–1.77 (m, 4H), 1.80–1.84 (m, 2H), 2.21 (d, 4H, J = 7.5 Hz), 2.51 (d, 4H, J = 7.5 Hz), 2.59 (d, 2H, J = 14.5 Hz), 2.88 (s, 2H), 2.99–3.03 (m, 2H), 3.34 (d, 2H, J = 12.5 Hz), 3.42–3.46 (m, 2H), 3.66–3.70 (m, 4H), 3.89 (s, 2H), 4.31 (d, 2H, J = 7 Hz), 4.55 (s, 4H), 4.69–4.73 (m, 2H), 5.33 (s, 2H), 7.07 (s, 2H), 7.14–7.50 (m, 34H), 7.78 (s, 4H), 7.93 (s, 2H). CD measurements cUV = 63 μM, cVIS = 0.63 mM, CD (CH2Cl2) λ/nm (Δε/M−1 cm−1): 239 (−3.51), 250 (2.15), 287 (−3.58), 365 (0.66), 457 (−0.17).
Catalysis at RT.
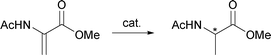
A two neck round bottomed flask (10 mL) was charged with the ligand (7.2 μmol, 2.4 mol%) dissolved in dichloromethane (3 mL; distilled, degassed in an ultrasonic bath for 15 min), followed by [(COD)Rh(CH3CN)2]BF4 (1.25 mg, 3 μmol, 1 mol%) dissolved in dichloromethane (2 mL). Instantaneously, the colorless ligand solution turned yellow and was stirred for several minutes. The flask was then flushed with hydrogen gas and the substrate (45 mg, 0.3 mmol, 100 mol%) was added in one portion and vigorously stirred. After 2 h/overnight, 0.5 mL of the solution was eluted through silica (150 mg) with ethyl acetate (25 mL), reduced to 1/5 of the volume by rotary evaporation and the enantiomeric excess (ee) was determined by chiral fused silica capillary column Beta Dex 225, at 130, 140, or 150 °C. From the remaining (crude) reaction solution, the solvent was removed by rotary evaporation and chemical yield was determined by 1H NMR (CDCl3).
Catalysis at low temperature.
Following the procedure for reactions at RT, before substrate addition, reaction flask was placed in a thermo-container filled with technical ethanol (or isopropanol), cooled by cryostat device to −5 °C, and stirred vigorously overnight.
Acknowledgements
These materials are based on work financed by the Unity Through Knowledge Fund (UKF, 36/08), the Croatian Science Foundation (HrZZ, 02.04/23) and the Ministry of Science, Education and Sports (MSES, 098-0982904-2946). We thank SME Chirallica for using their chiral analytics facility.
References
- P. J. Deuss, R. Heeten, W. Laan and P. C. J. Kamer, Chem.–Eur. J., 2011, 17, 4680 CrossRef CAS.
-
(a) S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 CrossRef CAS;
(b) A. Agarkov, S. Greenfield, D. Xie, R. Pawlick, G. Starkey and S. R. Gilbertson, Biopolymers, 2006, 84, 48 CrossRef CAS and references cited therein.
-
(a) X. Wu, C. He, X. Wu, S. Qu and C. Duan, Chem. Commun., 2011, 47, 8415 RSC;
(b) D. Coquiere, J. Bos, J. Beld and G. Roelfes, Angew. Chem., Int. Ed., 2009, 48, 5159 CrossRef CAS;
(c) C. A. Christensen and M. Meldal, Chem.–Eur. J., 2005, 11, 4121 CrossRef CAS;
(d) T. P. Clark and C. R. Landis, J. Am. Chem. Soc., 2003, 125, 11792 CrossRef CAS;
(e) P. Arya, N. V. Rao, J. Singkhonrat, H. Alper, S. C. Bourque and L. E. Manzer, J. Org. Chem., 2000, 65, 1881 CrossRef CAS;
(f) C. W. Edwards, M. R. Shipton and M. Wills, Tetrahedron Lett., 2000, 41, 8615 Search PubMed.
- R. S. Herrick, R. M. Jarret, T. P. Curran, D. R. Dragoli, M. B. Flaherty, S. E. Lindyberg, R. A. Slate and L. C. Thornton, Tetrahedron Lett., 1996, 37, 5289 CrossRef CAS.
-
(a) S. Cowdhury, D. A. R. Sanders, G. Schatte and H.-B. Kraatz, Angew. Chem., Int. Ed., 2006, 45, 751 CrossRef CAS;
(b) L. Barišić, M. Čakić, K. A. Mahmoud, Y.-n. Liu, H.-B. Kraatz, H. Pritzkow, S. I. Kirin, N. Metzler-Nolte and V. Rapić, Chem.–Eur. J., 2006, 12, 4965 CrossRef CAS;
(c) K. Heinze and M. Beckmann, Eur. J. Inorg. Chem., 2005, 3450 CrossRef CAS;
(d) T. Moriuchi, A. Nomoto, K. Yoshida, A. Ogawa and T. Hirao, J. Am. Chem. Soc., 2001, 123, 68 CrossRef CAS.
-
(a) T. Moriuchi and T. Hirao, Acc. Chem. Res., 2010, 43, 1040 CrossRef CAS;
(b) A. Lataifeh, S. Beheshti and H.-B. Kraatz, Eur. J. Inorg. Chem., 2009, 3205 CrossRef CAS;
(c) D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931 CrossRef CAS.
- S. I. Kirin, H.-B. Kraatz and N. Metzler-Nolte, Chem. Soc. Rev., 2006, 35, 348 RSC.
- A. C. Laungani and B. Breit, Chem. Commun., 2008, 844 RSC.
-
(a) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS;
(b) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355 RSC;
(c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS;
(d) B. Beit, Angew. Chem., Int. Ed., 2005, 44, 6816 CrossRef CAS;
(e) M. J. Wilkinson, P. W. N. M. van Leeuwen and J. N. H. Reek, Org. Biomol. Chem., 2005, 3, 2371 RSC.
-
(a) P. W. N. M. van Leeuwen, D. Rivillo, M. Raynal and Z. Freixa, J. Am. Chem. Soc., 2011, 133, 18562 Search PubMed;
(b) L. Pignataro, S. Carboni, M. Civera, R. Colombo, U. Piarulli and C. Gennari, Angew. Chem., Int. Ed., 2010, 49, 6633 CrossRef CAS;
(c) P.-A. R. Breuil, F. W. Patureau and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 2162 CrossRef CAS;
(d) A. C. Laungani, J. M. Slattery, I. Krossing and B. Breit, Chem.–Eur. J., 2008, 14, 4488 CrossRef CAS;
(e) J. M. Takacs, K. Chaiseeda, S. A. Moteki, D. S. Reddy, D. Wu and K. Chandra, Pure Appl. Chem., 2006, 78, 501 CrossRef CAS;
(f) A. C. Laungani and B. Breit, Tetrahedron: Asymmetry, 2003, 14, 3823 CrossRef CAS.
- Introducing additional chirality within the coordination sphere of the catalytic metal (double induction), Breit8 obtained up to 99% ee in the studied asymmetric hydrogenation.
- Ferrocene analogues with three amino acids per chain, [Fe(C5H4CO-Aa1-Aa2-Aa3-OMe)2], were studied in Ref. 5a.
- See for example:
(a) D. E. Dorman and F. A. Bovey, J. Org. Chem., 1973, 38, 2379 CrossRef;
(b) I. Žigrović and Š. Horvat, Eur. J. Org. Chem., 2001, 1533 CrossRef CAS.
- X-ray crystal structures of [Rh(COD)(PPh3)2] derivatives reveal a square-planar coordination geometry with P-Rh-P chelatation angles in the range 88.7–95.0:
(a) A. Preetz, H.-J. Drexler, S. Schulz and D. Heller, Tetrahedron: Asymmetry, 2010, 21, 1226 Search PubMed;
(b) L. Kaganovsky, K.-B. Cho and D. Gelman, Organometallics, 2008, 27, 5139 CrossRef CAS;
(c) J. Albers, E. Dinjus, S. Pitter and O. Walter, J. Mol. Catal. A: Chem., 2004, 219, 41 CrossRef;
(d) Y. Nishihara, K. Nara, Y. Nishide and K. Osakada, Dalton Trans., 2004, 1366 RSC;
(e) X. Zhang, K. Mashima, K. Koyano, N. Sayo, H. Kumobayashi, S. Akutagawa and H. Takaya, J. Chem. Soc., Perkin Trans. 1, 1994, 2309 RSC.
-
A. Rodger and B. Norden, Circular dichroism and linear dichroism, Oxford University Press, Oxford, 1997 Search PubMed.
- For CD spectra of [Rh(COD)(PPh3)2] derivatives see: J. Yu, T. V. Rajan Babu and J. R. Parquette, J. Am. Chem. Soc., 2008, 130, 7845 Search PubMed.
- For CD spectra of ferrocene peptides [Fe(C5H4CO-Aan-R)2] (Herrick-conformation) see Ref. 7 and
(a) S. I. Kirin, D. Wissenbach and N. Metzler-Nolte, New J. Chem., 2005, 29, 1168 RSC;
(b) S. I. Kirin, U. Schatzschneider, S. D. Koster, D. Siebler and N. Metzler-Nolte, Inorg. Chim. Acta, 2009, 362, 894 CrossRef CAS.
-
(a) G. Erre, S. Enthaler, K. Junge, G. Serafino and M. Beller, Coord. Chem. Rev., 2008, 252, 471 CrossRef CAS;
(b) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS;
(c) J. A. Osborn, F. H. Jardine, J. F. Young and G. Wilkinson, Chem. Commun., 1965, 131 RSC.
-
(a) N. B. Johnson, I. C. Lennon, P. H. Moran and J. A. Ramsden, Acc. Chem. Res., 2007, 40, 1291 CrossRef CAS;
(b) H. Shimizu, I. Nagasaki, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS.
- E. G. Hutchinson and J. M. Thornton, Protein Sci., 1994, 3, 2208 Search PubMed.
-
(a) N. T. Ross, W. P. Katt and A. D. Hamilton, Philos. Trans. R. Soc. London, Ser. A, 2010, 368, 98 Search PubMed;
(b) O. Khakshoor and J. S. Nowick, Curr. Opin. Chem. Biol., 2008, 12, 722 CrossRef CAS;
(c) C. K. Smith and L. Regan, Acc. Chem. Res., 1997, 30, 153 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.