Effect of chelate ring and rigidity on Se⋯N interactions: a computational study†
Raghu Nath
Behera
* and
Arunashree
Panda
*
Department of Chemistry, Birla Institute of Technology & Science, Pilani-K. K. Birla Goa Campus, Zuarinagar, 403726, Goa, India. E-mail: rbehera@bits-goa.ac.in; panda_arunashree@yahoo.com
First published on 25th May 2012
Abstract
The intramolecular Se⋯N interactions between the selenium and the nitrogen atom in three series of o-substituted organoselenium compounds has been studied using density functional theory (DFT). The nature and the strength of these interactions and their dependence on substituents, the chelate ring size as well as the rigidity are predicted using a B3LYP/6-31G(d)/LanL2DZ method. The strength of the Se⋯N interactions is found to be dependent on the nature of Se–X (X = Cl, Br, I, SPh, CH2Ph; Ph: Phenyl) acceptor orbitals and follow the order I > Br > Cl > SPh > CH2Ph. The natural bond orbital (NBO) analysis using DFT methods points to nN→σ*Se–X electron delocalization as the key contributing factor towards Se⋯N nonbonding interactions. Both NBO and atoms in molecules (AIM) methods suggest that the intramolecular interaction in the organoselenium compounds is dominantly covalent in nature. Studies on the effect of solvent on the Se⋯N interaction show that a polar solvent stabilizes the Se⋯N interactions by shortening the Se⋯N distances.
Introduction
The importance of nonbonding interactions has been widely recognized in chemistry, biochemistry, crystallography and material sciences. Nonbonding interactions involving a divalent selenium atom have an important influence on the structures, properties, and reactivities of organoselenium compounds and have received immense attention in recent times.1 Organochalcogen compounds having intramolecular E⋯Y interactions (where E = Se or Te; Y = O, N, S etc.) play a very important role in the areas of (1) enzyme mimetics;2–4 (2) asymmetric synthesis/catalysis;5,6 (3) isolation of monomeric metal chalcogenolates, which are useful precursors for the metal organic chemical vapour deposition (MOCVD) of semiconducting materials,7 where these complexes are generally polymeric with bridging chalcogenolate ligands; (4) ‘template-free’ synthesis of novel Schiff base macrocycles containing selenium and tellurium.8The main evidence for such intramolecular nonbonding E⋯Y interactions is the distance between the two heteroatoms determined by X-ray crystallography1 and 77Se/125Te NMR.9 Computational methods based on electronic structure calculations have also been proved to be very useful in understanding the nature and magnitude of such intramolecular interactions.10 The detailed studies on such systems suggest that the interaction between a heteroatom Y (Y = N, O, S, halogens etc.) lone pair orbital and a σ*E–X orbital (E = Se/Te) leads to a weak unsymmetrical hypervalent bond. The collinear geometry between the donor atom (Y) and the E–X σ* acceptor orbital helps in maximizing the orbital interaction between the donor lone pair and the E–X anti bonding orbital and the geometry around the divalent selenium/tellurium is distorted T-shaped. The Y⋯E–X angle ranges from 165 to 180° depending on the strength of the Y⋯E interaction. The stronger the interaction, the more linear the Y⋯E–X angle. The bonding scheme of this interaction is described as three-centre four-electron [3c–4e] bonding. A pair of electrons occupies the nonbonding orbital and another pair of electrons occupies the bonding orbital of the molecular orbital. As a result, the E–X bonds get elongated and Y⋯E length shortens. Thus, one of the reasons of the origin of the Y⋯E interaction is due to the hypervalent nature of E. Although the hypervalency in divalent chalcogens increases in the order O < S < Se < Te, the basic factors responsible for the origin and the strength of such interactions are not completely understood.
In view of this, we will present results for intramolecular Se⋯N nonbonding interactions in a series of ortho substituted arylchalcogenides as studied using DFT, NBO and the quantum theory of AIM analysis. The effect of factors like substituent, chelate ring and rigidity on these interactions will also be discussed.
Computational methods
Gaussian0911 was used as the source program for geometry optimization, for NBO12 calculations, for NBO deletion analysis and for wavefunction calculation for AIM13 analysis. All the geometries were fully optimized using the hybrid B3LYP exchange correlation functional14 with a 6-31G(d) basis set, except for iodine, where we used the LanL2DZ basis set. Frequency calculations were performed for all the compounds to check (no imaginary frequencies) the stationary points as minima on the potential energy surface. Solvent effects were studied using the polarized continuum model (PCM).15 The topological analysis of electron density with Badar's theory of AIM was analyzed using AIM200016 software. Since bond path cannot be traced to the nuclei of atoms described by effective core potential,13d for calculation of wavefunctions for all iodine compounds, we ran single point calculations at the optimized geometries (at B3LYP/LanL2DZ level) using a B3LYP/6-311G* method.System studied
We studied the Se⋯N nonbonding interactions in the three series of organoselenium compounds containing an sp2 hybridized nitrogen in the oxazoline and oxazine groups as the donor and the Se–X bond as the acceptor [X = Cl, Br, I, SPh and CH2Ph; Ph: Phenyl] (Fig. 1). In the first series (1a–e), the donor N atom is part of a six membered oxazine ring, while in the second (2a–e) and the third (3a–e) series it is part of a five membered oxazoline ring. In the third series, we have a rigid aromatic ring compared to the rest. Our aim is to study (a) the effect of different substituents, X, for a given series, (b) the effect of rigidity (series 2 and 3) and chelate ring (series 1 and 2) on Se⋯N nonbonding interactions in the gas phase as well as in the presence of solvent (CHCl3). | ||
| Fig. 1 The organoselenium compounds 1a–e, 2a–e and 3a–e investigated in this study. | ||
Results and discussion
In this work, we have employed the B3LYP/6-31G(d)/LanL2DZ level of theory to study the Se⋯N nonbonding interactions. This level of DFT has been successfully employed in a number of previous theoretical studies of organoselenium compounds.10d–f To check the accuracy of our results, we compare (in Table 1) the molecular structures of selected compounds with those determined by X-ray crystallography. All the angles considered are well reproduced (maximum deviation is 3.5°). For the cases of bond lengths, most of them are well reproduced (deviation below 0.08 Å). Relatively larger deviations for the bond lengths are obtained for 2c, 3c and 3e, which may be due to intermolecular interactions in the solid state.10g Overall, the important atomic distances and angles of all the compounds in the solid state are reasonably reproduced by our calculations and are in agreement with earlier DFT work.10d Selected structural parameters of the optimized geometries of the compounds are summarized in Table 2. From the dihedral angles for the Se–X and C–N bonds, with respect to the aromatic ring (ωX–Se–C–C(c) and ωN–C–C–C(Se), respectively), it is clear that compounds 1a–e, 2a–e and 3a–c possessed planar structures (between 175°–180° and 0°–6°), while non-planar structures were obtained for 3d (165° and 26°) and 3e (134° and 70°). The N⋯Se–X angle (Table 2) is almost linear (174°–178°) for all the compounds studied except 3e (152°). In addition, the relatively shorter atomic distances between Se and N atoms (rSe⋯N) compared to sum of their van der Waals radii [rvdw(Se) +rvdw(N) = 1.90 + 1.55 = 3.45 Å]17 indicates the presence of a hypervalent Se⋯N interaction for all the studied compounds.| Compounds | r Se–C (Å) | r Se⋯N (Å) | r Se–X (Å) | θ N⋯Se–X (°) | θ C–Se–X (°) | Remark |
|---|---|---|---|---|---|---|
| 1c | 1.965 | 2.204 | 2.907 | 178.2 | 99.1 | This work |
| (1.896) | (1.971) | (2.983) | (179.7) | (97.2) | (Ref. 20) | |
| 0.069 | 0.033 | −0.076 | −1.5 | 1.9 | Difference | |
| 1d | 1.946 | 2.476 | 2.277 | 177.2 | 100.7 | This work |
| (1.937) | (2.458) | (2.25) | (176.4) | (100.4) | (Ref. 20) | |
| 0.009 | 0.018 | 0.027 | 0.8 | 0.3 | Difference | |
| 1e | 1.924 | 2.653 | 2.014 | 176.4 | 99.7 | This work |
| (1.909) | (2.686) | (1.975) | (174) | (100.2) | (Ref. 20) | |
| 0.015 | −0.033 | 0.039 | 2.4 | −0.5 | Difference | |
| 2c | 1.977 | 2.259 | 2.881 | 178.3 | 99.4 | This work |
| (1.945) | (2.133) | (2.777) | (177.7) | (97.4) | (Ref. 21) | |
| 0.032 | 0.126 | 0.104 | 0.6 | 3.0 | Difference | |
| 2e | 1.923 | 2.756 | 2.005 | 175.3 | 99.9 | This work |
| (1.916) | (2.798) | (1.966) | (175.4) | (100.2) | (Ref. 21) | |
| 0.007 | −0.042 | −0.039 | −0.1 | 0.3 | Difference | |
| 3c | 1.983 | 2.2 | 2.908 | 178.2 | 99.5 | This work |
| (—) | (2.059) | (2.83) | (175.3) | (97.5) | (Ref. 19) | |
| — | 0.141 | 0.078 | 2.9 | 2.0 | Difference | |
| 3e | 1.926 | 3.26 | 1.987 | 151.9 | 99.8 | This work |
| (1.916) | (3.384) | (1.979) | (—) | (98) | (Ref. 19) | |
| 0.01 | 0.124 | 0.008 | — | 1.8 | Difference |
| Compounds | r Se–X (Å) | r Se⋯N (Å) | θ N⋯Se–X (°) | ω X–Se–C–C(C) (°) | ω N–C–C–C(Se) (°) |
|---|---|---|---|---|---|
| 1a | 2.362 | 2.204 | 176.4 | 178.7 | 1 |
| 1b | 2.487 | 2.229 | 177.3 | 178.7 | 1.1 |
| 1c | 2.907 | 2.204 | 178.2 | 179 | 0.5 |
| 1d | 2.277 | 2.476 | 177.2 | 178.4 | 3.5 |
| 1e | 2.014 | 2.653 | 174 | 174.5 | 5.5 |
| 2a | 2.339 | 2.259 | 176.2 | −179.4 | −0.8 |
| 2b | 2.462 | 2.294 | 177.2 | −179.4 | −0.8 |
| 2c | 2.881 | 2.259 | 178.3 | −180 | 0 |
| 2d | 2.25 | 2.608 | 177.5 | −179.8 | 1.9 |
| 2e | 2.005 | 2.756 | 175.3 | −178.3 | −1.3 |
| 3a | 2.358 | 2.211 | 175.9 | 178.5 | 2.1 |
| 3b | 2.481 | 2.236 | 176.9 | 178.6 | 2.6 |
| 3c | 2.908 | 2.2 | 178.2 | 179.9 | −0.1 |
| 3d | 2.254 | 2.585 | 174.7 | 165.3 | 26.2 |
| 3e | 1.987 | 3.26 | 151.9 | 133.6 | 70.1 |
It has been suggested that the Se⋯Y (Y = O, N, F) interactions consist of an electrostatic and a covalent contribution.10d,f Thus, to estimate (approximately) the extent of electrostatic contributions to Se⋯N interactions, we calculated the electrostatic interaction energy between Se and N atoms from their charges obtained by natural population analysis (NPA). In this calculation, we assumed a point charge model so that the charge is located at the respective nuclei.10d The calculated electrostatic contribution to the Se⋯N interaction (Eel) and the NPA charges on Se and N atoms (qSe and qN, respectively) are given in Table 3.The values of Eel follow the order SPh < CH2Ph < Br < I < Cl for series 1 and 2 and CH2Ph < SPh < Br < I < Cl for 3. For a given X, the order is 2 < 3 < 1 (for X = Cl, Br, I) and for X = SPh, CH2Ph the trend is 3 < 2 < 1. This trend is nearly according to the electronegativity of X attached to the Se atom. The NPA charge on the Se atom also increases with the Eel. We observe quite good correlation (R = 0.904) of Eel with qSe (Fig. 2). However, qN does not correlate so well with Eel. These observations are very similar to those of other intramolecular chalcogen interactions.10d,e
| Compounds | q N (e) | q Se (e) | E el (kcal mol−1) | E Se…N (kcal mol−1) | E del (kcal mol−1) | △q(σ*) (e) | △q(N) (e) | Covalency factor (χ) |
|---|---|---|---|---|---|---|---|---|
| 1a | −0.557 | 0.555 | 46.58 | 63.63 | 77.26 | −0.199 | 0.203 | 0.814 |
| 1b | −0.552 | 0.486 | 39.97 | 59.08 | 73.73 | −0.189 | 0.194 | 0.798 |
| 1c | −0.596 | 0.457 | 41.04 | 72.95 | 97.57 | −0.177 | 0.224 | 0.814 |
| 1d | −0.545 | 0.358 | 26.17 | 22.85 | 27.92 | −0.105 | 0.1 | 0.637 |
| 1e | −0.536 | 0.449 | 30.13 | 11.58 | 13.32 | −0.051 | 0.05 | 0.521 |
| 2a | −0.555 | 0.544 | 44.39 | 50.96 | 64.62 | −0.185 | 0.189 | 0.778 |
| 2b | −0.552 | 0.472 | 37.72 | 45.28 | 59.22 | −0.173 | 0.177 | 0.756 |
| 2c | −0.603 | 0.437 | 38.74 | 58.97 | 82.74 | −0.18 | 0.214 | 0.778 |
| 2d | −0.541 | 0.352 | 24.25 | 13.44 | 17.05 | −0.074 | 0.072 | 0.55 |
| 2e | −0.53 | 0.452 | 28.87 | 7.5 | 9.12 | −0.036 | 0.037 | 0.454 |
| 3a | −0.555 | 0.534 | 44.52 | 59.41 | 74.28 | −0.194 | 0.202 | 0.81 |
| 3b | −0.551 | 0.465 | 38.05 | 54.83 | 70.67 | −0.183 | 0.192 | 0.793 |
| 3c | −0.608 | 0.444 | 40.75 | 71.42 | 98.04 | −0.177 | 0.227 | 0.817 |
| 3d | −0.539 | 0.327 | 22.64 | 11.89 | 14.75 | −0.064 | 0.064 | 0.565 |
| 3e | −0.511 | 0.358 | 18.64 | — | — | — | — | 0.124 |
 | ||
| Fig. 2 Plot of the magnitude of the ionic contribution, Eel (calculated by considering point charges for Se and N atoms), with the charge on the selenium atom, (qSe). | ||
To estimate the covalent contribution to the Se⋯N interactions, we used NBO second order perturbation and NBO deletion analysis. The NBO method, based on local block eigenvectors of a one-particle density matrix, predicts bonds and lone pairs of an optimized Lewis structure of a molecule from ab initio wavefunctions.12 Delocalization of electron density between occupied (bond or lone pair) NBOs and formally unoccupied (antibonding or Rydberg) NBOs corresponds to a stabilizing interaction. In the NBO deletion technique, a selected set of NBOs from a donor–acceptor pair is deleted and the energy change (loss of delocalization energy) associated with this process provides information on the interaction between the respective orbitals.
We studied the orbital interactions between the lone pairs of the nitrogen atom (nN) and the antibonding orbital of the Se–X bond (σ*Se⋯X). Our NBO study indicated the absence of nN→σ*Se⋯X orbital interaction for the compound 3e. The NBO second order perturbation energies for nN→σ*Se⋯X orbital interactions (ESe⋯N) are presented in Table 3. The values of ESe⋯N range from 11.58 to 72.95 kcal mol−1 for 1a–e, 7.5 to 58.97 kcal mol−1 for 2a–e and 11.89 to 71.42 kcal mol−1 for 3a–d. Also, these interaction energies increase in the order CH2Ph < SPh < Br < Cl < I for all series and for a given X, the order is 2 < 3 < 1 (except for X = SPh where it is 3 < 2 < 1). This trend is more or less according to the electrophilicity of the Se–X bond and the nucleophilicity of the N atom for the Se⋯N interactions. The NBO deletion energies (Edel), which provide information on the orbital interaction energies associated with the Se atom and the chelating N atom, follow the same trend as those of the ESe⋯N. These factors clearly suggest that the nN→σ*Se⋯X orbital interactions contribute significantly to the stability of Se⋯N interactions in these compounds.
It is known that a strong Se⋯N interatomic distance causes shortening of the Se⋯N interatomic distance and an increase in the linearity of the N⋯Se–X alignment.18 This indeed is observed from the present calculations. However, the X-ray study of selenenyl halides shows that the bond angles N⋯Se–X (X = Cl, Br or I) deviate from 180° as the Se⋯N interaction increases (e.g.1b: Se⋯N = 2.052 Å and N⋯Se–Br = 175.15°; 1c: Se⋯N = 2.059 Å, N⋯Se–I = 175.26°).19–21 This is probably due to the presence of intermolecular interactions between Se and X (X = Cl, Br or I). The intermolecular interaction increases when the electronegativity of the halogen atom decreases and hence is greatest for X = I. The weakest Se⋯N interaction is observed in all the aryl benzyl selenides compared to the selenenyl halides and selenenyl sulphides, maybe due to the presence of the benzyl group, which cannot accept electron density from the selenium. This is in agreement with the experimental observations (Se⋯N; 1e: 3.3842 Å; 3e: 2.798 Å).19,21
The value of ESe⋯N depends on at least two factors: (a) the distance between the Se and N atom (rSe⋯N) and (b) the donor ability of the N atom. To find out the contribution of each of these factors, we reoptimized the geometries of the compounds at the fixed Se⋯N distances and then calculated the ESe⋯N of all the compounds (see Table 4). It is clear from Table 4 that the values of ESe⋯N are highly dependent on rSe⋯N and almost comparable ESe⋯N values were obtained for a fixed Se⋯N distance for series 1, 2 and 3 (for a given X). The relatively higher ESe⋯N values of compound 1, compared to those of 3 and 2, may be due to the difference in the size of chelate rings containing a donor nitrogen atom.
| X | r Se⋯N (Å) | E Se⋯N (kcal mol−1) | ||
|---|---|---|---|---|
| 1 | 2 | 3 | ||
| Cl | 2.211 | 62.4 | 59.04 | 59.41 |
| Br | 2.236 | 57.93 | 54.43 | 54.83 |
| I | 2.2 | 73.7 | 70.35 | 71.42 |
| SPh | 2.585 | 16.43 | 14.48 | 11.89 |
| CH2Ph | 3.26 | 0.81 | 0.85 | — |
The values for ESe⋯N (and Edel) decrease with an increase in Se⋯N distance (rSe⋯N) for all the compounds. The single correlation curve between ESe⋯Nversus rSe⋯N (Fig. 3) and Edelversus rSe⋯N (Fig. 4) clearly show that the nature of the interactions in all the studied compounds are the same. The changes in orbital occupancies of the lone pair of nitrogen (nN) and σ*Se⋯X orbitals (△q = qdel−q) during NBO deletion analysis are displayed in Table 3. The significant decrease of charge of the σ*Se⋯X orbitals and a similar increase of charge for the nN orbitals upon NBO deletion suggest that the nature of orbital interaction is electron delocalization from the lone pair of the N atom to the anti-bonding orbital of Se–X (i.e. nN→σ*Se⋯X). This dominant contribution of nN→σ*Se⋯X charge transfer in the intramolecular Se⋯N bonding is supported by relatively high values (up to 81%) of covalency factor χ for the intramolecular coordination Se⋯N bond (Table 3). The values of χ follow the same trend as ESe⋯N and Edel (CH2Ph < SPh < Br < Cl < I for all series and for a given X, the order is 2 < 3 < 1) and show a single correlation curve (Fig. 5) for all the compounds.
 | ||
| Fig. 3 Variation of NBO second order perturbation energies (ESe⋯N) with the Se⋯N distances (rSe⋯N). | ||
 | ||
| Fig. 4 Variation of NBO deletion energies (Edel) with the Se⋯N distances (rSe⋯N). | ||
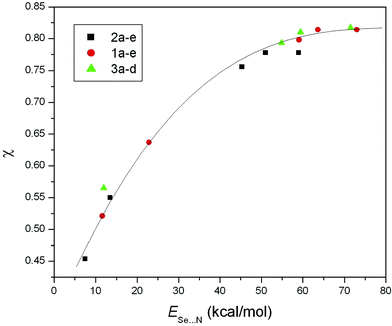 | ||
| Fig. 5 Plot of covalency factor (χ) with the NBO second order perturbation energies (ESe⋯N). | ||
The strength of the Se⋯N interactions has been observed in the order CH2Ph < SPh < Br < Cl ≈ I. The ionic contribution (Eel) decreases in the order Cl > I > Br > SPh > CH2Ph and the covalent contribution (ESe⋯N) decreases in the order I > Cl > Br > SPh > CH2Ph (Table 3). However, the total strength (ionic + covalent) decreases in the order I ≈ Cl > Br > SPh > CH2Ph. The observed trend for halides may be due to the negligence of intermolecular interaction during our theoretical calculations. These interactions, which are often present in the organoselenenyl halides in the solid state, have a significant affect on the Se⋯N interactions. However, the Se⋯N bond distances of organoselenenyl halides 1a [1.9695 Å(av.)],201b (1.975 Å)20 and 1c (1.971 Å)20 from the X-ray studies follow the trend Br > Cl ∼ I. These halides either have very weak or no intermolecular interactions. Also for organoselenenyl halides 3b (2.052 Å)19 and 3c (2.059 Å)19 (absence of intermolecular interactions due to the presence of a sterically more demanding group), Se⋯N bond distances are very close. Furthermore, for organoselenenyl halides of 2 (where the intermolecular interaction is greatest among the three series studied here), the trend of Se⋯N distances is Cl (2a, 2.052 Å) < Br (2b, 2.063 Å) < I (2c, 2.133 Å).
In summary, introduction of rigidity (for 3) onto the backbone of 2, or replacing the five membered oxazoline ring in 2 by a six membered oxazine ring in 1, enhances the strength of Se⋯N nonbonding interactions; the latter effect seems to be more dominant. This is in accordance with the experimental findings. For example, the Se–N distances in 1a, 1b and 1c are 1.9695 (average), 1.975 (average) and 1.971 Å, respectively, whereas for 2a, 2b and 2c they are 2.052(2), 2.063(3) and 2.133 Å, respectively. Similar trends have also been observed for similar organotellurium compounds.22
Atoms in molecules (AIM) analysis
As an alternative method to study the nonbonding Se⋯N interaction, we used Bader's theory of AIM,13 which involves topological analysis of electron density between two interacting atoms. According to this theory, atoms that are chemically bonded have their nuclei linked by a (single) bond path (a single line of locally maximum electron density) and they share a bond critical point (a point on the bond path with the lowest value of the electron density). The presence of a (3, −1) bond critical point (BCP) along the bond path (BP) is a universal indicator of chemical bonding. Chemical bonding interactions are characterized and classified according to the properties of the electron densities (ρb), Laplacian of electron density (∇2ρb) and the total energy densities (Hb) at the BCP. In general, ρb is greater than 0.20 au in a shared (covalent) bond and less than 0.10 au in a closed-shell interaction (ionic, van der Waals, hydrogen bonding, etc.). The Laplacian of electron density also provides valuable information. A positive value of ∇2ρb indicates a closed-shell bonding while a negative value indicates covalent bonding. In strongly polar bonding (e.g. C–X, where X = O, N, F) there is a significant accumulation of electron density between the nuclei, as in all shared interactions, but the Laplacian in this type of bonding can be of either sign.13c The total electronic energy density at the BCP (Hb) is negative for interactions with significant sharing of electrons, its magnitude reflecting the ‘‘covalence’’ of the interaction.The presence of BCP between Se and N atoms and the ring critical point (RCP) for the five membered ring formed with the phenyl ring due to Se⋯N interaction was observed for all the studied compounds, except 3e. The electron density at RCP (ρrcp), along with the other AIM parameters (ρSe⋯N, ∇2ρSe⋯N and HSe⋯N) for the Se⋯N interaction, are given in Table 5. The values of ρSe⋯N for the studied compounds range from 0.0237 to 0.0736 e Å−3, which are in between a typical covalent bond (e.g. ρC–C ≈ 0.24 e Å−3) and that of hydrogen bond (ρH–Bond ≈ 0.002–0.04 e Å−3). Similar values were reported for Se⋯N interactions by Sharma et al.10d The values of ρSe⋯N follow the trend CH2Ph < SPh < Br < Cl < I for all series, and 2 < 3 < 1 for a given X. This trend is similar to that for ESe⋯N and Edel. This is not surprising because a covalent interaction in AIM theory is related to orbital interactions in NBO analysis. The values of ESe⋯N and Edel correlate nicely with ρSe⋯N (Fig. 6 and 7, respectively), indicating a dominant covalent contribution in the Se⋯N interactions. The values of ρrcp lie in the range 0.014 to 0.0248 e Å−3 and increase in the order CH2Ph < SPh < I < Br < Cl for all three series, and for a given X: 2 < 3 < 1. The values of ∇2ρSe⋯N obtained (Table 5) are all positive (range: 0.07 to 0.138 e Å−5) and follow the overall trend CH2Ph < SPh < I < Br < Cl for series 2, 3 and CH2Ph < SPh < Br < Cl ≈ I for series 1; and for a given X: 1 < 2 < 3. All positive values indicate a dominant electrostatic character, which is in contrast to “dominant covalent contribution” as predicted by NBO analysis. Thus, we analyze the total energy density at the critical point (HSe⋯N), which is known to be more reliable in identifying the type of interactions (electrostatic or covalent).23,10d The calculated values of HSe⋯N (Table 5) are mostly negative (except 1d, 1e, 2d and 2e) indicating dominant covalent interaction, as predicted by NBO analysis. The values of HSe⋯N become more negative for stronger Se⋯N interactions (shorter rSe⋯N) and are positive with weaker Se⋯N interactions. This correlation is quite evident from Fig. 8.
| Compounds | ρ Se⋯N (e Å−3) | ρ rcp (e Å−3) | ∇2ρSe⋯N (e Å−5) | H Se⋯N (e Å−4) |
|---|---|---|---|---|
| 1a | 0.0736 | 0.0248 | 0.123 | −0.0076 |
| 1b | 0.0706 | 0.0242 | 0.115 | −0.0059 |
| 1c | 0.0752 | 0.0237 | 0.124 | −0.0121 |
| 1d | 0.043 | 0.0193 | 0.076 | 0.0006 |
| 1e | 0.0302 | 0.0163 | 0.07 | 0.0012 |
| 2a | 0.0641 | 0.023 | 0.133 | −0.0112 |
| 2b | 0.0602 | 0.022 | 0.127 | −0.0092 |
| 2c | 0.0659 | 0.0223 | 0.127 | −0.0179 |
| 2d | 0.0319 | 0.016 | 0.087 | 0.0002 |
| 2e | 0.0237 | 0.0139 | 0.085 | 0.0004 |
| 3a | 0.0706 | 0.0242 | 0.141 | −0.015 |
| 3b | 0.0677 | 0.0236 | 0.137 | −0.0132 |
| 3c | 0.0738 | 0.0234 | 0.132 | −0.0171 |
| 3d | 0.0338 | 0.0178 | 0.089 | −0.0001 |
 | ||
| Fig. 6 Correlation plot of the NBO second order perturbation energies (ESe⋯N) with the electron density (ρSe⋯N) at the bond critical point of the Se⋯N interaction. ρSe⋯N is in units of e Å−3. | ||
 | ||
| Fig. 7 Correlation plot of the NBO deletion energies (Edel) with the electron density (ρSe⋯N) at the bond critical point of the Se⋯N interaction. ρSe⋯N is in units of e Å−3. | ||
 | ||
| Fig. 8 Variation of AIM total energy density (HSe⋯N) at the bond critical point with the Se⋯N distances (rSe⋯N). HSe⋯N are given in units of e Å−4. | ||
The AIM dual-parameter analysis (plot of HSe⋯Nversus ∇2ρSe⋯N) for weak to strong interaction is shown in Fig. 9. Most of the points fall in the fourth quadrant (HSe⋯N < 0, ∇2ρSe⋯N > 0; more shared-shell character) indicating strong Se⋯N interaction. A few points lie on first quadrant (HSe⋯N > 0, ∇2ρSe⋯N > 0), indicating that the electron density at BCP is neither stabilized nor concentrated, i.e. weak Se⋯N interactions.
 | ||
| Fig. 9 Variation of AIM total energy density (HSe⋯N in units of e Å−4) with the Laplacian of the electron density (in units of e Å−5) at the bond critical point. | ||
Detailed inspection of the AIM analysis reveals some interesting facts. Several intramolecular interactions have been observed for all the systems discussed in the present study (Fig. 1) from the AIM analysis through BCP and RCP.24 These interactions have not been reported for the solid state structures of these compounds19–21 and are discussed below.
(A) The common features that have been observed for all cases i.e. for 1–3 (except 3e) are; (1) the presence of Se⋯N interactions and (2) the presence of interaction between the substituents X and the aryl hydrogen ortho to the Se–X group. Similar observations have been made by Sunoj et al. earlier.10e A representative diagram of 1a showing both Se⋯N and Se⋯H interactions is given in Fig. 10.24
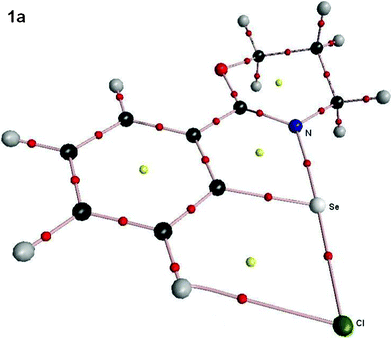 | ||
| Fig. 10 Molecular plot of 1a (X = Cl) showing Se⋯N and Se⋯H bond critical points (small red spheres) and the corresponding ring critical points (yellow spheres) arising due to intramolecular interactions. | ||
(B) Besides these above interactions, the existence of an additional interaction between the Se atom and the aryl hydrogen ortho to the S atom from the SPh group could be identified through Se⋯H BCPs and the corresponding RCPs for compounds 1d, 2d and 3d. One of the representative cases is shown in Fig. 11.
 | ||
| Fig. 11 Molecular plot of 2d (X = SPh) showing the Se⋯N, S⋯H and Se⋯H bond critical points (small red spheres) and the corresponding ring critical points (yellow spheres) arising due to intramolecular interactions. | ||
(C) Furthermore, another interaction in addition to (A) between oxygen and aryl hydrogen ortho to the oxazoline ring is predicted for the compound 1e.24
(D) Interestingly, along with the interactions described in (A), interactions between the oxygen and carbon atom (of the phenyl ring attached to the naphthyl ring) have been observed for the sterically hindered compounds 3a–d.24
(E) More interestingly, although compound 3e (Fig. 12) does not have a Se⋯N interaction, existence of several other intramolecular interactions have been predicted by our AIM studies. (1) The interaction between the hydrogen atom of the methyl group of the oxazoline ring and the hydrogen atom of the methyl group of the phenyl ring perpendicular to the naphthyl ring results in the formation of a 12 membered chelate ring. (2) The hydrogen atom of the other methyl group of the oxazoline ring interacts with the selenium atom forming a eight membered chelate ring. (3) A 6-membered chelate ring arises from the interaction of the aryl hydrogen ortho to the –CH2– group and the oxygen atom of the oxazoline ring. (4) Additionally, another 5-membered chelate ring results from the interaction of the same aryl hydrogen ortho to the –CH2– group and the selenium atom.
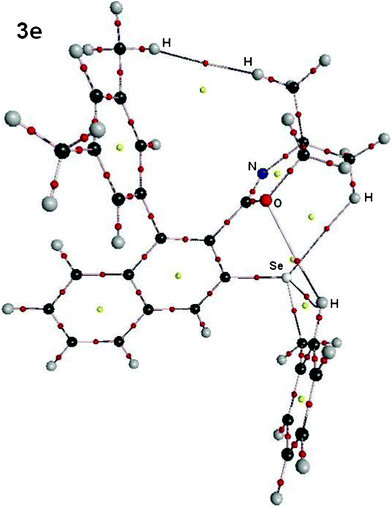 | ||
| Fig. 12 Molecular plot of 3e (X = CH2Ph) showing the H⋯H, O⋯H, Se⋯H bond critical point (small red spheres) and the corresponding ring critical points (yellow spheres) arising due to intramolecular interactions. | ||
To sum up, the AIM analysis reveals that the electron density at the Se⋯N BCP shows a very good correlation with the computed intramolecular interaction energies (from NBO analysis) and hence suggests a dominant covalent nature of the intamolecular interactions. Also, several types of intramolecular interactions other than Se…N are predicted.
Solvent effect
In order to study the effect of a polar solvent on Se…N interactions, we use the Tomasi's PCM at the B3LYP/6-31G(d)/LanL2DZ level in CHCl3 solvent. The values reported in Table 6 show that the Se…N distances are much shorter and the values of ESe…N are larger than those of the gas phase values. This indicates that the Se⋯N interactions in polar solvent become stronger than those of the gas phase. This is in contrast to the usual belief that a compound having an intramolecular Se⋯N interaction (having less exposed polar surface in solution) would destabilize the structure by weakening the Se⋯N interaction. A similar observation was also obtained recently by Sharma et al.10d| Compounds | r Se⋯N (Å) | r Se⋯N (CHCl3) (Å) | E Se⋯N (kcal mol−1) | E Se⋯N (CHCl3) (kcal mol−1) |
|---|---|---|---|---|
| 1a | 2.204 | 2.092 | 63.63 | 93.36 |
| 1b | 2.229 | 2.13 | 59.08 | 82.02 |
| 1c | 2.204 | 2.088 | 72.95 | 105.48 |
| 1d | 2.476 | 2.409 | 22.85 | 28.31 |
| 1e | 2.653 | 2.665 | 11.58 | 11.16 |
| 2a | 2.259 | 2.123 | 50.96 | 81.62 |
| 2b | 2.294 | 2.171 | 45.28 | 68.85 |
| 2c | 2.259 | 2.103 | 58.97 | 97.63 |
| 2d | 2.608 | 2.574 | 13.44 | 15.06 |
| 2e | 2.756 | 2.776 | 7.5 | 7.05 |
| 3a | 2.211 | 2.092 | 59.41 | 90.52 |
| 3b | 2.236 | 2.132 | 54.83 | 78.25 |
| 3c | 2.2 | 2.074 | 71.42 | 108.15 |
| 3d | 2.585 | 2.536 | 11.89 | 14.32 |
| 3e | 3.26 | 3.151 | — | 0.68 |
Conclusions
A comparison of the Se⋯N interaction in the organoselenium compounds derived from 2-phenyl-5,6-dihydro-4H-1,3-oxazine and 4,4-dimethyl-2-phenyl-oxazoline and 2-[1-(3,5-dimethylphenyl)-2-naphthyl]-4,5-dihydro-4,4-dimethyl-1,3-oxazole have been made to study the effect of rigidity and ring size on Se⋯N interaction. In line with earlier studies, the origin of Se⋯N interaction was identified to be due to orbital interaction involving the delocalization of nitrogen lone pair electrons into the suitably aligned Se–X antibonding orbital (nN→σ*Se–X) and both electrostatic and covalent contributions are important for this interaction. Computed interaction energies (NBO second order perturbation and NBO deletion energies) revealed that the extent of interaction depends strongly on the nature of substituent attached to selenium (i.e. on the nature of the Se–X acceptor orbital) and roughly follows the order CH2Ph < SPh < Br < Cl < I. For a given X, the strength of Se⋯N interaction follows the order 2 < 3 < 1. Thus, introduction of a naphthyl ring in place of phenyl (3versus2) as well as introduction of an oxazine ring in place of an oxazoline ring (1versus2) favors (strengthens) the Se⋯N interaction. However, the latter (ring size) effect is stronger than the former (steric effect) one. The strength of this interaction is highly dependent on the distance between the Se and N atoms (almost similar values of interaction energies are obtained for same Se⋯N distances), which is the outcome of the specific structure of a compound. The quantified interactions were found to correlate well with the structural parameters as well as topological properties of electron density at the Se⋯N BCP obtained using AIM analysis. The AIM analysis suggests that the nature of Se⋯N interaction is predominantly covalent. Larger Se⋯N interaction energies and shorter Se⋯N distances in polar solvent (CHCl3) compared to those of gas phase values indicate that the Se⋯N interaction is favorable in polar solvent.Acknowledgements
AP thanks DST, New Delhi for financial support under the WOS-A scheme (SR/WOS-A/CS-23/2010). The support from BITS, Pilani-K. K. Birla Goa Campus is gratefully acknowledged. We thank Dr A. Chattopadhyay for providing part of the computing resources.References
- A. J. Mukherjee, S. S. Zade, H. B. Singh and R. B. Sunoj, Chem. Rev., 2011, 110, 4357–4416 CrossRef and references therein.
- (a) T. G. Chasteen and R. Bentley, Chem. Rev., 2003, 103, 1 CrossRef CAS; (b) G. Mugesh, W. W. du Mont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS.
- (a) G. Mugesh and H. B. Singh, Chem. Soc. Rev., 2000, 29, 347 RSC; (b) G. Mugesh, A. Panda, N. S. Punekar, H. B. Singh and R. J. Butcher, Chem. Commun., 1998, 2227 RSC; (c) G. Mugesh, A. Panda, N. S. Punekar, H. B. Singh and R. J. Butcher, J. Am. Chem. Soc., 2001, 123, 839 CrossRef CAS; (d) M. Iwaoka and S. Tomoda, J. Chem. Soc., Chem. Commun., 1992, 1165–1167 RSC; (e) M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1994, 116, 2557–2561 CrossRef CAS; (f) T. Wirth, S. Häuptli and M. Leuenberger, Tetrahedron: Asymmetry, 1998, 9, 547–550 CrossRef CAS; (g) T. G. Back and Z. Moussa, J. Am. Chem. Soc., 2002, 124, 12104–12105 CrossRef CAS; (h) C. A. Bayse, R. A. Baker and K. N. Ortwine, Inorg. Chim. Acta, 2005, 358, 3849–3854 CrossRef CAS; (i) B. K. Sarma and G. Mugesh, Inorg. Chem., 2006, 45, 5307–5314 CrossRef CAS; (j) K. P. Bhabak and G. Mugesh, Chem.–Eur. J., 2007, 13, 4594–4601 CrossRef CAS; (k) B. K. Sarma and G. Mugesh, Org. Biomol. Chem., 2008, 6, 965–974 RSC; (l) B. K. Sarma and G. Mugesh, Chem.–Eur. J., 2008, 14, 10603–10614 CrossRef CAS; (m) G. Mugesh and W.-W. du Mont, Chem.–Eur. J., 2001, 7, 1365 CrossRef CAS; (n) L. Engman, D. Stern, I. A. Cotgreave and C. M. Andersson, J. Am. Chem. Soc., 1992, 114, 9737 CrossRef CAS; (o) Y. You, K. Ahsan and M. R. Detty, J. Am. Chem. Soc., 2003, 125, 4918 CrossRef CAS; (p) L. Engman, D. Stern, M. Pelcman and C. M. Andersson, J. Org. Chem., 1994, 59, 1973 CrossRef CAS; (q) K. Vessman, M. Ekstorm, M. Verglund, C. M. Andersson and L. Engman, J. Org. Chem., 1995, 60, 4461 CrossRef CAS.
- G. Mugesh, A. Panda, S. Kumar, S. D. Apte, H. B. Singh and R. Butcher, Organometallics, 2002, 21, 884 CrossRef CAS.
- (a) K. Fujita, M. Iwaoka and S. Tomoda, Chem. Lett., 1994, 923–926 CrossRef CAS; (b) K. Fujita, K. Murata, M. Iwaoka and S. Tomoda, Tetrahedron, 1997, 53, 2029–2048 CrossRef CAS; (c) M. Iwaoka and S. M. Tomoda, J. Chem. Soc., Chem. Commun., 1992, 1165 RSC; (d) T. Wirth, Tetrahedron Lett., 1995, 36, 7849 CrossRef CAS; (e) R. Kaur, H. B. Singh and R. P. Patel, J. Chem. Soc., Dalton Trans., 1996, 2719 RSC; (f) S. Fukuzawa, K. Takahashi, H. Kato and H. Yamazaki, J. Org. Chem., 1997, 62, 7711 CrossRef CAS; (g) S. Uemura, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 219, 136–138 Search PubMed; (h) K. Hiroi, Y. Suzuki and I. Abe, Tetrahedron: Asymmetry, 1999, 10, 1173 CrossRef CAS; (i) H. Ehara, M. Noguchi, S. Sayama and T. Onami, J. Chem. Soc., Perkin Trans. 1, 2002, 1429 Search PubMed; (j) C. Bolm, M. Kesselgraber, A. Grehz, N. Hermanns and J. P. Hilderbrand, New J. Chem., 2001, 25, 13 RSC; (k) A. L. Braga, M. W. Paixao, D. S. Ludtke, C. C. Silveira and O. E. D. Rodrigues, Org. Lett., 2003, 5, 2635 CrossRef CAS.
- (a) Organoselenium Chemistry: A Practical Approach, T. G. Back., Ed.; Oxford University Press, New York, 1999 Search PubMed; (b) T. Wirth, Tetrahedron, 1999, 55, 1 CrossRef CAS; (c) T. Wirth, Angew. Chem., Int. Ed., 2000, 39, 3740 CrossRef CAS; (d) Organoselenium Chemistry: Modern Development in Organic Synthesis, T. Wirth, Ed; Springer-Verlag: Berlin, Germany, 2000 Search PubMed; (e) Y. Nishibayashi and S. Uemura, Top. Curr. Chem., 2000, 208, 201 CrossRef CAS Also see.; (f) L. Uehlin and T. Wirth, Org. Lett., 2001, 3, 2931 CrossRef CAS; (g) M. Uchiyama, M. Oka, S. Harai and A. Ohta, Tetrahedron Lett., 2001, 42, 1931 CrossRef CAS; (h) T. G. Back, Z. Moussa and M. Parvez, J. Org. Chem., 2002, 67, 499 CrossRef CAS; (i) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Tetrahedron: Asymmetry, 2002, 11, 4645 CrossRef; (j) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Chem.–Eur. J., 2002, 8, 1118 CrossRef CAS; (k) L. Uehlin, G. Fragale and T. Wirth, Chem.–Eur. J., 2002, 8, 1125 CrossRef CAS; (l) S. S. Khokhar and T. Wirth, Angew. Chem., Int. Ed., 2004, 43, 631 CrossRef CAS; (m) G. Fragale and T. Wirth, Eur. J. Org. Chem., 1998, 1361–1369 CrossRef CAS; (n) G. Fragale, M. Neuburger and T. Wirth, Chem. Commun., 1998, 1867–1868 RSC; (o) M. Tiecco, L. Testaferri, C. Santi, C. Tomassini, F. Marini, L. Bagnoli and A. Temperini, Angew. Chem., Int. Ed., 2003, 42, 3131 CrossRef CAS.
- (a) A. N. Gleizes, Chem. Vap. Deposition, 2000, 6, 155 CrossRef CAS; (b) M. Lazell, P. O'Brien, D. J. Otway and J.-H. Park, J. Chem. Soc., Dalton Trans., 2000, 4479 RSC; (c) J. D. Sandman, L. Li, S. Tripathy, J. C. Stark, L. A. Acampora, B. M. Foxman and L. A. Foxman, Organometallics, 1994, 13, 348 CrossRef; (d) P. J. Bonasia and J. Arnold, J. Organomet. Chem., 1993, 449, 147 CrossRef CAS; (e) Y. Cheng, T. J. Emge and G. Brennan, Inorg. Chem., 1994, 33, 3711 CrossRef CAS; (f) Y. Cheng, T. J. Emge and G. Brennan, J. Inorg. Chem., 1996, 35, 3342 Search PubMed; (g) Y. Cheng, T. J. Emge and G. Brennan, Inorg. Chem., 1996, 35, 7339 CrossRef CAS; (h) G. Mugesh, H. B. Singh and R. J. Butcher, Inorg. Chem., 1998, 37, 2663 CrossRef CAS; (i) G. Mugesh, H. B. Singh and R. J. Butcher, J. Organomet. Chem., 1999, 577, 243 CrossRef CAS; (j) K. Kandasamy, H. B. Singh and G. Wolmershäuser, Inorg. Chim. Acta, 2005, 358, 207 CrossRef CAS.
- (a) A. Panda, Coord. Chem. Rev., 2009, 253, 1056–1098 CrossRef CAS; (b) A. Panda, Coord. Chem. Rev., 2009, 253, 1947–1965 CrossRef CAS; (c) A. Panda, S. Panda, K. Srivastava and H. B. Singh, Inorg. Chim. Acta, 2011, 372, 17–31 CrossRef CAS.
- A. Panda and H. B Singh, NMR of Organoselenium and Organotellurium Compounds, Ed. Z. Rappoport, Chichester, Wiley, 2012 (under revision) Search PubMed.
- (a) M. Iwaoka and S. Tomoda, J. Am. Chem. Soc., 1996, 118, 8077 CrossRef CAS; (b) H. Komatsu, M. Iwaoka and S. Tomoda, Chem. Commun., 1999, 205–206 RSC; (c) M. Iwaoka, H. Komatsu and S. Tomoda, Chem. Lett., 1998, 969 CrossRef CAS; (d) B. K. Sharma and G. Mugesh, ChemPhysChem, 2009, 10, 3013 CrossRef; (e) D. Roy and R. B. Sunoj, J. Phys. Chem. A, 2006, 110, 5942 CrossRef CAS; (f) M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2004, 126, 5309 CrossRef CAS; (g) M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 1902 CrossRef CAS; (h) S. K. Tripathi, U. Patel, D. Roy, R. B. Sunoj, H. B. Singh, G. Wolmershäuser and R. J. Butcher, J. Org. Chem., 2005, 70, 9237 CrossRef CAS.
- Gaussian 09, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2010.
- (a) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss, J. E. Carpenter and F. Weinhold, NBO version 3.1 Search PubMed.
- (a) R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed; (b) P. Popelier, Atoms in Molecules: An Introduction, Pearson, Harlow, 2000 Search PubMed; (c) R. J. Gillespie, P. L. A. Popelier, Chemical Bonding and Molecular Geometry, Oxford University Press, New York, 2001 Search PubMed; (d) C. F. Matta, R. J. Boyd, The Quantum Theory of Atoms in Molecules, Wiley-VCH, 2007 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- F. Biegler-Konig, J. Schonbohm and D. Bayles, J. Comput. Chem., 2001, 22, 545–559 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- M. Iwaoka and S. Tomoda, J. Org. Chem., 1995, 60, 5299 CrossRef CAS.
- K. Kandasamy, S. Kumar, H. B. Singh, R. J. Butcher and K. T. Holman, Eur. J. Inorg. Chem., 2004, 1014 CrossRef CAS.
- S. Kumar, K. Kandasamy, H. B. Singh, G. Wolmershäuser and R. J. Butcher, Organometallics, 2004, 23, 4199 CrossRef CAS.
- G. Mugesh, A. Panda, H. B. Singh and R. J. Butcher, Chem.–Eur. J., 1999, 5, 1411 CrossRef CAS.
- (a) S. D. Apte, S. S. Zade, H. B. Singh and R. J. Butcher, Organometallics, 2003, 22, 5473 CrossRef CAS; (b) T. A. Hamor, H. Chen, W. R. McWhinnie, S. L. W. McWhinnie and Z. Majeed, J. Organomet. Chem., 1996, 523, 53 CrossRef CAS; (c) S. Kumar, H. B. Singh and G. Wolmershäuser, J. Organomet. Chem., 2005, 690, 3149 CrossRef CAS.
- D. Cremer and E. Kraka, Angew. Chem., 1984, 96, 612–614 CrossRef CAS.
- See Supporting Information for further details†.
Footnote |
| † Electronic supplementary information (ESI) available: B3LYP/6-31G(d)/LanL2DZ optimized geometries and AIM molecular plots of all the studied compounds. See DOI: 10.1039/c2ra20591b |
| This journal is © The Royal Society of Chemistry 2012 |