DOI:
10.1039/C2RA20592K
(Paper)
RSC Adv., 2012,
2, 6049-6056
Lithiation of η6-fluorobenzenetricarbonylchromium(0) and reaction with dialkyl disulfides: synthesis of η6-1,2-bis- and 1,2,3-tris-alkylsulfanylbenzenetricarbonylchromium(0) complexes†
Received
31st March 2012
, Accepted 24th April 2012
First published on 25th April 2012
Abstract
Directed lithiation of η6-fluorobenzenetricarbonylchromium(0) and reaction with dialkyl disulfides affords η6-1,2-bis-alkylsulfanylbenzenetricarbonylchromium(0) complexes by electrophilic addition of an alkylthio group at C-2 of the benzene ring, and subsequent SNAr reaction with displacement of fluoride by the alkanethiolate generated in the first step. 1,2,3-tris-Alkylsulfanylbenzene complexes are formed as by-products. The structures of four of the new complexes have been confirmed by X-ray crystallography, one using synchrotron radiation.
1. Introduction
Naturally occurring benzopentathiepins such as varacin 1a and lissoclinotoxin 1b (Fig. 1) exhibit potent anti-fungal and anti-tumour activity.1 Substituted benzopentathiepins such as 2, exhibit planar chirality due to the slow inversion of the puckered ring of sulfur atoms,2 making such compounds interesting targets for synthetic and mechanistic study. Varacin has been synthesised by three different approaches to date,3 and a number of methods have been developed for the preparation of substituted benzopentathiepins.4 We have been interested in developing convenient methods to synthesise arenes substituted with adjacent sulfur substituents which could be employed as precursors to benzopentathiepins, and as part of this investigation we have examined the use of η6-arenetricarbonylchromium(0) complexes as substrates to allow the controlled introduction of two adjacent sulfur containing substituents onto the arene ring.
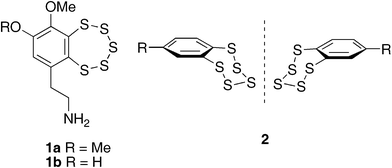 |
| | Fig. 1 Structures of planar chiral benzopentathiepins; varacin 1a, lissoclinotoxin A 1b, and side-on view showing puckered ring 2. | |
2. Results and discussion
In this paper we report our initial results on functionalising arenetricarbonylchromium(0) complexes with sulfur-based electrophiles. Our first experiments involved the use of the t-butylsulfanylbenzene complexes 4 (Scheme 1). It was hoped that deprotonation ortho to the t-butylthio group could be effected with a strong base at low temperature as is well known5 for a range of donor substituted benzenechromium complexes. Treatment with a sulfur electrophile such as a dialkyl disulfide was then expected to introduce a second alkylsulfanyl substituent forming complexes such as 5a. It was envisaged that the t-butyl group could be removed at a later stage, enabling construction of a pentathiepin ring. Complexes 4a and 4b were formed in good yields (76% and 68% respectively) by heating the t-butylthioethers 3a or 3b with hexacarbonylchromium(0) in 1,4-dioxane for 4 days in the dark. Attempts to effect deprotonation with butyllithium and electrophilic addition at the ortho-position in 4a were, however, unsuccessful, and it was not possible to introduce deuterium or a methyl group (to form 5b or 5c) under conditions that were successful with the simple η6-benzenetricarbonylchromium complex 7 (Scheme 2). This may be due to the steric bulk of the t-butylthio group preventing the base from reaching the ortho-hydrogen atoms.
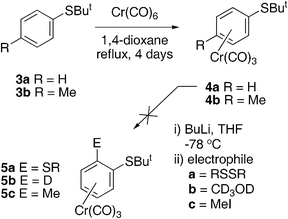 |
| | Scheme 1 Inert behaviour of the t-butylsulfanylbenzenechromium complex. | |
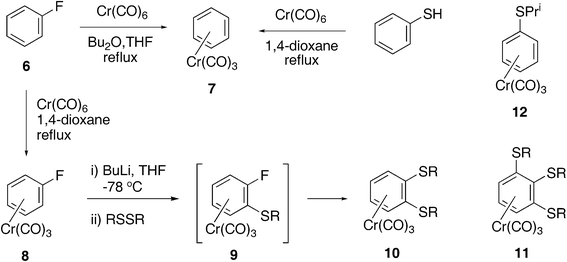 |
| | Scheme 2 Formation of di- and tri-alkylsulfanylbenzenechromium complexes 10–12 from the fluorobenzene complex 8. Conditions: step i) nBuLi (1.6 M) 1.2 eq, THF, −78 °C, 90 min; step ii) disulfide (1.5 eq.), −78 °C to room temp, 16 h. | |
We then turned to the use of the fluorobenzene complex68 (Scheme 2), in which substitution adjacent to fluorine is well documented.7 Preparation of 8 was found to be best carried out in 1,4-dioxane as solvent. Reaction of fluorobenzene with chromium hexacarbonyl in the di-n-butyl ether/THF mixtures commonly employed, caused loss of fluorine and production of the unsubstituted benzene complex 7 (32%) (identified by NMR spectroscopy and from the unit cell dimensions of crystals produced8). Interestingly η6-benzene complex 7 was also formed in 72% yield in an attempt to form a thiophenol chromium complex by heating thiophenol with chromium hexacarbonyl in 1,4-dioxane. Attempts to displace the fluorine atom in complex 8 by treatment with NaSH in aqueous THF or in DMF were unsuccessful, while reaction in ethanol gave a 19% yield of the corresponding ethoxybenzene complex and 25% starting material. No evidence of the formation of a η6-thiophenoltricarbonylchromium complex was obtained.
The reaction of the η6-fluorobenzene complex 8 with dialkyl disulfides was then investigated. Treatment of the complex 8 with n-butyllithium in THF at low temperature followed by addition of a dialkyl disulfide and warming to room temperature afforded, not the expected η6-1-alkylsulfanyl-2-fluorobenzenetricarbonylchromium(0) complexes 9, but the η6-1,2-bis-alkylsulfanylbenzene complexes 10, in which the fluorine atom has been replaced by an alkylthio group. The reaction was successful for dimethyl, diethyl, diisopropyl and dibenzyl disulfides (Table 1).
Table 1
| Entry |
R |
Conversion of 8a (%) |
Ratio of productsa mono![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :di:tri and isolated yield (%) :di:tri and isolated yield (%) |
|
Conversion of starting material 8, and product ratio obtained by integration of the 1H NMR spectrum of the crude product mixture.
All three products were characterised as a mixture as they could not be separated by flash chromatography or recrystallisation.
|
| a |
Me |
70 |
0:100 (10a, 46%):0 |
| b |
Et |
82 |
0:71 (10b, 42%):11 (11b) |
| cb |
i
Pr |
70 |
40 (12):21 (10c):9 (11c) |
| d |
t
Bu |
0 |
n/a |
| e |
CH2Ph |
67 |
0:50 (10e):17 (11e) |
The reaction with di-t-butyl disulfide was unsuccessful with only a mixture of starting material and uncomplexed decomposition products obtained. In the reactions involving diethyl-, diisopropyl- and dibenzyl-disulfides the corresponding tri-substituted benzenechromium(0) complexes 11b, 11c and 11d were formed as by-products. The reaction with diisopropyl disulfide also afforded the mono-substituted complex 12 as the major product in 40% yield.
We propose the mechanism shown in Scheme 3 to account for the formation of the di-substituted compounds 10a–e.
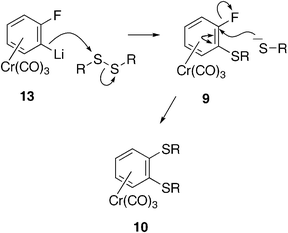 |
| | Scheme 3 Proposed mechanism of formation of disubstituted complexes 10a–e. | |
Attack by the lithiated fluorobenzene complex 13 on the disulfide will release the corresponding alkylthiolate anion which can then effect nucleophilic aromatic substitution (SNAr) of the fluorine atom in the first formed product 9. A similar mechanism has been proposed by Widdowson in reactions with lactones and isocyanates which lead to bicyclic complexes by intramolecular SNAr reaction.9 It is less obvious how the tri-substituted complexes 11 are formed, but excess base may deprotonate the di-substituted product 10ortho to one of the alkylthio groups, which are not bulky enough to restrict attack by the base, unlike the t-butylthio group which appeared to block proton abstraction in the unsuccessful attempts to lithiate 4a. Alternatively, the thiolate released in the first step may diffuse away from the intermediate product 14 before it can effect the SNAr reaction, leaving the fluorine in place to activate the deprotonation of the remaining ortho-hydrogen. The thiolate generated in a second reaction with further disulfide would then effect the necessary SNAr substitution of the fluorine atom forming 11. The formation of the mono-substituted complex 12, generated in the reaction with diisopropyl disulfide, can be accounted for if the ortho-lithio complex 13, which is known to be highly basic,10 abstracted the H-2 proton from the diisopropyl disulfide, eliminating isopropylthiolate, which could then effect SNAr reaction on the regenerated complex 8, displacing the fluorine atom.
In an attempt to extend the scope of the reaction, the lithiated fluorobenzene complex 13 (Scheme 4) was treated with 1,2-dithiane in the hope of forming the benzo[b]-1,4-dithiocane complex 14. This reaction, and an attempt to react 13 with elemental sulfur to form the benzopentathiepin complex 15, were both unsuccessful, and no evidence for the formation of either complexes 14 or 15 was obtained.
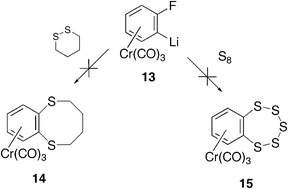 |
| | Scheme 4 No reaction occurred with a cyclic disulfide or S8. | |
3. X-Ray crystal structures
X-Ray crystal structures were determined for disubstituted complexes 10a (two polymorphs), 10aα (Fig. 2–4) and 10aβ (Fig. 5–7), 10b (Fig. 8 and 9), and 10e (Fig. 10 and 11), as well as for trisubstituted compound 11e11 (Fig. 12 and 13). Selected geometrical parameters are presented in Table 2.
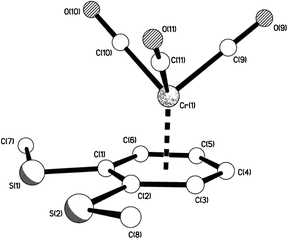 |
| | Fig. 2 The molecular structure of 10aα showing the piano-stool, half-sandwich arrangement. | |
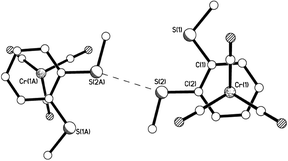 |
| | Fig. 3 View of a pair of neighbouring molecules of 10aα showing an S⋯S intermolecular interaction and staggered arrangement of arene ring vs. carbonyl groups. S⋯S = 3.330 Å. | |
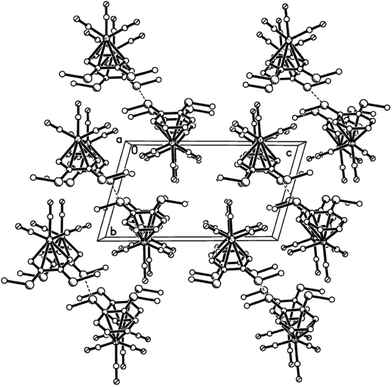 |
| | Fig. 4 Packing plot of 10aα showing columns of S⋯S paired molecules in a parallel arrangement. | |
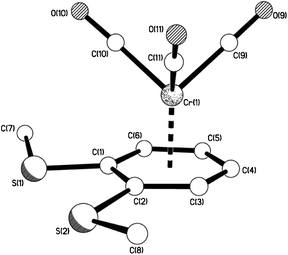 |
| | Fig. 5 The molecular structure of 10aβ showing the piano-stool, half-sandwich arrangement. | |
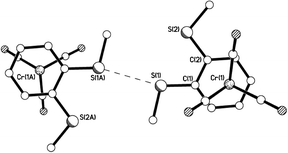 |
| | Fig. 6 View of a pair of neighbouring molecules of 10aβ showing an S⋯S intermolecular interaction and staggered arrangement of arene ring vs. carbonyl groups. S⋯S = 3.315 Å. | |
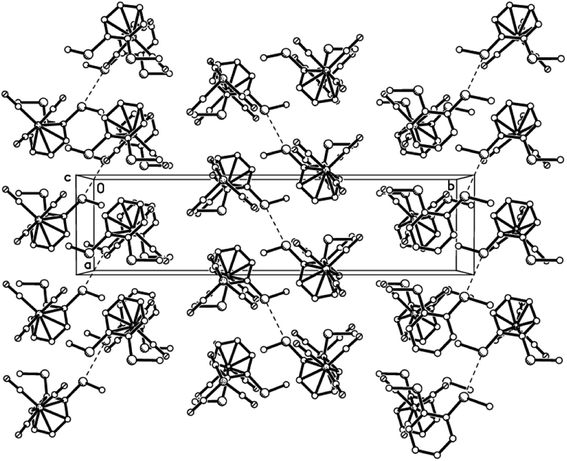 |
| | Fig. 7 Packing plot of 10aβ showing columns of S⋯S paired molecules in a herring-bone arrangement. Although molecules of this polymorph still pair up, the packing is different to polymorph 10aα; here the columns are stacked in an anti fashion. | |
 |
| | Fig. 8 The molecular structure of 10b showing the piano-stool, half-sandwich arrangement. | |
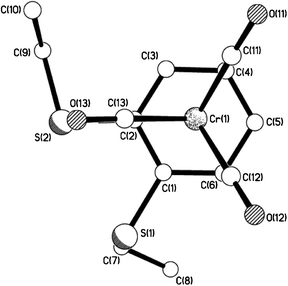 |
| | Fig. 9 View of 10b showing the eclipsed arrangement of arene ring vs. carbonyl groups. | |
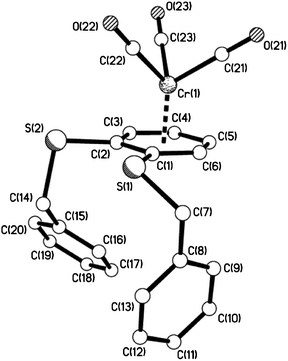 |
| | Fig. 10 The molecular structure of 10e showing the piano-stool, half-sandwich arrangement. | |
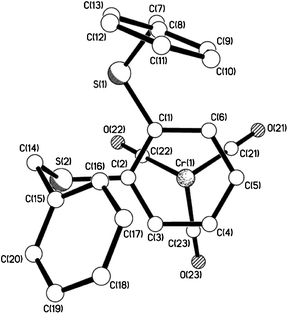 |
| | Fig. 11 View of 10e showing the staggered arrangement of arene ring vs. carbonyl groups. | |
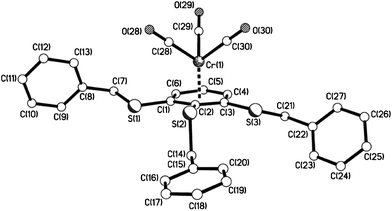 |
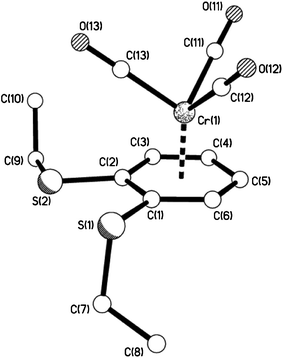
| | Fig. 12 The molecular structure of 11e showing the piano-stool, half-sandwich arrangement. | |
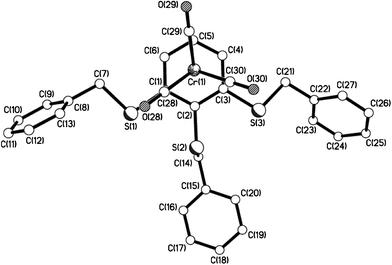 |
| | Fig. 13 View of 11e showing the almost eclipsed arrangement of arene ring vs. carbonyl groups. | |
Table 2 Selected lengths (Å) and angles (°) for 10aα, 10aβ, 10b, 10e, and 11e
| |
10aα
|
10aβ
|
10b
|
10e
|
11e
|
| Cr(1)–CCO range |
1.831(2)–1.846(2) |
1.814(14)–1.836(15) |
1.8357(12)–1.8486(13) |
1.8436(19)–1.855(2) |
1.847(2)–1.860(2) |
| Cr(1)–Carene range |
2.217(2)–2.291(2) |
2.211(15)–2.278(13) |
2.1816(11)–2.2717(10) |
2.2038(19)–2.2517(17) |
2.185(2)–2.254(2) |
| av. of alternate η6-arene C–C, long |
1.420(3) |
1.422(19) |
1.4172(17) |
1.421(3) |
1.420(3) |
| av. of alternate η6-arene C–C, short |
1.402(3) |
1.415(19) |
1.4110(17) |
1.404(3) |
1.412(3) |
| Carene–S |
1.762(2), 1.764(2) |
1.747(12), 1.759(13) |
1.7560(11), 1.7677(11) |
1.7701(18), 1.7792(18) |
1.767(2), 1.769(2), 1.769(2) |
| Cr(1)⋯Cg {(C(1) > C(6)} |
1.7442(9) |
1.731(6) |
1.7175(5) |
1.7134(7) |
1.7131(9) |
Each of the five structures adopts the piano-stool arrangement. A staggered arrangement of carbonyl groups relative to the carbon atoms of the η6-arene is observed for 10aα, 10aβ, while the others are eclipsed. Analysis of the geometrical data in Table 2 reveals considerable consistency among the five structures. There is some variation in Cr(1)–Carene distances, with a difference of ca. 0.05–0.09 Å between longest and shortest in any one structure. The bonds from the metal to the substituted carbons tend to be longest, but this is not universally true, with Cr(1)–C(1) in 10b being among the shorter lengths. The ranges observed are consistent with the mean value of 2.22(3) Å observed among 1191 examples of ArCrCO fragments in the CSD.12 The Cr(1)–CCO distances also show some small variation, but with a difference no greater than 0.02 Å in any one structure. This observation is consistent with the narrow range observed among the 1191 examples reported in the CSD12 which have a mean distance of 1.83(3) Å. The Carene–S distances also fall into a narrow range. There is no compelling evidence for η6-aromatic bond length alternation, with the average of the three longest C–C bonds not statistically significantly different from the three shorter lengths. Finally, the Cr(1) to η6-arene centroid distances also fall into a narrow range between 1.7131(9) and 1.7442(9) Å and are consistent with the CSD mean of 1.73(2) Å.12 The two polymorphs of 10a both exhibit very similar S⋯S interactions between pairs of molecules, but differ in the way these pairs arrange in space (Fig. 3, 4, 6 and 7). A search of the CSD revealed that these interactions, at ca. 3.32 Å, are towards the lower end of S⋯S interactions, which are generally no shorter than 3.2 Å, and are often 3.5–3.6 Å or longer.12
Future work is aimed at developing an asymmetric version of this reaction by desymmetrisation of a 4-substituted η6-fluorobenzenechromium complex with a chiral base13 as this would allow construction of enantiomerically enriched, planar, chiral benzopentathiepins after decomplexation of the arene from chromium.
4. Conclusions
In conclusion we have shown that lithiation of η6-fluorobenzenechromium(0) tricarbonyl and reaction with disulfides is an effective method to introduce two adjacent sulfur substituents onto an arene ring, and that η6-arenechromium complexes are stable towards normally strongly metal binding sulfur reagents.
5. Experimental
5.1 Material and methods
All reactions, unless otherwise stated, were run under nitrogen, using glassware that had been dried in an oven at 150 °C for 2 h prior to use. Room temperature refers to ambient temperature (18–22 °C). A temperature of 0 °C implies use of an ice-slush bath, and −78 °C refers to use of a dry ice–acetone bath. Heated experiments were conducted using thermostatically controlled oil baths. Reactions were monitored by TLC using aluminium backed silica gel F254 plates (Merck) and visualised using UV254 nm or KMnO4 dip. Rf values are given under these conditions. Flash column chromatography was carried out using 60 Å silica gel (Fluorochem).
“Petrol” refers to petroleum ether of the 40–60 °C boiling range and was distilled prior to use. Anhydrous THF and anhydrous Bu2O were dried by refluxing over Na-benzophenone prior to distillation. Anhydrous 1,4-dioxane was purchased from Aldrich and not further purified. Other reagents were from commercial sources and were used without purification unless otherwise stated. All chromium-based compounds were stored in the dark. All chromium-based reactions were carried out in foil-wrapped glassware. Deoxygenated solvents were prepared by bubbling a stream of nitrogen through the solvent for at least 20 min prior to use.
NMR spectra were recorded on a Bruker DPX-400 MHz spectrometer operating at 100.61 MHz for carbon and 400.13 MHz for proton spectra, employing a high-resolution broad-band HX probe. The 13C and 1H chemical shifts are reported in δ units, parts per million downfield from TMS. All NMR samples of chromium-based materials were filtered through a plug of silica and charcoal prior to running the sample. Coupling constants (J) are measured in hertz (Hz). IR spectra were recorded using a Perkin Elmer, Paragon 1000 Spectrometer using NaCl plates. The sample was loaded onto the plates as a solution in dichloromethane and the solvent was allowed to evaporate prior to data acquisition.
5.2 General procedure for the preparation of [η6-(1,2-bis-alkylthio)benzene]-tricarbonylchromium(0) complexes 10
A stirred solution of freshly recrystallised (η6-fluorobenzene)tricarbonylchromium(0)68 (100 mg, 0.43 mmol) in anhydrous THF (5.0 ml) was cooled to −78 °C with vigorous stirring in the dark. nBuLi (0.33 ml of a 1.6 M solution in hexanes, 0.43 mmol) was added dropwise at a moderate rate and the mixture was stirred at −78 °C for 90 min. After this time the appropriate disulfide (0.65 mmol) was added to the flask in one portion. The mixture was then allowed to warm to room temperature overnight with stirring. The mixture was quenched by the addition of saturated aqueous NH4Cl solution (25 ml) and extracted with Et2O (2 × 50 ml). The combined organic extracts were dried (MgSO4) and evaporated to give an orange oil, which was purified as described below.
5.2.1 [η6-1,2-Bis(methylthio)benzene]tricarbonylchromium(0) 10a.
Dimethyl disulfide (0.058 ml, 0.65 mmol) was used. 1H NMR analysis of the crude reaction mixture showed a 70% conversion of starting material 8. This mixture was purified by flash chromatography over silica, eluting with 0% → 5% EtOAc–petrol. The product 10a and starting material 8 co-eluted. This fraction was then recrystallised from CH2Cl2–petrol, giving pure [η6-1,2-bis(methylthio)benzene] tricarbonylchromium(0) 10a (61 mg, 46%) as a yellow-orange solid, mp 124–125 °C (CH2Cl2–petrol); δH(CDCl3; 400 MHz) 5.47 (2H, m, AA′BB′ system, 2 × ortho-ArH), 5.27 (2H, m, AA′BB′ system, 2 × meta-ArH), 2.47 (6H, s, 2 × CH3) ppm; δC(CDCl3; 100 MHz) 232.3 (3 × CO), 112.3 (2 × ipso-ArC), 91.3 (2 × ortho- or meta-ArC), 89.9 (2 × ortho- or meta-ArC), 17.8 (2 × CH3) ppm; νmax (NaCl plate, neat) 1937 and 1844 (CO), 1417 cm−1; m/z (EI) 306 (31%, 32M+), 307 [8%, M+ (32S + 33S)], 308 (5%, 33M+), 309 [1%, M+ (33S+34S)], 250 (40%, M−2CO), 222 (59%, M − 3CO), 207 [100%, M − (3CO + CH3)], 192 [68%, M − (3CO + 2CH3)] (Found: 305.9475, C11H10CrO3S2 requires 305.9471).
5.2.2 [η6-1,2-Bis(ethylthio)benzene]tricarbonylchromium(0) 10b and [η6-1,2,3-tris(ethylthio)benzene]tricarbonyl chromium(0) 11b.
Diethyl disulfide (0.08 ml, 0.65 mmol) was employed. After the usual work-up, a 71:18:11 mixture of [η6-1,2-bis(ethylthio)benzene]tricarbonylchromium(0) 10b, (η6-fluorobenzene)tricarbonylchromium(0) 8 and a second product, [η6-1,2,3-tris(ethylthio)benzene]tricarbonylchromium(0) 11b was evident in the 1H NMR spectrum of the crude mixture, giving an overall 82% conversion of starting material. The crude mixture was purified by flash column chromatography to remove baseline material, eluting with 0% → 1% → 2% EtOAc–petrol. The three compounds co-eluted in this solvent system, but recrystallisation of the mixture obtained using CH2Cl2–petrol gave the major product [η6-1,2-bis(ethylthio)benzene]tricarbonylchromium(0) 10b (61 mg isolated pure, 42%, with some remaining in the mother liquor) as an orange-yellow solid, mp 59–60 °C (CH2Cl2–petrol); δH(CDCl3; 400 MHz) 5.49 (2H, AA′BB′ system, 2 × ortho-ArH), 5.25 (2H, m, AA′BB′ system, 2 × meta-ArH), 2.91 (4H, m, 2 × CH2), 1.38 (6H, t, J 7.6, 2 × CH3) ppm; δC(CDCl3; 100 MHz) 220.1 (3 × CO), 128.1 (2 × ipso-ArC), 92.8 (2 × ortho or meta-ArC), 90.1 (2 × meta-or ortho-ArC), 28.9 (2 × CH2), 13.8 (2 × CH3) ppm; νmax (NaCl plate, neat) 1962 and 1886 (CO) cm−1. Evidence for the presence of the minor product [η6-1,2,3-tris(ethylthio)benzene]tricarbonylchromium(0) 11b was given by the following data: δH(CDCl3; 400 MHz) 5.64 (1H, t, J 6.4, ArH), 4.95 (2H, d, J 6.4, 2 × ortho-ArH), 2.96–2.84 (6H, m, AB system obscured by peaks from disubstituted compound 10b, 3 × CH2), 0.91–0.80 (9H, m, obscured by peaks from di-substituted compound 10b, 3 × CH3) ppm; m/z (CI) 395 [5%, 32MH+], 335 (30%, MH+ − SCH2CH3), 259 [88%, 32MH+ − Cr(CO)3], 260 [14%, MH+ − Cr(CO)3, 32S + 33S], 199 [100%, MH2+ − Cr(CO)2SCH2CH3], 61 (52%, SCH2CH3) (Found: 394.9900, C15H19Cr32S3O3 requires 394.9896).
5.2.3 [η6-(iso-Propylthio)benzene]tricarbonylchromium(0), 12ii [η6-1,2-bis-(iso-propyl thio)benzene]tricarbonylchromium(0) 10c and [η6-1,2,3-tris-(iso-propylthio)-benzene]tricarbonyl chromium(0) 11c.
Following the general procedure, iso-propyldisulfide (0.10 ml, 0.65 mmol) was used as the disulfide. After the usual work-up, 1H NMR analysis of the crude reaction mixture showed 70% conversion of starting material η6-(fluorobenzene)-tricarbonylchromium(0) 8 to a mixture of [η6-(iso-propylthio)benzene]tricarbonyl chromium(0) 12, [η6-1,2-bis-(iso-propylthio)benzene]tricarbonyl-chromium(0) 10c and [η6-1,2,3-tris-(iso-propylthio)benzene]tricarbonylchromium(0) 11c in a 40:21:9 ratio. The mixture of products was purified by flash column chromatography, eluting with 0% → 1% → 2% → 5% EtOAc–petrol, to remove baseline material. This also gave a small portion of major product 12 as a yellow solid, with the majority remaining as part of a mixture with the other two products 10c and 11c. Data for [η6-(iso-propylthio)benzene]tricarbonylchromium(0) 12ii: δH(CDCl3; 400 MHz) 5.43 (2H, br dd, J 6.8 and 1.2, 2 × ortho-ArH), 5.34 (2H, t, J 6.8, 2 × meta-ArH), 5.20 (1H, br tt, J 6.4 and 1.0, para-ArH), 3.25 [1H, septet, J 6.6, CH(CH3)2], 1.34 [6H, d, J 6.6, CH(CH3)2] ppm; δC(CDCl3; 100 MHz) 109.5 (ipso-ArC), 95.13 (2 × ortho-ArC), 91.43 (2 × meta-ArC), 89.29 (para-ArC), 38.75 (CH), 22.2 (2 × CH3) ppm; νmax (NaCl plate, neat) 2923 (CH), 1964 and 1887 (CO), 1454 (S−C). The two minor products 10c and 11c could not be further separated by flash chromatography or recrystallisation and were characterised as far as possible as a mixture. Data for [η6-1,2-bis-(iso-propylthio)benzene]tricarbonylchromium(0) 10c: δH(CDCl3; 400 MHz) 5.43 (2H, AA′BB′ system, 2 × ortho-ArH), 5.19 (2H, AA′BB′ system, 2 × meta-ArH), 3.25 [2H, sept, J 6.8, 2 × CH(CH3)2], 1.32 [6H, d, J 6.4, 2 × CH(CH3)2] ppm; δC(CDCl3; 100 MHz) 106.2 (2 × ipso-ArC), 93.0 (2 × ortho-ArC0, 89.3 (2 × meta-ArC), 37.3 (2 × CH), 21.7 (4 × CH3) ppm; νmax (NaCl plate, neat, mixture of products 10c and 11c) 2952 (CH) 1964, 1887 (CO), 1455 (C–S), 1448 (C–S) cm−1; m/z (CI) 363 [12%, (M + H)+], 227 [100%, 32MH+− Cr(CO)3], 228 [13%, MH+ − Cr(CO)3, 32S + 33S], 229 [11%, 33MH+ − Cr(CO)3][ES (M + H) Found: 363.0178, C15H19CrS2O3 requires 363.0175]. Data for [η6-1,2,3-tris-(iso-propylthio)benzene]tricarbonylchromium(0) 11c: δH(CDCl3; 400 MHz) 5.67 (1H, t, J 6.4, ArH), 5.04 (2H, d, J 6.4, 2 × ortho-ArH), 3.60–3.46 [3H, m, 3 × CH(CH3)2], 1.46–1.41 [18H, m, 3 × CH(CH3)2] ppm; m/z (CI) 437 [6%, (M + H)+], 301 [64%, 32MH+ − Cr(CO)3], 302 [12%, MH+ − Cr(CO)3, 32S + 33S], 303 [11%, 33MH+ − Cr(CO)3][ES, (M + H) Found: 437.0363, C18H24CrS3O3 requires 437.0365]. Further analysis of this compound was not possible due to contamination with products 10c and 12 and starting material 8.
5.3 Synthesis of chromium complexes 4a16 and 4b
Anhydrous dioxane (20 ml) was transferred via syringe to a dry single neck flask under N2. The solvent was then degassed by bubbling with N2 for 20 min. The arene 3a14 or 3b15 (typically 4.0 mmol) and hexacarbonylchromium(0) (10 mmol) were then added to the flask and the flask was equipped with an air condenser leading to a water condenser (to prevent blockage of the water condenser with Cr(CO)6 sublimate). The mixture was then heated under reflux under N2 in the dark for 3–5 days. After this time a brown-yellow solution was present and the reaction was allowed to cool to ambient temperature. The mixture was filtered through celite, eluting with Et2O and the filtrate evaporated to dryness. The crude product was purified by flash chromatography to give the following compounds:
(η6-tert-Butylthiobenzene)tricarbonylchromium(0) 4a16.
(η6-tert-Butylthiobenzene)tricarbonylchromium(0) 4a was obtained as a yellow solid after refluxing the reaction mixture in the dark for 5 days. Chromatography, eluting with 0% → 5% EtOAc–petrol (increasing in 1% increments) to give (η6-tert-butylthiobenzene)tricarbonylchromium(0) 4a16 (920 mg, 76%) as yellow needles; δH(CDCl3; 400 MHz) 5.47 (2H, br d, J 6, 2 × o-ArH), 5.23 (3H, br m, 3 × ArH), 1.27 [9H, s, C(CH3)] ppm; m/z (EI) 304 (3%, 34M+), 303 (7, 33M+), 302 (26, 32M+), 248 (3, 34MH − tBu), 247 (6, 33MH − tBu), 246 (22, 32MH − tBu), 218 (38, 32M − 3CO), 162 (100, PhCrSH); νmax(NaCl plate, neat) 3083 and 2959 (CH), 1962, 1889 and 1860 (CO), 1394 and 1366 [C(CH3)3] cm−1.
[η6-(4-Methyl-tert-butylthio)benzene]tricarbonylchromium(0) 4b.
Using the general method described above, and heating for 3 days, the crude mixture obtained was purified by flash chromatography, eluting with 0% → 5% EtOAc–petrol (increasing in 1% increments. This gave [η6-(4-methyl-tert-butylthio)benzene]tricarbonylchromium(0) 4b (860 mg, 68%) as a yellow solid, mp 92–93 °C (DCM-petrol); δH(CDCl3; 400 MHz) 5.55 (2H, d, J 6.5, 2 × ArH, ortho-StBu), 5.02 (2H, d, J 6.4, 2 × ArH, ortho-Me), 2.13 (3H, s, CH3-Ar), 1.25 [9H, s, C(CH3)3] ppm; δC(CDCl3; 100 MHz) 109.2 (1 × ArC, ipso-to StBu), 103.4 (2 × ArC, ortho- to StBu), 96.9 (1 × ArC, ipso- to Me), 91.9 (2 × ArC, ortho- to Me), 47.2 [SC(CH3)3], 30.7 [3C, SC(CH3)3], 20.5 (1C, CH3Ar) ppm; νmax (NaCl plate, neat) 2958 (CH), 1964, 1910 and 1884 (CO) cm−1.
5.4 X-Ray crystallography
X-Ray quality crystals were grown from CH2Cl2–petrol for 10aα, 10b, 10e, and 11e, and from THF–ether for 10aβ. Crystal data were collected on a Bruker SMART 1000 CCD diffractometer using narrow slice 0.3° ω-scans for 10aα, 10aβ, 10e, and 11e.17 Data for 10b were collected at Daresbury Laboratory SRS Station 9.8 using silicon 111 monochromated X-radiation due to small, thin crystal habit. Diffractometer used at Daresbury was a Bruker Apex 2 CCD. Data were corrected for Lp effects and for absorption, based on repeated and symmetry equivalent reflections, and solved by direct methods (Patterson synthesis for 10b).18 Structures were refined by full matrix least squares on F2.18 All non-H atoms were refined anisotropically. H atoms were included in a riding model. Hydrogen atom Uiso values were constrained to be 120% of that of the carrier atom except for methyl H (150%). The structure refinements were routine with key parameters given in Ref. 10. CCDC 830653–830657 contain the supplementary crystallographic data for this paper.†
Acknowledgements
We thank the EPSRC, STFC, and Loughborough University for support, and the EPSRC National Mass Spectrometry service at the University of Swansea. We also thank Drs J. E. Warren and T. J. Prior for scientific support at Daresbury Laboratory and acknowledge the use of the EPSRC's Chemical Database Service at Daresbury.
References
- B. S. Davidson, T. F. Molinski, L. R. Barrows and C. M. Ireland, J. Am. Chem. Soc., 1991, 113, 4709 CrossRef CAS.
- B. S. Davidson, P. W. Ford and M. Wahlman, Tetrahedron Lett., 1994, 35, 7185 CrossRef CAS.
-
(a) V. Behar and S. J. Danishefsky, J. Am. Chem. Soc., 1993, 115, 7017–7018 CrossRef CAS;
(b) P. W. Ford, M. R. Narbut, J. Belli and B. S. Davidson, J. Org. Chem., 1994, 59, 5955 CrossRef CAS;
(c) F. D. Toste and I. W. Still, J. Am. Chem. Soc., 1995, 117, 7261 CrossRef CAS.
- B. L. Chenard, R. L. Harlow, A. L. Johnson and S. A. Vladuchick, J. Am. Chem. Soc., 1985, 107, 3871 CrossRef CAS.
-
(a) R. A. Ewin, A. M. MacLeod, D. A. Price, N. S. Simpkins and A. P. Watt, J. Chem. Soc., Perkin Trans. 1, 1997, 401 RSC;
(b) M.J. Dickens, J. P. Gilday, T. J. Mowlem and D. A. Widdowson, Tetrahedron, 1991, 47, 8621–8634 CrossRef CAS;
(c) S. E. Gibson, P. Ham, G. R. Jefferson and M. H. Smith, J. Chem. Soc., Perkin Trans. 1, 1997, 2161 RSC;
(d) S. E. Gibson, P. O'Brien, E. Rahimian and M. H. Smith, J. Chem. Soc., Perkin Trans. 1, 1999, 909 RSC.
- C. A. L. Mahaffy and P. L. Pauson, Inorg. Synth., 1990, 28, 136 CAS.
- P. J. Dickens, J. P. Gilday, J. T. Negri and D. A. Widdowson, Pure Appl. Chem., 1990, 62, 575 CAS.
- Y. Wang, K. Angermund, R. Goddard and C. Kruger, J. Am. Chem. Soc., 1987, 109, 587 CrossRef CAS.
-
(a) M. Ghavshou and D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1, 1983, 3065 RSC;
(b) J. P. Gilday and D. A. Widdowson, Tetrahedron Lett., 1986, 27, 5525 CrossRef CAS;
(c) P. J. Beswick and D. A. Widdowson, Synthesis, 1985, 492 Search PubMed;
(d) J. P. Gilday and D. A. Widdowson, J. Chem. Soc., Chem. Commun., 1986, 1235 RSC.
-
J. Tsuji, Transition Metal Reagents and Cataysts, John Wiley & Sons, Chichester, 2000, p. 371 Search PubMed.
- Crystal data: For 10aα: C11H10CrO3S2, M = 306.31, triclinic, a = 6.3675(9), b = 7.5155(11), c = 13.1748(19) Å, α = 105.757(2) β = 94.059(2), γ = 91.292(2)°, U = 604.69(15) Å3, T = 150(2)K, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, μ(Mo-Kα) = 1.281 mm−1, 5042 reflections measured, 2727 unique (Rint = 0.0198), R1 for 2313 data with I > 2σ(I) = 0.0299, wR2 for all data = 0.0790. For 10aβ: C11H10CrO3S2, M = 306.31, monoclinic, a = 6.349(3), b = 25.347(11), c = 7.531(3) Å, β = 91.629(6)°, U = 1211.5(9) Å3, T = 150(2)K, space group P21/n, Z = 4, μ(Mo-Kα) = 1.279 mm−1, 8036 reflections measured, 1899 unique (Rint = 0.1360), R1 for 1205 data with I > 2σ(I) = 0.0971, wR2 for all data = 0.2693. For 10b: C13H14CrO3S2, M = 334.36, monoclinic, a = 10.6871(12), b = 7.2672(8), c = 18.787(2) Å, β = 97.4396(13)°, U = 1446.8(3) Å3, T = 150(2)K, space group P21/n, Z = 4, synchrotron radiation, λ = 0.6719 Å, μ = 1.078 mm−1, 16190 reflections measured, 4899 unique (Rint = 0.0589), R1 for 4581 data with I > 2σ(I) = 0.0372, wR2 for all data = 0.1058. For 10e: C23H18CrO3S2, M = 458.49, triclinic, a = 9.0130(9), b = 10.2094(10), c = 12.8603(13) Å, α = 106.7566(15) β = 99.0147(16), γ = 109.5597(15)°, U = 1024.82(18) Å3, T = 150(2)K, space group P, Z = 2, μ(Mo-Kα) = 0.784 mm−1, 9059 reflections measured, 4714 unique (Rint = 0.0151), R1 for 3947 data with I > 2σ(I) = 0.0315, wR2 for all data = 0.0819. For 11e: C30H24CrO3S3, M = 580.67, monoclinic, a = 11.7886(7), b = 24.6453(15), c = 10.2118(6) Å, β = 115.6251(9)°, U = 2675.1(3) Å3, T = 150(2)K, space group P21/c, Z = 4, μ(Mo-Kα) = 0.693 mm−1, 23396 reflections measured, 6468 unique (Rint = 0.0223), R1 for 5276 data with I > 2σ(I) = 0.0383, wR2 for all data = 0.1109.
, Z = 2, μ(Mo-Kα) = 1.281 mm−1, 5042 reflections measured, 2727 unique (Rint = 0.0198), R1 for 2313 data with I > 2σ(I) = 0.0299, wR2 for all data = 0.0790. For 10aβ: C11H10CrO3S2, M = 306.31, monoclinic, a = 6.349(3), b = 25.347(11), c = 7.531(3) Å, β = 91.629(6)°, U = 1211.5(9) Å3, T = 150(2)K, space group P21/n, Z = 4, μ(Mo-Kα) = 1.279 mm−1, 8036 reflections measured, 1899 unique (Rint = 0.1360), R1 for 1205 data with I > 2σ(I) = 0.0971, wR2 for all data = 0.2693. For 10b: C13H14CrO3S2, M = 334.36, monoclinic, a = 10.6871(12), b = 7.2672(8), c = 18.787(2) Å, β = 97.4396(13)°, U = 1446.8(3) Å3, T = 150(2)K, space group P21/n, Z = 4, synchrotron radiation, λ = 0.6719 Å, μ = 1.078 mm−1, 16190 reflections measured, 4899 unique (Rint = 0.0589), R1 for 4581 data with I > 2σ(I) = 0.0372, wR2 for all data = 0.1058. For 10e: C23H18CrO3S2, M = 458.49, triclinic, a = 9.0130(9), b = 10.2094(10), c = 12.8603(13) Å, α = 106.7566(15) β = 99.0147(16), γ = 109.5597(15)°, U = 1024.82(18) Å3, T = 150(2)K, space group P, Z = 2, μ(Mo-Kα) = 0.784 mm−1, 9059 reflections measured, 4714 unique (Rint = 0.0151), R1 for 3947 data with I > 2σ(I) = 0.0315, wR2 for all data = 0.0819. For 11e: C30H24CrO3S3, M = 580.67, monoclinic, a = 11.7886(7), b = 24.6453(15), c = 10.2118(6) Å, β = 115.6251(9)°, U = 2675.1(3) Å3, T = 150(2)K, space group P21/c, Z = 4, μ(Mo-Kα) = 0.693 mm−1, 23396 reflections measured, 6468 unique (Rint = 0.0223), R1 for 5276 data with I > 2σ(I) = 0.0383, wR2 for all data = 0.1109.
- The United Kingdom Chemical Database Service
(a) D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Comput. Sci., 1996, 36, 746 CrossRef;
(b) F. H. Allen, Acta Cryst., 2002, B58, 380–388 CAS . CSD version 5.33 + 2 updates, Feb 2012.
-
(a) S. E. Gibson and E. G. Reddington, Chem. Commun., 2000, 989 RSC;
(b) A. Griffin, A. L. Blake, R. A. Ewin, W.-S. Li and N. S. Simpkins, J. Chem. Soc., Perkin Trans. 1, 1999, 3177 RSC.
- S. E. Gibson (née Thomas), N. Guillo, A. J. P. White and D. J. Williams, J. Chem. Soc., Perkin Trans. 1, 1996, 2575 RSC.
- G. Mann, D. Boranao, J. F. Hartwig, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 9205 CrossRef CAS.
- A. Alemagna, P. Del Buttero, C. Gorini, D. Landini, E. Licandro and S. Maiorana, J. Org. Chem., 1983, 48, 605 CrossRef CAS.
- SMART (2001), SAINT (2001 & 2009), and APEX 2 (2009) software for CCD diffractometers. Bruker AXS Inc., Madison, USA.
- G.M. Sheldrick, Acta Crystallogr., 2008, A64, 112 CrossRef CAS.
Footnotes |
| † CCDC reference numbers 830653–830657. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20592k |
| ‡ Author to whom correspondence about X-ray crystallography should be addressed; Tel: +44 (0)1509 228751; E-mail: m.r.j.elsegood@lboro.ac.uk. |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.