Stability and phase transition of nanoporous rutile TiO2 under high pressure†
Quanjun
Li
a,
Ran
Liu
a,
Bingbing
Liu
*a,
Lin
Wang
a,
Kai
Wang
a,
Dongmei
Li
a,
Bo
Zou
a,
Tian
Cui
a,
Jing
Liu
b,
Zhiqiang
Chen
c and
Ke
Yang
d
aState Key Laboratory of Superhard Materials, Jilin University, Changchun, China. E-mail: liubb@jlu.edu.cn
bBeijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing, China
cGeoScience Department, Stony Brook University, Stony Brook, NY, USA
dShanghai Synchrotron Radiation Facility, Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, China
First published on 30th July 2012
Abstract
The high-pressure behavior of nanoporous rutile TiO2 was studied at room temperature using in situ synchrotron X-ray diffraction and Raman spectroscopy. It was found that the nanoporous rutile TiO2 starts to transform to the baddeleyite phase at a pressure of 10.8 GPa. The phase transition pressure is obviously different from those of rutile nanoparticles and the bulk solid. The rutile phase transforms into the baddeleyite phase completely when the pressure reaches beyond 26.1 GPa. Upon decompression, the baddeleyite phase transforms into the α-PbO2 phase. The bulk modulus obtained for nanoporous rutile TiO2 is 204(4) GPa. The nanoporous structure remains structurally intact during the compression–decompression cycle and thus shows excellent stability. We suggest that these high-pressure behavior characteristics of nanoporous rutile TiO2 are due to its unique nanoporous microstructure. Our study indicates that high pressure may be a powerful tool for researching the physicochemical properties of nanoporous materials, and also provides a potential method for preparing novel high-pressure phase nanoporous materials.
Introduction
Nanoporous materials, which possess high specific surface area, large pore volume, uniform pore size, and rich surface chemistry, are an important class of nanostructural materials.1–6 These materials show great opportunities for a new generation of functional materials with improved and tailorable properties for applications in energy storage, sensors, catalysis, photocatalysis, biotechnology, etc. Among these materials, nanoporous TiO2 materials have proved to possess excellent properties in terms of photocatalysis, lithium ion batteries, solar cells, etc.7–10In general, the properties of nanoporous materials largely depend on its crystal phase, porosity, surface area, morphology and architecture. Therefore, structural stability of nanoporous materials is one of the crucial factors for their application. It is well known that high pressure provides a powerful method for researching the mechanical properties and structural phase transitions of materials. Recently, a high-pressure technique has been employed to study nanoporous materials.11–14 Mechanical properties of the dense zinc framework Zn(Im)2 and its lithium–boron analogue LiB(Im)4 have been compared by using high-pressure synchrotron X-ray diffraction, nanoindentation and density functional calculations.11 Porous metal–organic frameworks (MOFs) such as Zn(2-methylimidazole)2 (ZIF-8) have been investigated under modest and industrially accessible pressures (∼1 GPa), which indicated ZIF-8 is highly compressible with an irreversible pressure-induced amorphization at low pressures. These amorphized MOFs retain some structural order.13 These studies provided a new route to modify the structure and properties of nanoporous materials. In addition, pressure-induced amorphization and an amorphous–amorphous transition were found in nanoporous Si, which is very different from that of the corresponding bulk solid and routine nanomaterials.14–16 This indicates that the unique nanoporous microstructure plays an important role in the high-pressure phase transitions of nanoporous materials.
TiO2 not only is an important functional material, but also possesses rich phase diagrams under high pressure.17–20 The high-pressure phase transitions of TiO2 depend on its starting phases and particle sizes. Bulk anatase TiO2 undergoes a series of phase transitions from the anatase to the α-PbO2-type structure, and then to the baddeleyite structure under high pressure. There exist particle size dependent pressure-induced phase transitions in nanocrystalline anatase.19–22 Pressure-induced amorphization is observed in small size (less than ∼12 nm) anatase nanocrystals. Anatase transforms directly into baddeleyite in larger size (12–50 nm) nanocrystals while for macrocrystals (larger than 40–50 nm), anatase transforms into the α-PbO2 structure. For rutile TiO2, different phase transition sequences of rutile–α-PbO2–baddeleyite and rutile–baddeleyite were both found in the bulk solid.18 Wang et al. reported the phase transition from rutile to baddeleyite at ∼8.7 GPa for anatase/rutile nanocrystals in which the transition pressure is lower than the pressure of 13 GPa observed in its bulk counterpart.17 On the contrary, the rutile–baddeleyite phase transition was observed at ∼20 GPa in rutile nanoparticles.23 There are still some controversies for the high-pressure phase transition behaviors of rutile TiO2 and so further high-pressure studies of rutile TiO2 are needed. Recently, the shape-dependent compressibility was also studied in sphere-, rod- and rice-shaped TiO2 nanoparticles.24 These studies indicated that the size and morphology of nanomaterials play important roles in the phase transitions of TiO2. However, there is no high-pressure research for nanoporous TiO2 materials. It is known that the excellent properties of nanoporous TiO2 depend on its crystal structure and nanoporous morphology. The structural and morphological stabilities of nanoporous TiO2 are significant for their applications. The high-pressure study of nanoporous TiO2 not only helps to confirm the structural stabilities for nanoporous materials but also is useful to explore their properties. It also possibly may lead to novel functional nanomaterials with high-pressure phases that are difficult to prepare by routine methods.
In this paper, we have studied the structural stability and the phase transitions of nanoporous rutile TiO2 under high pressure using X-ray diffraction, Raman spectroscopy and transmission electron microscopy (TEM). The phase transition from rutile to baddeleyite was observed at 10.8–31.3 GPa while the nanoporous α-PbO2 phase TiO2 was obtained upon decompression. TEM results indicate that the nanoporous architecture remains structurally intact during the compression–decompression cycle and shows excellent stability. This also provides an effective route for preparing α-PbO2 phase nanoporous TiO2.
Experimental
Nanoporous rutile TiO2 were synthesized by a simple ethylene glycol-mediated synthesis route as described in our previous work.25 The morphology of the sample was characterized using TEM (200 kV, HITACHI, H-8100IV). Brunauer–Emmett–Teller (BET) specific surface area and pore size distribution was determined using a Micromeritics instrument ASAP 2420. In situ angle-dispersive synchrotron XRD measurements under high pressure were carried out at the X17C beamline of the Brookhaven National Laboratory with a wavelength of 0.4066 Å. The beam size is confined to 25 × 30 μm2. Part of the X-ray diffraction experiments were performed at the 4W2 beamline of Beijing Synchrotron Radiation Facility (BSRF) (wavelength 0.6199 Å, beam size 30 × 30 μm2) and the BL15U1 beamline of Shanghai Synchrotron Radiation Facilities (SSRF) (wavelength 0.6199 Å, beam size 4 × 7 μm2). High-pressure Raman experiments were carried out at room temperature by a Renishaw inVia Raman spectrometer with an Ar+ 514.5 nm laser. High pressure was generated by a diamond anvil cell (DAC) with 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol–ethanol mixture as the pressure-transmitting medium. The pressures were determined from the pressure-dependent shift of the R1 line fluorescence of ruby. After release to ambient pressure, the sample in the DAC was transferred to a carbon-coated copper grid for TEM and HRTEM observation.
:1 methanol–ethanol mixture as the pressure-transmitting medium. The pressures were determined from the pressure-dependent shift of the R1 line fluorescence of ruby. After release to ambient pressure, the sample in the DAC was transferred to a carbon-coated copper grid for TEM and HRTEM observation.
Results and discussion
The pressure evolution of the angle-dispersive XRD patterns of the nanoporous rutile TiO2 are shown in Fig. 1. The diffraction peaks are all indexed to the rutile phase except for two peaks of Au (marked with Au). All the peaks of rutile TiO2 shift to smaller d-spacings with increasing pressure. As the pressure increases to 10.8 GPa, a new diffraction peak appears at ∼1.91 Å, characteristic of the baddeleyite phase. This indicates that the rutile-to-baddeleyite phase transition takes place. The onset phase transition pressure is higher than that of rutile nanoparticles and lower than that of bulk materials.17,18 As the pressure further increases, the intensity of the new peak increases. When the pressure increases to 24.2 GPa, another new peak of the baddeleyite phase occurs at ∼2.84 Å. Most peaks of the rutile phase are retained up to ∼20.4 GPa. When the pressure further increases to ∼24.2 GPa, the intensities of these peaks became dramatically broader and weaker. These peaks of the rutile phase disappeared when the pressure was increased beyond 26.1 GPa. This suggests that the nanoporous rutile TiO2 transformed into the baddeleyite phase completely. It is worth noting that the peaks of the baddeleyite are excessively broad and weak above ∼31.3 GPa. This indicates that the poor crystallinity of the baddeleyite phase in nanoporous TiO2 under high pressure, which may arise from imminent pressure-induced amorphization as seen in TiO2 nanocrystals, and the effect of nonhydrostatic stress at these elevated pressures.19–21,26,27 During decompression, the intensity of the peaks of the baddeleyite phase are enhanced gradually with decreasing pressure (Fig. 1b). This indicates that the order degree of the baddeleyite structure recovers with the release of pressure. When the pressure is released to ∼6.3 GPa, two new diffraction peaks are observed which are assigned to the (111) and (110) planes of the α-PbO2 phase.15 All peaks of the baddeleyite phase completely disappear at 4.8 GPa. Obviously, the baddeleyite form transforms to the α-PbO2 structure at the range of 7.7–4.8 GPa. The α-PbO2 structure is retained upon release back to ambient pressure.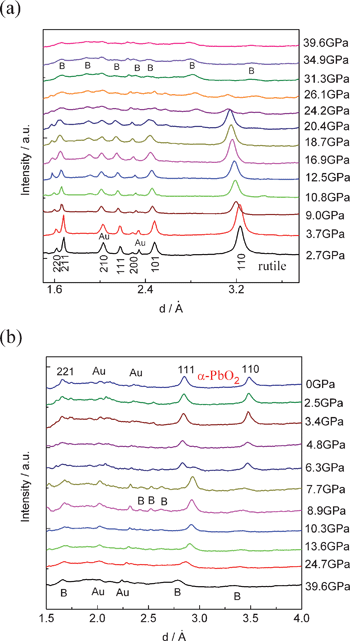 | ||
| Fig. 1 High-pressure powder X-ray diffraction patterns of nanoporous TiO2: (a) compression, (b) decompression. Reflections marked B originate from baddeleyite-TiO2. | ||
The pressure dependence of the d-spacings is illustrated in Fig. 2a. All of the d-spacings decrease linearly at an almost similar rate with increasing pressures. At pressures above 10.8 GPa, a phase transformation is evident by the appearance of additional peaks. When the pressure is beyond 26.1 GPa, all the peaks of the rutile phase disappeared and three discernable weak peaks were maintained up to 39.6 GPa. The lattice parameter ratios (a/a0 and c/c0) for the rutile phase are shown in Fig. 2b as a function of pressure. The a/a0 and c/c0 radios decrease with increasing pressure. It can be seen that the a axis is more compressible than the c axis. This is in good agreement with a previous study.18 The evolution of the volume with pressure at room temperature is reported in Fig. 3. The pressure–volume data of the rutile phase was fitted to the third-order Birch–Murnaghan equation of state:
| P = 3/2B0[(v/v0)−7/3 − (v/v0)−5/3]{1 + 3/4(B0′ − 4)[(v/v0)−2/3 − 1]} |
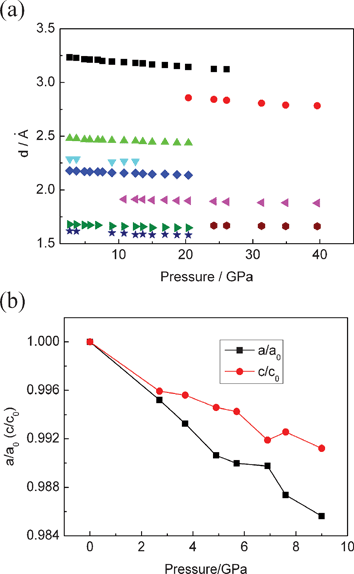 | ||
| Fig. 2 (a) Pressure dependence of the d-spacings. (b) Cell parameter ratios as a function of pressure for nanoporous rutile TiO2. | ||
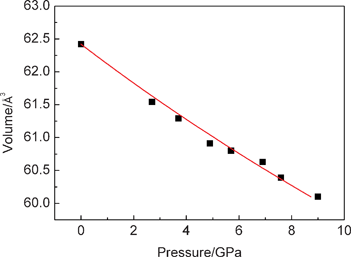 | ||
| Fig. 3 Pressure–volume relations of nanoporous rutile TiO2. | ||
To further verify the pressure-induced phase transition for the nanoporous rutile TiO2, we also carried out high-pressure Raman measurements. Fig. 4 shows the Raman spectra of TiO2 measured during compression and decompression. As is well known, rutile has five Raman active modes, which are at 143 (B1g), 237 (two-phonon scattering), 447 (Eg), 612 (A1g) and 826 (B2g) cm−1.28 As shown in Fig. 4a, two Raman peaks are observed at 450 and 613 cm−1, and a weak Raman peak is located at 243 cm−1, at atmospheric pressure. These Raman bands at 243, 450 and 613 cm−1 can be ascribed to the two-phonon scattering, Eg and A1g modes of the rutile phase, respectively. The B1g and B2g modes of the rutile phase can not be observed in our case because their weak signal. Obviously, the Eg (450 cm−1) and the A1g (613 cm−1) Raman bands of nanoporous rutile show significant broadening and blue shifting. The blue shifts of peak positions and broadening of linewidths of the rutile Raman features can be attributed to both size and residual stress effects. With increasing the pressure, Eg and A1g Raman bands show a blue shift and become weaker. When the pressure reaches ∼25.8 GPa, all Raman modes of the rutile TiO2 disappear, and several very weak peaks of the baddeleyite TiO2 appear. The intensities of these baddeleyite peaks increase first with increasing the pressure, then decrease when the pressures reaches beyond 31.3 GPa. Up to 38.8 GPa, all the baddeleyite peaks become substantially broadened and more weak. This result is in good agreement with our XRD results. This indicates that the baddeleyite phase under a high pressure of 38.8 GPa is a low ordered structure. Upon decompression, the intensities of these Raman bands of the baddeleyite grow gradually with decreasing pressure to 7.8 GPa, as shown in Fig. 4b. When the pressure is released to 9.1 GPa, new Raman bands (312, 372 and 612 cm−1) appear which indicate that a phase transition from baddeleyite to α-PbO2 occurs. At 4.8 GPa, other Raman bands (165, 180, 290, 368, 438, 463, 557 and 628 cm−1) of the α-PbO2 phase are observed and the Raman bands of the baddeleyite phase disappeared. From these results, the baddeleyite structure begins to transform into the α-PbO2 structure at about 9.1 GPa and this is complete at about 4.8 GPa upon decompression. The intensities of those Raman bands of the α-PbO2 phase increased significantly when the pressure was released to ambient pressure. It is clear that the α-PbO2 phase remains at ambient pressure, which is in agreement with the XRD results.
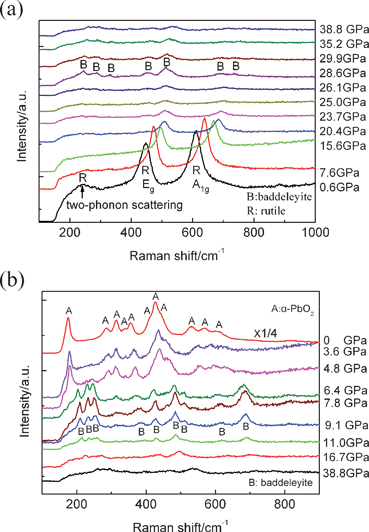 | ||
| Fig. 4 Raman spectra of nanoporous TiO2 at various pressures: (a) compression, (b) decompression. | ||
Raman mode frequencies of the nanoporous rutile TiO2 as a function of pressure are shown in Fig. 5. The pressure dependences of the Raman frequencies (dv/dP) have been obtained by linear fits over the pressure range before and after the structural transition that occurs at ∼22 GPa. The obtained pressure coefficients of Eg and A1g modes are 3.07 cm−1/GPa (0.09) and 3.81 cm−1/GPa (0.07), respectively. The pressure coefficient of the Eg mode for nanoporous rutile TiO2 is less than 3.48 cm−1/GPa of bulk material, but the pressure coefficient of the A1g mode is similar to 3.82 cm−1/GPa of the bulk material.29 This may be caused by the nanoporous microstructure and nanosize effect of rutile TiO2.
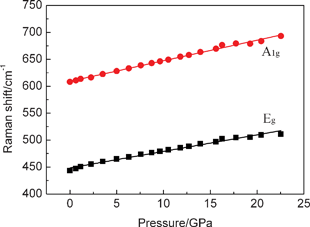 | ||
| Fig. 5 Pressure dependence of the peak frequencies of Raman spectra of nanoporous rutile TiO2. | ||
To investigate the microstructure change of the nanoporous TiO2 under high pressure, we carried out TEM and HRTEM observations for both pristine and quenched samples. Fig. 6a and b show the TEM and HRTEM images of the pristine nanoporous TiO2 (further information for our sample can be seen in our previous work25 and Fig. S1 and S2, ESI†). Obviously, the morphology of the sample is rod-like with a polygonal section which is 1–20 μm in diameter and 80–100 μm in length, as shown in Fig. S1 and S2 (ESI†). It is worth noting that these TiO2 microrods consist of a number of ∼10 nm nanoparticles constructing a nanoporous structure (Fig. 6a). The SAED pattern (inset in Fig. 6a) shows the pristine nanoporous TiO2 is a rutile structure. The typical HRTEM image (Fig. 6b) indicates the nanoparticles in nanoporous TiO2 are single crystals. These results thus demonstrate that nanoporous TiO2 consists of large numbers of single crystalline nanoparticles. As shown in Fig. 6c, the pressure-treated TiO2 sample still retains its nanoporous microstructure after being released from 38.8 GPa, and the size of these nanoparticles is almost unchanged. It is worth noting that the SAED pattern (inset in Fig. 6c) indicates the pressure-released sample is of α-PbO2 structure. This is in good accordance with the XRD and Raman results. Fig. 6d shows the typical HRTEM image of the recovered sample. The crystal structure of the sample is indexed as the α-PbO2 structure. It is worth noting that the small nanocrystals are dramatically distorted and even partly disordered. As we know, the α-PbO2 structure originates from the baddeleyite form upon decompression. Thus, it is reasonable to believe that the baddeleyite structure in nanoporous rutile is more distorted and disordered under higher pressures. We suggest that the nanoporous architecture and the small particle sizes play important roles in the phase transition of the nanoporous rutile.
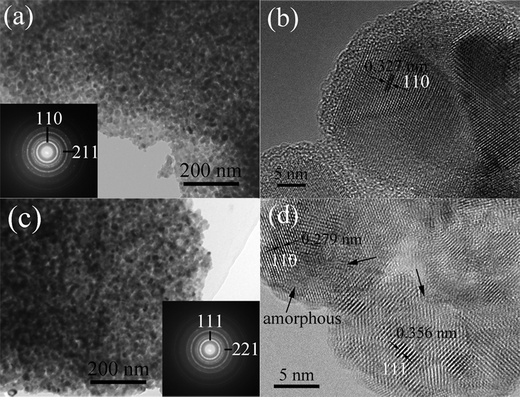 | ||
| Fig. 6 (a) TEM and (b) HRTEM images of pristine nanoporous TiO2, (c) TEM and (d) HETEM images of pressure-treated nanoporous TiO2 after being released from 38.8 GPa. The arrows denote the distorted and disordered areas. | ||
Nanophase rutile particles (∼30 nm) appear to transform to the baddeleyite structure at an externally applied pressure of ∼8.7 GPa according to previous studies.18 The phase transition pressure of rutile TiO2 nanomaterial is lower than the pressure of this transition for the bulk material (∼12–13 GPa). The reduction of transition pressure in rutile nanomaterials was explained by a large volume collapse upon phase transition. However, a higher phase transition pressure (∼20 GPa) from rutile to baddeleyite was observed in the smaller nanoparticles (∼10 nm).23 The increase of the phase transition pressure with decreasing nanocrystal size was explained by the increase of surface energy. In our case, the transition pressure of ∼10.8 GPa is higher than that of ∼30 nm nanoparticles previously reported and lower than those of ∼10 nm nanoparticles and bulk material.18,19,23 We believe that the surface energy plays a more important role than the volume collapse in the phase transition for the small size rutile TiO2 nanoparticles. Therefore, we suggest that two main factors perform important functions in the phase transition process. One is the smaller size of nanoparticles in the porous rutile TiO2 while the other is the unique nanoporous microstructure. The smaller sized (∼10 nm) nanoparticles possess higher surface energy than the previously studied ∼30 nm particles. Because of the existence of a higher energy hindrance in small sized nanoparticles, a higher phase transition pressure is observed in our nanoporous rutile TiO2 compared to that of other larger nanoparticles. In addition, the nanoporous structure of the rutile TiO2 may induce very high stress at the contact points between individual grains at low applied pressures leading to so-called microstrains. Thus, the onset transition pressure of the nanoporous rutile is lower than that of ∼10 nm nanoparticles.23 The high stress results in the baddeleyite phase becoming disordered or distorted further under high pressure. On the contrary, the stress gradually decreases upon decompression, which leads to improved crystallinity for TiO2 high-pressure phases. This indicates that the phase transition behaviors of the rutile nanomaterials are dominated by both their ultrafine grain sizes and nanoporous microstructure.
The structural durability of nanoporous materials is very important for their applications. For nanoporous rutile TiO2, the porous architectures show excellent structural durability under high pressure. The starting crystal phase is stable up to ∼10.8 GPa and transforms to the baddeleyite phase under higher pressures. After quenching to ambient pressure, nanoporous α-PbO2-type TiO2 nanomaterials were obtained that consist of a number of nanoparticles with diameters about 10 nm, the same as our pristine nanoporous rutile samples (as shown in Fig. 6). Although a series of phase transitions (rutile–baddeleyite–α-PbO2) occur during the compression–decompression cycle, the porous architectures still remain structurally intact. To further characterize the starting nanoporous microstructure, the nitrogen adsorption–desorption isotherms and pore size distribution of the nanoporous rutile TiO2 are presented in Fig. 7. The sample shows typical IV isotherms, indicating the presence of mesopores in the sample, which give a BET surface area of 62 m2 g−1 and average pore diameter around 8 nm. Obviously, there is accessible space for the transmitting media (methanol–ethanol 4:1) in the nanoporous TiO2. Thus, the nanoporous structure and the inside transmitting media assemble into a compound nanostructure under high pressure, which exhibits excellent tenacity. This is similar to the behavior of nanocrystalline ceramics.30 The enhanced fracture toughness may be due to the creep of nanoparticles upon compression. This further indicates that the unique phase transition behaviors of the nanoporous rutile TiO2 are dominated by both their ultrafine grain sizes and nanoporous microstructure.
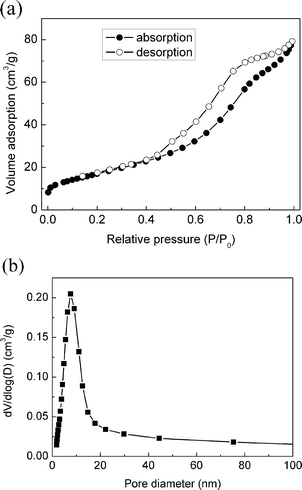 | ||
| Fig. 7 Nitrogen adsorption–desorption isotherms (a) and pore size distribution (b) of nanoporous rutile TiO2. | ||
Conclusions
In summary, the high-pressure behavior of nanoporous rutile TiO2 was studied at room temperature using in situ synchrotron X-ray diffraction and Raman scattering. The results show that the transition from nanoporous rutile phase to the baddeleyite phase takes place at a pressure of ∼10.8 GPa. The onset phase transition pressure is lower than that of ∼30 nm rutile nanoparticles (∼8.7 GPa) but lower than that of ∼10 nm rutile nanoparticles (∼20 GPa) and bulk material (12–13 GPa). The rutile phase transforms into the baddeleyite phase completely beyond 26.1 GPa, but with poor crystallinity. Upon decompression, the baddeleyite phase transforms into the α-PbO2 phase with improved crystallinity. A bulk modulus 204(4) GPa for the nanoporous rutile TiO2 is derived from the P–V data, which is close to the corresponding value of 230(20) GPa for the bulk counterpart. The nanoporous TiO2 remains its pristine porous microstructure after compression–decompression cycle shows excellent stability and mechanical property. We suggest that the unique high-pressure behavior of nanoporous rutile TiO2 is due to their nanoporous microstructure. Our study provides an effective route for preparing porous α-PbO2-type TiO2 nanomaterials that possibly can be employed to obtain various functional materials with high-pressure phases.Acknowledgements
This work was supported financially by the National Basic Research Program of China (2011CB808200), the NSFC (10979001, 51025206, 51032001, 21073071, 11004075, 11004072), the Cheung Kong Scholars Programme of China, This research was partially supported by COMPRES, the Consortium for Materials Properties Research in Earth Sciences under NSF Cooperative Agreement EAR 06-49658. Portions of this work were performed at the 4W2 beamline, Beijing Synchrotron Radiation Facility (BSRF) which is supported by Chinese Academy of Sciences (No. KJCX2-SW-N20, KJCX2-SW-N03).References
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169 CrossRef CAS.
- R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966 CrossRef CAS.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897 CrossRef CAS.
- C. X. Xu, J. X. Su, X. H. Xu, P. P. Liu, H. J. Zhao, F. Tian and Y. Ding, J. Am. Chem. Soc., 2007, 129, 42 CrossRef CAS.
- C. Y. Sun, S. X. Liu, D. D. Liang, K. Z. Shao, Y. H. Ren and Z. M. Su, J. Am. Chem. Soc., 2009, 131, 1883 CrossRef CAS.
- M. Signoretto, E. Ghedini, V. Nichele, F. Pinna, V. Crocella and G. Cerrato, Microporous Mesoporous Mater., 2011, 139, 189 CrossRef CAS.
- J. F. Ye, W. Liu, J. G. Cai, S. Chen, X. W. Zhao, H. H. Zhou and L. M. Qi, J. Am. Chem. Soc., 2011, 133, 933 CrossRef CAS.
- J. Y. Kim, S. H. Kang, H. S. Kim and Y. E. Sung, Langmuir, 2010, 26, 2864 CrossRef CAS.
- J. G. Yu, J. J. Fan and L. Zhao, Electrochim. Acta, 2010, 55, 597 CrossRef CAS.
- G. G. Zhang, H. T. Huang, Y. H. Zhang, H. L. W. Chan and L. M. Zhou, Electrochem. Commun., 2007, 9, 2854 CrossRef CAS.
- T. D. Bennett, J. C. Tan, S. A. Moggach, R. Galvelis, C. Mellot-Draznieks, B. A. Reisner, A. Thirumurugan, D. R. Allan and A. K. Cheetham, Chem.–Eur. J., 2010, 16, 10684 CrossRef CAS.
- D. Liu, W. W. Lei, Z. X. Liu and Y. J. Lee, J. Phys. Chem. C, 2010, 114, 18819 CAS.
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546 CrossRef CAS.
- S. K. Deb, M. Wilding, M. Somayazulu and P. F. McMillan, Nature, 2001, 414, 528–530 CrossRef CAS.
- S. H. Tolbert, A. B. Herhold, L. E. Brus and A. P. Alivisatos, Phys. Rev. Lett., 1996, 76, 4384–4387 CrossRef CAS.
- Y. J. Wang, J. Z. Zhang, J. Wu, J. L. Coffer, Z. J. Lin, S. V. Sinogeikin, W. G. Yang and Y. S. Zhao, Nano Lett., 2008, 8, 2891–2895 CrossRef CAS.
- Z. W. Wang, S. K. Saxena, V. Pischedda, H. P. Liermann and C. S. Zha, J. Phys.: Condens. Matter, 2001, 13, 8317 CrossRef CAS.
- L. Gerward and J. Staun Olsen, J. Appl. Crystallogr., 1997, 30, 259 CrossRef CAS.
- V. Swamy, A. Kuznetsov, L. S. Dubrovinsky, R. A. Caruso, D. G. Shchukin and B. C. Muddle, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 184302 CrossRef.
- V. Pischedda, G. R. Hearne, A. M. Dawe and J. E. Lowther, Phys. Rev. Lett., 2006, 96, 035509 CrossRef CAS.
- V. Swamy, A. Kuznetsov, L. S. Dubrovinsky, P. F. McMillan, V. B. Prakapenka, G. Y. Shen and B. C. Muddle, Phys. Rev. Lett., 2006, 96, 135702 CrossRef.
- G. R. Hearne, J. Zhao, A. M. Dawe, V. Pischedda, M. Maaza, M. K. Nieuwoudt, P. Kibasomba, O. Nemraoui, J. D. Comins and M. J. Witcomb, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 134102 CrossRef.
- J. S. Olsen, L. Gerward and J. Z. Jiang, High Pressure Res., 2002, 22, 385 CrossRef.
- S. W. Park, J. T. Jang, J. Cheon, H. H. Lee, D. R. Lee and Y. Lee, J. Phys. Chem. C, 2008, 112, 9627 CAS.
- Q. J. Li, B. B. Liu, Y. A. Li, R. Liu, X. L. Li, D. M. Li, S. D. Yu, D. D. Liu, P. Wang, B. Li, B. Zou, T. Cui and G. T. Zou, J. Alloys Compd., 2009, 471, 477 CrossRef CAS.
- V. Swamy, A. Y. Kuznetsov, L. S. Dubrovinsky, A. Kuznetsov and V. B. Prakapenka, Phys. Rev. Lett., 2009, 103, 075505 CrossRef.
- Q. J. Li, B. B. Liu, L. Wang, D. M. Li, R. Liu, B. Zou, T. Cui, G. T. Zou, Y. Meng, H. K. Mao, Z. X. Liu, J. Liu and J. X. Li, J. Phys. Chem. Lett., 2010, 1, 309 CrossRef CAS.
- V. Swamy, B. C. Muddle and Q. Dai, Appl. Phys. Lett., 2006, 89, 163118 CrossRef.
- H. Arashi, J. Phys. Chem. Solids, 1992, 53, 355 CrossRef CAS.
- I. A. Ovid'ko and A. G. Sheinerman, Rev. Adv. Mater. Sci., 2011, 29, 105–125 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ra20586f/ |
| This journal is © The Royal Society of Chemistry 2012 |