Isolation, biological activity and synthesis of the natural product ellipticine and related pyridocarbazoles
Charlotte M.
Miller
and
Florence O.
McCarthy
*
Department of Chemistry, Analytical and Biological Chemistry Research Facility, University College Cork, Western Road, Cork, Ireland
First published on 9th May 2012
Abstract
The tetracyclic natural product ellipticine (5,11-dimethyl-6H-pyrido[4,3-b]carbazole) was first isolated from the plant material of Ochrosia elliptica Labill in 1959. Woodward et al. reported the first synthesis of ellipticine later the same year, and this was followed by many different synthetic strategies in subsequent decades. Investigation of the biological activity of ellipticines uncovered potent anti-cancer properties and several ellipticine derivatives have been the subject of clinical trials. The ellipticine family of compounds exert their biological activity via several modes of action, the most well established of which are intercalation with DNA and topoisomerase II inhibition. In recent times other modes of action have been revealed, including kinase inhibition, interaction with p53 transcription factor, bio-oxidation and adduct formation. The scope of this review covers key features of the biological activity of ellipticine, with emphasis on new modes of action, followed by synthetic routes to ellipticine, including key early syntheses of pyrido[4,3-b]carbazoles and comprehensive coverage of the literature since the late 1980s, along with more recent syntheses of ellipticine analogues and substituted ellipticines.
 Charlotte M. Miller | Charlotte Miller obtained her Bachelor of Science degree (Hons) in Chemistry at University College Cork, Ireland being awarded the title of College Scholar. In 2012, she was awarded a Ph.D. in organic and medicinal chemistry, in the Department of Chemistry and Analytical and Biological Chemistry Research Facility (ABCRF) at UCC, for her work on the synthesis and evaluation of novel ellipticine derivatives and analogues as anti-cancer agents. |
 Florence O. McCarthy | Florence McCarthy graduated from the School of Pharmacy, University of Sunderland, UK winning the Pfizer prize for Excellence in Medicinal Chemistry and continued to PhD studies in anticancer research with Prof. Paul Groundwater. He subsequently joined Prof. Bill Denny at the ACSRC, University of Auckland, New Zealand in conjunction with Pfizer Global R&D working on a series of kinase inhibition programmes. He joined the Department of Chemistry at University College Cork in 2005 and currently leads a team of researchers in medicinal and pharmaceutical chemistry investigating the synthesis and evaluation of diverse bioactive molecules from steroids to complex heterocycles. |
1. Introduction
The tetracyclic natural product ellipticine 1 (5,11-dimethyl-6H-pyrido[4,3-b]carbazole, Fig. 1) was isolated in 1959.1 Woodward et al.2 reported the first synthesis of ellipticine later the same year, and this was followed by many different synthetic strategies in subsequent decades. Naturally the initial goal was an efficient synthesis of ellipticine itself, however substitution at C-9 was soon found to significantly increase the cytotoxic activity of ellipticine and as a result, 9-hydroxy and 9-methoxy ellipticines became attractive targets.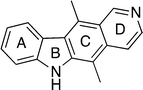 | ||
| Fig. 1 Ellipticine 1. | ||
2. Isolation and biological activity
Ellipticine 1 was first isolated from the leaf material of Ochrosia elliptica Labill by Goodwin et al. in 1959.1 This small tropical evergreen tree belonging to the Apocynaceae family also contained several other alkaloids, including 9-methoxyellipticine. Ellipticine has since been isolated from several other plants of the Apocynaceae family (Ochrosia vieillardii, Ochrosia acuminate and Ochrosia moorei)3–5 and from Strychnos dinkagei of the Loganiaceae family.6,72.1 Intercalation
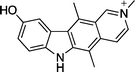
The first reported mechanism of action of ellipticine was intercalation with DNA, several early studies used UV absorption and circular dichroism to study the interaction of DNA and ellipticine.8–12 Most studies proposed a binding mode parallel to the base pairs of ellipticine with strong DNA binding constants. Dodin et al. reported DNA affinity constants of 8.3 × 105 M−1 and 3.3 × 105 M−1 at pH 5 & 9 respectively.9 The same group later used fluorescence spectroscopy in deuterated buffer solutions to study interactions of N-methylellipticinium 2 and 9-hydroxy-N-methylellipticinium 3 with DNA (Fig. 2).10N-Methylellipticinium 2 showed no preference for intercalation sites, while 3 was found to interact with a site containing at least one G–C base pair, inferring that the 9-hydroxy group confers G–C preference.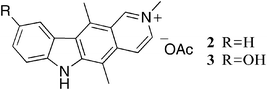 | ||
| Fig. 2 | ||
In 2005, Canals et al. published a crystal structure determination of ellipticine in complex with a six base-pair oligionucleotide–d(CGATCG)2, with 1.5 Å resolution.13 This showed ellipticine aligned with the major axis, parallel to the hydrogen bonds of the base pairs. The d(CGATCG)2 hexanucleotide was intercalated by two ellipticine molecules, with the pyridine nitrogen orientated towards the major groove in both cases. A preference for G–C base pairs was also evident, since the AT–TA site remained empty. Overall, there was good correlation with an earlier dinucleotide structure published by Jain et al.14 An unwinding angle of 14° with respect to standard B–DNA was observed for each intercalation site. Both intercalation sites were separated by 6.9 Å, compared to 3.3 Å for normal base pairs.
2.2 Topoisomerase II inhibition
Ross and Bradley were the first researchers to associate ellipticine induced DNA strand breaks with topoisomerase II inhibition.15 Ellipticine was found to induce strand breaks in DNA from L1210 cells, in concentrations of 1.25–5.0 μg mL−1 and the authors proposed that the strand breaks were due to topoisomerase II activity.Froelich–Ammon et al. published a major study of ellipticine-topoisomerase activity in 1995, identifying topoisomerase II as the primary cellular target of the drug.16 This was determined by employing yeast cells with a temperature sensitive strain of topo II (at 25 °C enzyme activity is 100% and at 30 °C activity decreases to 10%). At 25 °C, 90% of the cell culture was killed by a 200 μg dose of ellipticine, while at 30 °C, no cell death was observed at any concentration of ellipticine used. In a second experiment, a 10 μg dose of ellipticine induced a 6-fold increase in topoisomerase II-mediated strand breakage, but the same concentration of ellipticine had no effect on the rate of religation. Thus, ellipticine may be considered to be a catalytic inhibitor of topo II as opposed to a topo II poison, i.e. ellipticine acts primarily by increasing the forward rate of cleavage of DNA.
Fluorescence spectroscopy was then used to study interaction of ellipticine with DNA, topo II and the DNA-topo II complex. It was established that protonated ellipticine binds to DNA, whereas deprotonated ellipticine binds to topoisomerase II and is also the major species present in the ternary complex. Thus the enzyme determines the protonation state of ellipticine in the ternary complex. Formation of this ternary complex results in increased levels of covalently bound topoisomerase II-DNA cleavage complex.
2.3 Bio-oxidation and adduct formation
The bio-oxidation pathway was originally proposed by Auclair and Paoletti, who postulated that ellipticine could serve as a substrate for peroxidises in vivo.17 They employed a horse radish peroxidise (HRP)–hydrogen peroxide oxidizing system as a model of bio-oxidation and studied the ability of various ellipticines to undergo oxidation. Under these conditions, 9-hydroxyellipticine 4 was oxidised to the quinone imine 5 (Scheme 1). The quinone imine 5 was shown to be a strong electrophile, oxidizing NADH to NAD+. It is also highly susceptible to nucleophilic attack at the C-10 position, and bound irreversibly to bovine serum albumin.17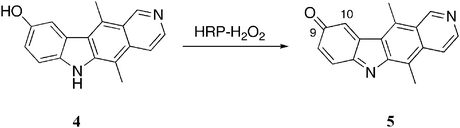 | ||
| Scheme 1 | ||
In the last decade, Stiborová et al. have extensively studied the bio-oxidation and adduct formation pathways of ellipticine in vitro and in vivo. The group began by investigating the potential of ellipticine to form DNA adducts after activation by cytochrome P450 (CYP) and have isolated and identified several metabolites formed by human cytochrome P450 enzymes (Scheme 2).18 13-Hydroxyellipticine and the ellipticine N-oxide were identified as the metabolites responsible for two DNA-ellipticine adducts.
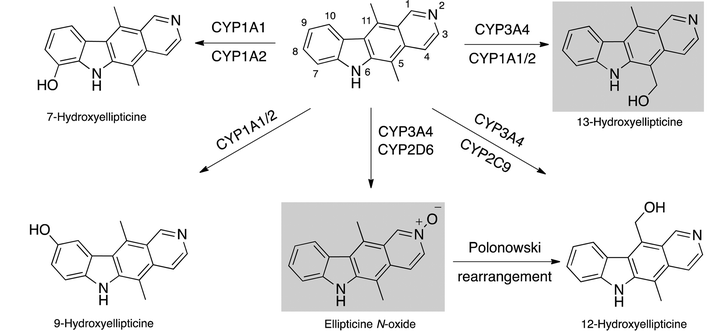 | ||
| Scheme 2 Ellipticine metabolites formed by human cytochrome P450 enzymes.18 | ||
The group have also studied peroxidase-mediated oxidation of ellipticine and performed experiments with peroxidases (human myeloperoxidase, human and ovine cyclooxygenases, bovine lactoperoxidase and horseradish peroxidase).19 In this case, four DNA adducts were formed, two major and two minor. The two major adducts corresponded to those previously identified from CYP-mediated oxidation, derived from 13-hydroxyellipticine and ellipticine N-oxide. The same adducts were formed in leukemia HL-60 and CCRF-CEM cells and a good correlation between cytotoxicity and the levels of DNA adducts was observed.20
The key on-going question in this area is whether adduct formation is responsible for the cytotoxicity of ellipticines. A recent publication by Stiborová et al. sought to evaluate the contribution of DNA adduct formation to cytotoxicity and found plausible correlation in six out of seven cancer cell lines tested.21 However, further work is required in this area in order to conclusively understand this mechanism and its overall contribution to ellipticine cytotoxicity.
2.4 Kinase inhibition
In the last decade, ellipticine interactions with several enzymes have been studied, including inhibition of c-Kit kinase and AKT.c-Kit kinase is a type III receptor tyrosine kinase (RTK), a class of enzymes which regulate signalling pathways that control cell growth and proliferation. c-Kit plays a key role in mast cell survival, differentiation, maturation and function.22 It is expressed by and critical for the development and growth of mast cells, melanocytes, hematopoetic stem cells and the interstitial cells of Cajal.23 Mutations in the gene encoding for c-Kit kinase are associated with some highly malignant cancers and therefore both wild-type and mutated c-Kit are viable drug targets in anticancer chemotherapy.24
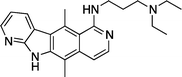
The most common enzymatic pocket mutation in c-Kit kinase is the D816V point mutation, which is associated with germ cell tumours, adult mastocytosis and a small proportion of atypical paediatric mastocytosis.25,26 In 2005, Vendome et al. reported that several ellipticine derivatives exhibited c-Kit kinase inhibition.27 A range of derivatives significantly inhibited both wild type and D816V mutated c-Kit kinase (Table 1), with Imatinib 6 as a reference compound.
| Compound | IC50 WT μM | IC50 D816V μM | Compound | IC50 WT μM | IC50 D816V μM |
|---|---|---|---|---|---|
 6
6
|
0.1 | 10 |
 7
7
|
2.3 | 1.9 |
 4
4
|
0.4 | 0.4 |
 8
8
|
2.5 | 2.0 |
 3
3
|
0.4 | 0.4 |
 9
9
|
2.5 | 3.6 |
 10
10
|
0.45 | 0.3 |
 1
1
|
> 10 | > 10 |
 11
11
|
0.8 | 0.6 |
 12
12
|
> 10 | > 10 |
 13
13
|
1.4 | 1.2 |
9-Hydroxyellipticine 4 and 9-hydroxy-N-methylellipticinium 3, were the most active of the series, with equal inhibition of wild type and D816V mutated c-Kit. These were closely followed by the N-alkylamino-9-hydroxyellipticinium 10 which showed a slight preference for D816V mutated c-Kit. Of the rest of the series, compounds which were unsubstituted at C-9 were generally inactive and those with a C-9 methoxy group were only moderately active. Salt formation at the N-2 position did not seem to affect the c-Kit inhibitory activity, suggesting that the N-2 position is not directly involved in binding to c-Kit. Finally, addition of bulky alkylamino side chains at C-1 was significantly unfavourable for activity.
Molecular mechanics simulations were employed in order to identify possible binding sites, with docking studies carried out using the GOLD program. It was found that all the active compounds docked similarly in part of the ATP binding pocket, which is made favourable by three hydrogen bonding interactions. The docked position of the most active compounds is shown in Fig. 3, along with hydrogen bonding interactions for 9-hydroxyellipticine 4.27
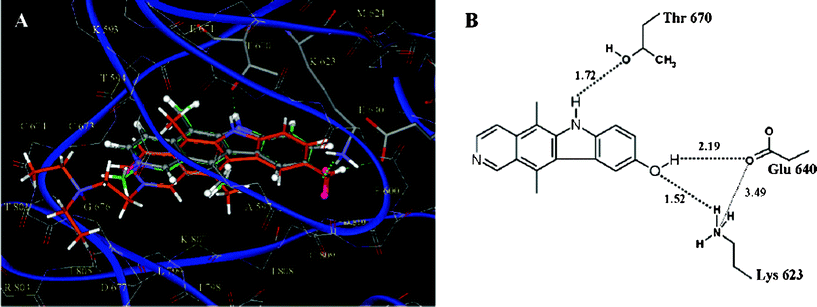 | ||
| Fig. 3 (A) Detail of the docked position of the most active compounds (9-hydroxyellipticine 4 is shown in gray, 9-hydroxy-N-methylellipticinium 3 in green, and N-alkylamino-9-hydroxyellipticinium 10 in orange). Conserved interactions are represented by dashed lines. (B) Map of the main interactions for 9-hydroxyellipticine 4.27 | ||
In our research group, extensive molecular dynamics simulations of five different binding modes for 9-hydroxyellipticine 4 suggested a different binding mode to that proposed by Vendome et al.27,28 Initially, Poisson–Boltzmann/Linear Response Approximation pKa calculations indicated a strong preference for protonation at N-2. The CHARMM force field was used for protein, ADP and Mg2+, while new force field parameters were developed for 9-hydroxyellipticine and two nanoseconds of molecular dynamics were performed for each of five different c-Kit-ellipticine complexes.
The preferred binding mode for 9-hydroxyellipticine 4 (Fig. 4) involves two key hydrogen bonding interactions, C(9)OH–Glu671 and N(2)H–Glu640, with the C(11) methyl group facing the protein backbone and the C(5) methyl group exposed to solvent. Poisson–Boltzmann Free Energy (PBFE) calculations indicated that this binding mode was preferred across a diverse range of substituted ellipticines.
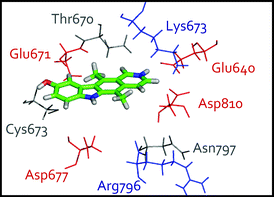 | ||
| Fig. 4 Proposed binding mode of protonated 9-hydroxyellipticine 4 in the c-Kit kinase active site, determined from molecular dynamics simulations.28 | ||
In 2004, Jin et al. reported the inhibition of AKT by 9-methoxy-N-methylellipticinium acetate 14.29 AKT is a serine/threonine kinase (i.e. it phosphorylates the hydroxyl group of serine and threonine residues of other proteins), which is activated in response to growth factors or cytokines by a mechanism involving PI3-K (phosphoinoside 3-kinase).30,31 It provides a survival signal that protects cells from apoptosis induced by various stresses.
In 40–50% of endometrial cancers, AKT is overexpressed due to mutation of tumour suppressor PTEN (phosphatase and tensin homolog). 9-Methoxy-N-methylellipticinium acetate 14 (Fig. 5) was tested against four endometrial cancer cell lines in a 12 μM or 24 μM single dose. Inhibition of AKT kinase activity and apoptosis was observed in the two cell lines with high AKT levels (RL95-2 and Ishikawa cells) but not in the cell lines with normal AKT activity. Further investigation suggested direct inhibition of AKT and not upstream kinases that phosphorylate AKT.
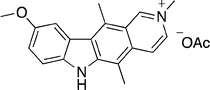 | ||
| Fig. 5 9-Methoxy-N-methylellipticinium acetate 14. | ||
Later, Tang et al. found that the same compound (9-methoxy-N-methylellipticinium acetate 14) inhibited AKT kinase activity in ovarian cancer cell lines with overactivation of AKT.32 In this case, 9-methoxy-N-methylellipticinium acetate 14 appeared to prevent phosphorylation of AKT at Ser473, so it is possible that inhibition of an upstream kinase such as PDK2 may be involved. Overall the compound selectively induced apoptosis in ovarian cancer cell lines with overactivation of AKT but had minimal effect on normal cells.
In 2009, Fang et al. reported that growth of non-small cell lung cancer (NSCLC) epithelial cells A549 was inhibited by ellipticine 1.33 They proposed that ellipticine induced cytotoxicity by modulating the signalling pathways and subcellular redistribution of AKT and p53.
2.5 Interaction with p53 tumour suppressor
Known as the ‘guardian of the genome’, p53 is the key transcription factor which orchestrates coordinated changes in proliferation and apoptosis. On activation, it increases transcription of genes involved in inhibition of DNA replication e.g. p21 and Gadd45, but has opposing effects on BAX and Bcl2 which help regulate apoptosis.34 In the last twenty years, the pivotal role of p53 in cancer has been elucidated. The gene TP53, encoding p53 protein, is mutated or deleted in approximately 55% of human cancers, mainly by point mutations in the core DNA binding domain.35,36In 1998, Shi et al. performed cluster analysis of 112 ellipticines from the NCI 60-cell line screen and found that the ellipticinium salts, but not the ellipticines, were more potent on average against p53 mutant cell lines over wild type p53 lines.37 This analysis opened up a new area of ellipticine research in the last decade. Sugikawa et al. found that 9-hydroxyellipticine 4 induced apoptosis in the G1 phase of the cell cycle of mutant p53 cells and suggested that this activity may be via restoration of wild type p53 function.38 Mizumoto et al. then examined the impact of 9-hydroxyellipticine 4 on the cellular responses to various antineoplastic agents in pancreatic cancer cells.39 Exposure of mutant p53 expressing cells to 1 μM of 9-hydroxyellipticine resulted in restoration to wild type p53 activity without producing apoptosis. This pre-treatment with 9-hydroxyellipticine sensitised cells to treatment with cisplatin and mitomycin C, but not 5-fluorouracil, etoposide or vincristine. The effect was limited to cells expressing mutant p53.
In 2003, Peng et al. definitively showed that ellipticine and several derivatives can activate the transcription function of p53, increasing the function of some mutant p53 types by 5–6 fold.40 In mutant p53-transfected H1299 cells, ellipticine induced MDM2 and p21 expression indicated activation of p53 function at an optimal dose of 8 μM. Immunoprecipitation experiments demonstrated that treatment with ellipticine induced a shift of mutant p53 conformation towards that of wild type p53, thus restoring function.
Kuo et al.41 studied the molecular mechanism of ellipticine activity in human breast MCF-7 cancer cells and showed that induction of p53, Fas/Fas ligand death receptor activation and the mitochondrial proapoptotic pathway were all involved in ellipticine action on these cells. An IC50 value of 1.52 μM was recorded, with cell cycle arrest at the G2/M phase. Upregulation of p53, KIPI/p27 was observed, along with increased expression of the Fas ligands, mFasL and sFasL. In the mitochondrial proapoptotic pathway, increased expression of BAX and decreased Bcl2 and Bcl-XL induced the release of cytochrome C from mitochondria to cytoplasm, which then activated caspase-9. The same group carried out a similar study on ellipticine action in hepatocellular carcinoma (HCC) cells.42 On treatment with ellipticine (IC50 = 4.1 μM), p53 levels increased in a dose dependent manner and reached maximum levels at 12 h.
In 2008, Xu et al. developed a high-content screen for compounds which increase localisation of p53 to the nucleus of cytoplasm.43 Ellipticine was among several hits resulting from this screen and was subjected to further testing. In HCT116 colon cancer cells expressing wild type p53, ellipticine was found to increase overall levels of p53 and induce localisation in the nucleus, resulting in increased p21 expression. This result was in contrast to previous work, which reported that ellipticine had no effect of cells expressing wild type p53.38,40 It was also shown that p53 localisation was unrelated to ellipticine action on DNA or topoisomerase II, since p53 localisation in the nucleus was observed within one hour, whereas DNA damage was not observed until 16 hours after treatment.
Lu et al.44 identified the ellipticine derivative, 3-(9-methoxy-5,11-dimethyl-6H-pyrido[4,3-b]carbazol-6-yl)propan-1-aminium chloride 15 (Fig. 6) as a potential lead compound for p53 activation and studied its effects on three cancer cell lines. HCT116, a wild type p53 line; SW620 with mutant p53 and HCT116 p53−/−, a p53 deficient cell line were treated with 15, with GI50 values between 0.5 and 1 μM. Induction of p53 activity was observed in all three cell lines, but was highest (7.5 fold activity) in HCT116 and much less in SW620 (1.5 fold). Two p53 target proteins, p21 and DR5 were also upregulated in all three cell lines. Another transcription factor p73, with similar function to p53, was also found to be critical in the anti-tumour effects of the derivative 15.
![3-(9-Methoxy-5,11-dimethyl-6H-pyrido[4,3-b]carbazol-6-yl)propan-1-aminium chloride 15.](/image/article/2012/RA/c2ra20584j/c2ra20584j-f6.gif) | ||
| Fig. 6 3-(9-Methoxy-5,11-dimethyl-6H-pyrido[4,3-b]carbazol-6-yl)propan-1-aminium chloride 15. | ||
Recent studies have focused on combination treatments of ellipticine with existing well known anti-cancer agents. Huang et al.45 found that combined treatment of DLD1 cancer cells with 5-fluorouracil and ellipticine resulted in increased cell death compared to treatment with either agent alone. Wang et al.46 recently reported that ellipticine alone did not alter p53 expression levels in mutant p53 Raji cells. However, in chemo-resistant mutant p53 Ramos cells, ellipticine treatment sensitised cells towards doxorubicin induced apoptosis.
2.6 Clinical use of ellipticine derivatives
An early clinical trial of 9-hydroxyellipticine 4 was terminated due to solubility problems (Fig. 7),47 however, in 1994 it was used against metastatic breast cancer in combination with etoposide, methotrexate and chlorambucil, producing 48% partial response (PR) and 1.8% complete response (CR).48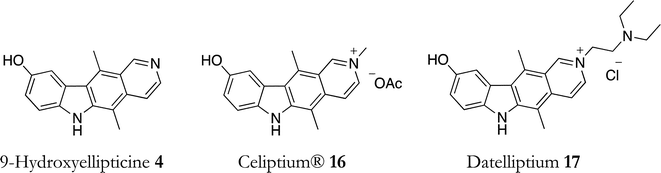 | ||
| Fig. 7 Ellipticine derivatives used in clinical trials. | ||
Clinical trials of 9-hydroxy-N-methylellipticinium acetate (Celiptium®) 16 began in 1977 and no favourable therapeutic response was observed in patients with advanced stages of gastric adenocarcinoma, lung carcinoma, metastatic soft tissue carcinoma, lymphoma or ovarian cancer. When administered to patients suffering from advanced metastatic breast cancer (refractory to all other treatment), 9-hydroxy-N-methylellipticinium acetate 16 induced objective remissions in 25% of patients, lasting up to 18 months.47 The dosage was 80–100 mg m−2/week, via 1 h i.v. infusion, for 4 weeks. The treatment was particularly effective in patients with oesteolytic breast cancer metastasis. 9-Hydroxy-N-methylellipticinium acetate 16 also gave 1 CR, 2 PR and 7 stabilisations out of 22 metastatic renal cancers.48 In another phase II study of 9-hydroxy-N-methylellipticinium acetate 16 (100 mg m−2/week) in advanced breast cancer, an objective response rate (CR & PR) of 19% was observed out of 79 patients (30% in soft tissue metastases).49
The clinical success of Celiptium® prompted extensive studies into the synthesis of ellipticinium analogues and several of these progressed to clinical trials. In 1992, 2-(diethylamino-2-ethyl)-9-hydroxyellipticinium chloride (Datelliptium) 17 was found to be active in previously treated metastatic breast cancer and devoid of the toxicities reported with 9-hydroxy-N-methylellipticinium acetate 16. Dosage was 150 mg m−2 day−1 i.v. for 5 days, every 3 weeks, and the responses were 1 CR, 4 PR in 30 patients.48 The toxicity of datelliptium was mainly hepatic, and it induced rare and mild leukopenia and severe fatigue.
In recent years, patent applications for ellipticine based cancer treatments have continued, along with patents in new areas such as treatments for genetic diseases and obesity.50–55
2.7 Summary of Biological Activity
Since its discovery, several key mechanisms of action have been found to contribute to ellipticine anticancer activity. These include DNA intercalation and topoisomerase II inhibition, two closely related mechanisms which have been well established in the literature. More recent investigations have shown that ellipticines induce multifaceted biological responses, including interaction with kinases, p53 tumour suppressors and the mitochondrial apoptotic pathway. The relative contributions of these interactions towards the overall bioactivity of ellipticines require further research, however it seems clear that the ellipticine family of compounds are truly multi-modal anticancer agents.While ellipticine and 9-hydroxyellipticine exhibit potent anticancer activity, properties such as low solubility and bioavailability have impeded their use in a clinical setting. To this end, a vast range of ellipticine derivatives have been prepared and evaluated for potential improvement in cytotoxicity, including ellipticinium salts,56–60 ellipticine glycosides,61–63 dimers,64 hybrids65–68 and conjugates.69–72
3. Classification of synthetic routes
There have been several reviews on the synthesis of ellipticine: Sainsbury73 (1977), Hewlins et al.74 (1984), Gribble and Saulnier75 (1985), Kansal et al.76 (1986), Álvarez et al.77 (2001), and finally, Knölker and Reddy78 (2002). Of these, Sainsbury, Hewlins and Gribble comprehensively reviewed the literature published to date and in particular, Gribble and Saulnier's review was extensive and detailed. Kansal focused on selected key syntheses to date, along with proposed biogenetic pathways to ellipticine and Álvarez covered a range of related alkaloids in a relatively short review. Knölker and Reddy's comprehensive review of biologically active carbazole alkaloids included a section on the synthesis of pyrido[4,3-b]carbazoles.The early reviews categorised the syntheses of ellipticine by the last ring to be cyclised (B, C or D–Fig. 1), giving three sub-groups: B-type, C-type and D-type.73,74 Gribble and Saulnier opted for a more detailed system based on the last bond to be formed, resulting in eight different categories.75 Kansal et al. used the earlier, ring-based system, adding a new B+C type category for syntheses in which the B and C rings are formed simultaneously.76 Finally, Knölker and Reddy categorised by the key reactions in the route (e.g. palladium catalysed, condensation, Diels–Alder, cycloaddition etc.).78
In this review, the ring-based system is used, giving four main categories: B-type, C-type, D-type and B+C type. Within these sections, the syntheses are generally grouped by the key reaction type. The scope of this review covers key early syntheses of pyrido[4,3-b]carbazoles (pre-1985), followed by comprehensive coverage of the literature since the late 1980s, along with more recent synthesis of ellipticine analogues and substituted ellipticines.
4. B-type syntheses
B-type syntheses may be categorised by the key B-ring forming reaction, which has been accomplished by, a) Fisher indolization, b) triazole formation, c) nitrene insertion and d) other B-type cyclisations.4.1 Fischer indolization reactions
The first B-type synthesis was developed by Stillwell and Woodward with the key B-ring formation via Fischer indolization (Scheme 3).79,73 Condensation of (Z)-pent-3-en-2-one and 1-methyl-4-piperidone in the presence of sodium hydride gave 18. This was converted to 2,5,8-trimethyloctahydroisoquinolin-6(2H)-one 19via reductive Stork alkylation using methyl iodide in the presence of lithium and liquid ammonia. The phenylhydrazone derivative 20 was formed and then subjected to Fischer indolisation to afford 2-methyl-1,2,3,4,5,5a,11,11a-octahydroellipticine 21 (82%). Finally, dehydrogenation with palladium on carbon gave ellipticine 1 in a very low yield of 0.3%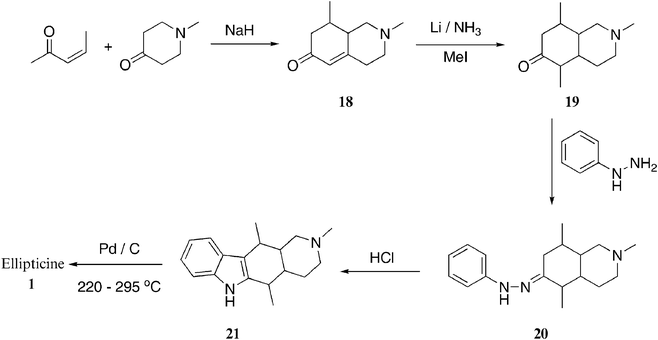 | ||
| Scheme 3 | ||
Archer et al. utilized a similar strategy in their synthesis of 5,11-demethylellipticines (Scheme 4).80 Catalytic hydrogenation of hexahydroisoquinolin-6-one 22 gave the cis and trans isomers 23 and 24, which were easily separable by chromatography. The trans isomer 24 was subjected to Fischer indolization with para-methoxyphenylhydrazine to give octahydro-1H-pyrido[4,3-b]carbazole 25 in 61% yield. The cis isomer 23 gave a mixture of 25 and a non linear isomer, which were readily separated. Debenzylation and dehydrogenation were effected with palladium on charcoal to give 9-methoxy-6H-pyrido[4,3-b]carbazole 26 (73%). This was converted to 6H-pyrido[4,3-b]carbazol-9-ol 27 with pyridine hydrochloride in 52% yield.
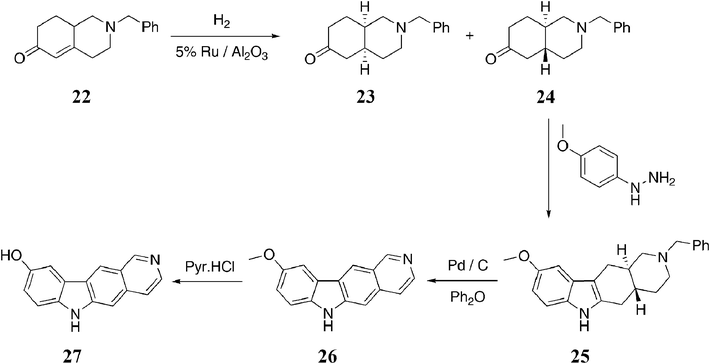 | ||
| Scheme 4 | ||
4.2 Triazole formation
Bisagni et al. were the first to use a triazole intermediate in their synthesis of the ellipticine analogue, 9-azaellipticine 28 (Scheme 5).81 The intermediate 29 cyclised under thermal conditions to give 28 in 41% yield.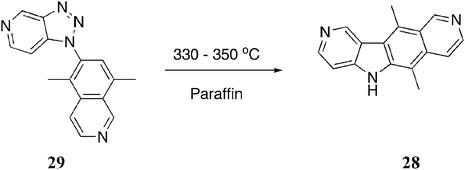 | ||
| Scheme 5 | ||
When this reaction was applied to the synthesis of 1-chloro-9-azaellipticines, only a trace of the desired product was detected. The route was successfully adapted by changing the triazole intermediate82–84 and this area is extensively reviewed by Gribble et al.75
In 1980, Miller et al. published a route involving Goldberg coupling of an acetanilide with an isoquinoline, followed by B-ring cyclisation with palladium acetate.85 Unfortunately this final cyclisation step cyclisation was rather low yielding (15–25%). As a result, a triazole intermediate was incorporated into the synthesis, providing a much more attractive route (Scheme 6).86 Goldberg coupling of o-nitroaniline or 4-methoxy-2-nitroaniline with 6-bromo-5,8-dimethylisoquinoline 30 gave the diarylamines 31 in good yields (54% and 53%). Reduction of the nitro group gave the diamines 32, which were diazotized without purification to benzotriazoles 33 (97% and 94%). After studying several methods of triazole decomposition, pyrolysis at 500 °C gave ellipticine 1 (69%) and 9-methoxyellipticine 11 (62%).
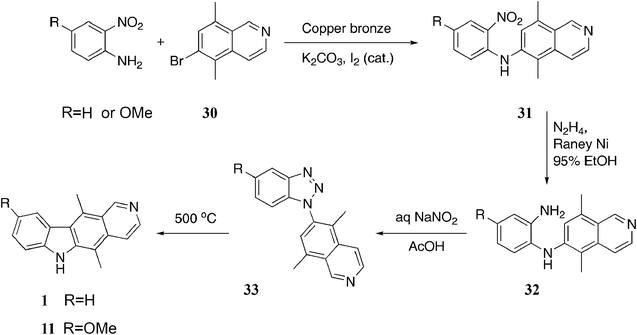 | ||
| Scheme 6 | ||
While the inclusion of a triazole intermediate significantly improved the overall yield of the route, a four step synthesis of 6-bromo-5,8-dimethylisoquinoline 30 was also required, with overall yields of 30–35%. Any alteration of the C and D rings would require adaptation of this route and, as Bisagni et al. previously discovered with 1-chloro-9-azaellipticines, could pose problems in the thermal cyclization step.82–84
In a modern application of triazole intermediates, Vera-Luque et al. prepared the 5-aza-2-deazaellipticine analogues (α-carbolines), under microwave conditions (Scheme 7).87 Commercially available benzotriazoles were reacted with 2-chloroquinoline or 2-chloro-4-methylquinoline under optimized microwave conditions to give the pyridylbenzotriazoles 34a–d in yields of 73–94%. Cyclization was carried out under microwave conditions in the presence of pyrophosphoric acid to give 5-aza-2-deazaellipticines 35a–d (19–54%).
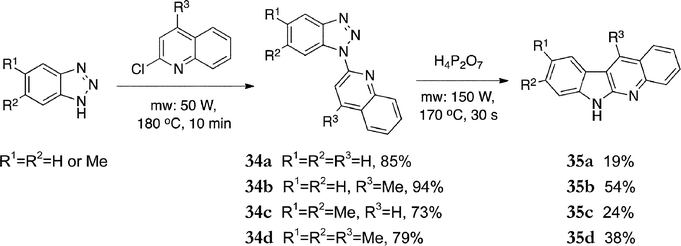 | ||
| Scheme 7 | ||
4.3 Nitrene insertion
In 1989, Miller et al. published the first synthesis to utilise nitrene insertion as the B-ring forming reaction (Scheme 8).88 7-Bromo-5,8-dimethylisoquinolin-6-amine 36 was prepared in four steps from N-(2,5-dimethylphenyl)acetamide in 50% overall yield. This was subjected to Suzuki coupling with phenylboronic acid to give the amine 37 in 99% yield, followed by conversion to the azide 38 (85%). Finally the azide was heated in dodecane to give ellipticine 1 in 96% yield after preparative thin layer chromatography.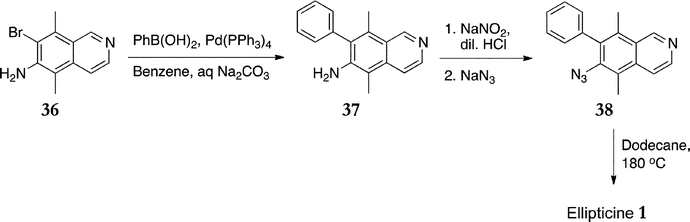 | ||
| Scheme 8 | ||
The overall yield of ellipticine, including the four step synthesis of isoquinoline 36, was 40% which is comparable with most of the highest yielding routes. Any variation in the C and D rings would require a separate synthesis of the isoquinoline, which lessens its attractiveness in this area. Nevertheless, the route would be amenable to the synthesis of A-ring substituted elllipicines or A-ring analogues due to the availability of a wide range of arylboronic acids.
In their recent synthesis of the ellipticine analogue, indazolo[2,3-b]isoquinoline 39, Timári et al. applied similar chemistry, including Suzuki coupling and nitrene insertion (Scheme 9).89 3-Hydroxyisoquinoline 40 was converted to its triflate 41 (90%) and coupled with the boronic acid 42 under Suzuki conditions to give 43 (94%). Hydrolysis to the free amine 44 and conversion to the azide 45 proceeded smoothly and in good yields (89% and 90% respectively). Ring closure was effected by heating the azide in 1,2-dichlorobenzene to give indazolo[2,3-b]isoquinoline 39 in 66% yield.
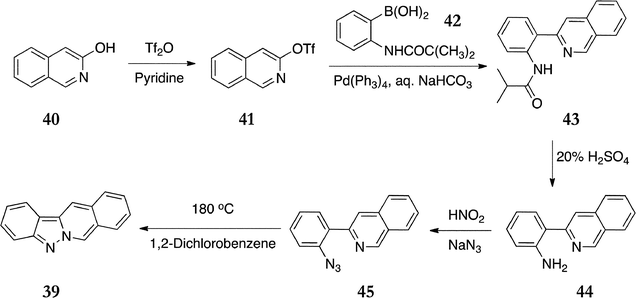 | ||
| Scheme 9 | ||
Liu and Knochel published an alternative nitrene insertion synthesis in 2007.90 Attracted by the convenient conversion of aryl triazines to azides under mild conditions, they first optimised the preparation of a wide variety of polyfunctional aryl triazines. Triazines 46 and 47 were prepared from 2-iodoaniline and 2-iodo-4-methoxyaniline in excellent yields (92% and 94% respectively, Scheme 10). Negishi coupling with 7-bromo-5,8-dimethylisoquinoline 48 gave the aryl triazines 49 (75% and 63%). Conversion to the azides 50 (78% & 94%), followed by refluxing in mesitylene gave ellipticine 1 (57%) and 9-methoxyellipticine 11 (68%).
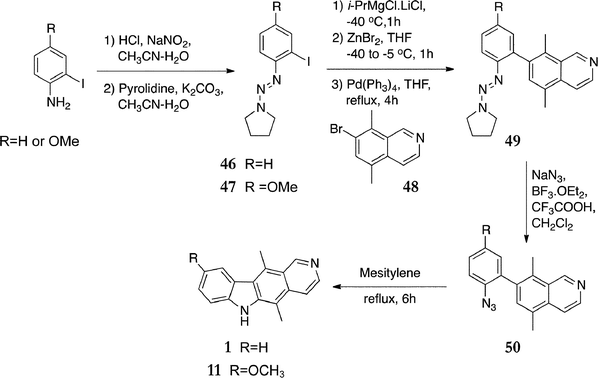 | ||
| Scheme 10 | ||
Overall yields for ellipticine and 9-methoxyellipticine were 31% and 38%. 7-Bromo-5,8-dimethylisoquinoline 48 was prepared from 1,4-dibromo-2,5-dimethylbenzene in eight steps, with an overall yield of 43%. Isoellipticine 51 and ethyl 5,11-dimethyl-10H-pyrido[3,4-b]carbazole-7-carboxylate 52 (Fig. 8) were prepared via the same route from 6-bromo-5,8-dimethylisoquinoline 30, in similar yields.
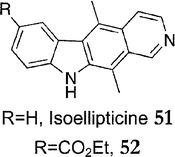 | ||
| Fig. 8 | ||
4.4 Other applications of B-type routes
Asche et al. utilized a modified Nenitzescu reaction in their synthesis of the ellipticine analogues, 5H-benzo[b]carbazoles and their corresponding quinones (Scheme 11).91 2-Aminomethylene-1-indanones 53 were prepared in two steps from 1-indanone with twenty different R2 groups (yields varying from 49% to 98%). Reaction with benzoquinones 54 in glacial acetic acid for 30 min gave 5H-benzo[b]carbazoles 55 (50%) and 56 (60–70%). The 1-substituted compounds 56, readily oxidised to the corresponding quinones 57 in 70–90% yields. The synthesis was limited to 2-aminomethylene-1-indanones 53 bearing electron donating substituents. When electron withdrawing substituents were present the reaction did not proceed at room temperature and heating resulted in the formation of spirocyclic benzofuran derivatives.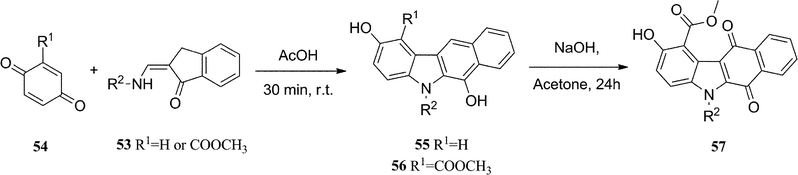 | ||
| Scheme 11 | ||
Recently, Bouclé and Guillard published their synthesis of a new pyrido[3,2-b]carbazole as an ellipticine–makaluvamine hybrid.92 The synthesis features a Hartwig–Buchwald coupling followed by a Heck reaction to form the carbazole ring system (Scheme 12). The aminotetrahydroquinoline 58 was prepared in five steps from 2,5-dimethoxyaniline in 48% yield. Pd-catalyzed Hartwig–Buchwald coupling of 58 with 2-bromoiodobenzene gave the diarylamine 59 (74%), which was cyclised to the corresponding carbazole 60 under Heck conditions (80%). Methylation of the indole nitrogen was achieved under standard conditions to give 61 in quantitative yield. Treatment of 61 with LDA followed by quenching the resulting enolate with benzenesulfonyl chloride afforded the chloroamide 62 (40%). This was treated with sodium azide followed by reduction of the amide to give 63 (62% over 2 steps). Finally, oxidation with PIFA gave the quinone, the azide was reduced by catalytic hydrogenation and cyclised to give the imine target compound 64 in 35% yield.
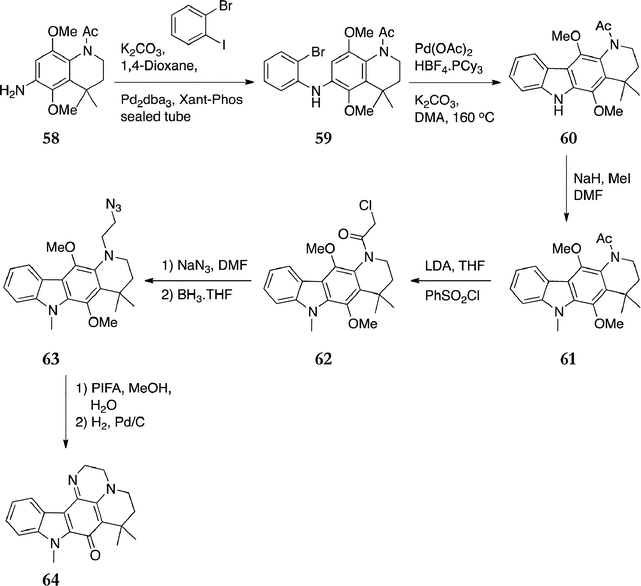 | ||
| Scheme 12 | ||
5. C-type syntheses
C-Type syntheses are some of the most versatile and highly exploited routes to ellipticine, typically involving coupling of indole and pyridine subunits. In this section, the syntheses are categorised based on the coupling reaction: C-3 coupling of indoles with substituted pyridines, C-2 coupling of indoles with substituted pyridines and cycloaddition reactions.5.1 C-3 coupling of indoles with substituted pyridines
The first synthesis of ellipticine 1 was published by Woodward et al. in 1959 along with their elucidation of the structure (Scheme 13).2 Condensation of indole with 3-acetylpyridine gave the bis-indoyl derivative 65, which was reduced with zinc and acetic anhydride to the N,γ-diacetyldihydropyridine derivative 66. This was subjected to pyrolysis to give ellipticine 1.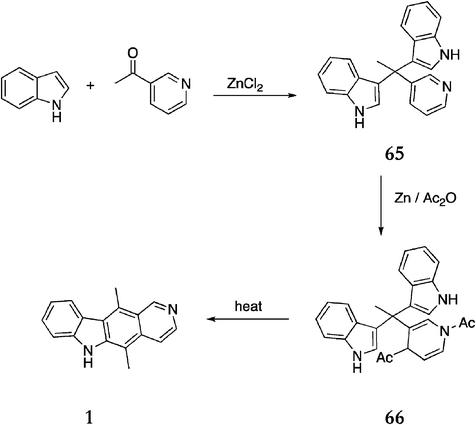 | ||
| Scheme 13 | ||
Despite being attractively simple, this route had little practical value as the overall yield was minute. The bulky bis-indoyl derivative 65 was a poor substrate for the reduction step and the final oxidative cyclisation required severe pyrolytic conditions, resulting in an overall yield of 2%.
Zee and Su were attracted by the simplicity of Woodward's original route and attempted to adapt the route to overcome the problematic reactions and low yields (Scheme 14).93 The initial condensation of indole and 3-acetylpyridine was catalysed by HCl instead of zinc chloride, which improved the yield of bis-indoyl derivative 65 (81%). Vacuum pyrolysis of 65 gave 67 (50%), which was hydrogenated to give the key intermediate 68 (90%). When reductive acetylation was carried out, the expected product (1,4 acetylation of the pyridine ring) was not isolated, as intramolecular condensation at C-2 had occurred simultaneously to give 2-acetyl-1,2-dihydroellipticine 69 in 48% yield. This was hydrolysed and aromatised by refluxing with 10% sulfuric acid in an oxygen rich environment to produce ellipticine 1 in 68% yield.
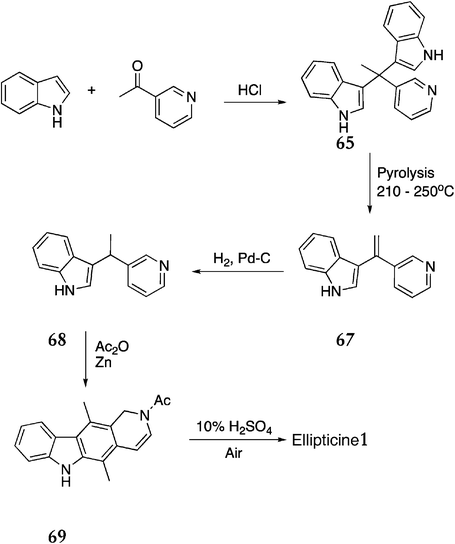 | ||
| Scheme 14 | ||
The overall yield of ellipticine from indole was 12%, a significant improvement on that of Woodward et al. (2% overall). However, two additional steps were required and the problematic pyrolysis step was still low yielding. Although this route has not been used to synthesise any substituted ellipticines, it is feasible that substitution on the indole would be tolerated.
In the course of their work on Friedel–Crafts acylation of N-protected indoles, Ketcha and Gribble developed a novel route to the ellipticine quinone 70 (Fig. 9).94 First reported in 1979 by Talyor and Joule, the ellipticine quinone has proved a popular and convenient ellipticine precursor.95 Treatment of 70 with two equivalents of methyllithium, followed by sodium borohydride, affords ellipticine in yields of over 90% and it is a key intermediate in a variety of C-type routes.
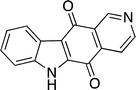 | ||
| Fig. 9 Ellipticine quinone 70. | ||
In the Gribble route, 3,4-pyridinedicarboxylic anhydride 71 was converted to the monomethylester 72 (73%), followed by conversion to the acid chloride 73 with thionyl chloride in 78% yield.94 Friedel–Crafts acylation of 1-phenylsulfonylindole with 73 gave the keto ester 74 (50%), which was cyclised employing Comins' methodology to the ellipticine quinone 70 (47%) (Scheme 15).
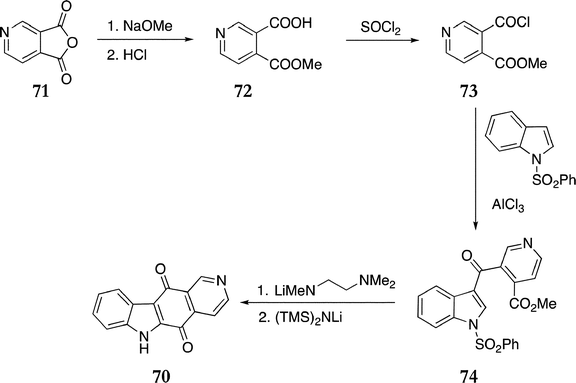 | ||
| Scheme 15 | ||
The analogous quinones 75, 76 (Fig. 10) were also synthesised via this route. The deazaellipticine quinone 75 was synthesised in three steps from phthalic anhydride (21% overall). The isoellipticine quinone 76 was accessible via direct reaction of 3,4-pyridinedicarboxylic anhydride 71 with 1-phenylsulfonylindole (19% over three steps).
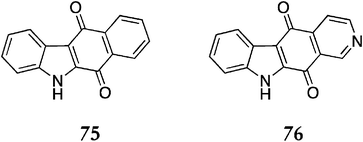 | ||
| Fig. 10 | ||
While not the best yielding route to ellipticine (13% overall yield for the ellipticine quinone 70), this route does provide access to a variety of ellipticine analogues.
In 2005, Bennasar et al. published a radical cascade route to the ellipticine quinone (Scheme 16).96 Reaction of the N-benzyl-2,3-disubstituted indole 77 with 3-pyridylmagnesium bromide gave the coupled product 78, which was reduced with triethylsilane to the methyl ester 79 (55% from 77). Hydrolysis followed by phenylselenation of the carboxylic acid yielded the acyl selenide 80 in 76% yield. The key cyclisation step was carried out with irradiation and hexabutylditin, affording the N-benzylellipticine quinone 81 in a 42% yield.
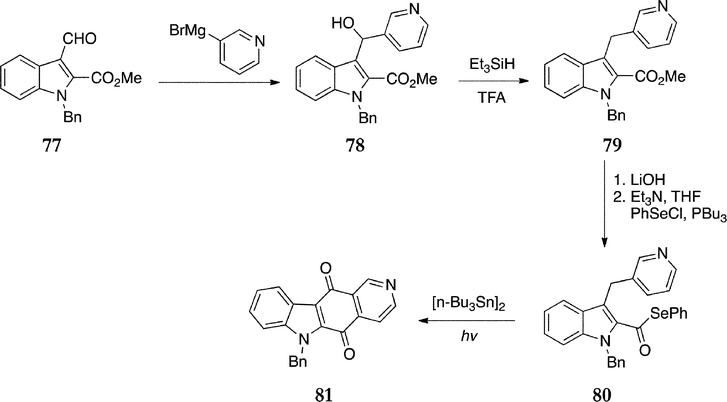 | ||
| Scheme 16 | ||
The N-benzylellipticine quinone 81 may be converted to ellipticine in two steps; treatment with methyllithium followed by sodium borohydride would give N-benzylellipticine. However, subsequent removal of the benzyl group has been shown to be problematic, Birch reduction effects deprotection but also reduces the pyridine ring which must then be re-aromatised with 10% palladium on carbon and decalin.97 These additional steps significantly reduce the overall yields for the route and impact on its versatility. Other indole protecting groups (Me, MOM) were used, but with little success.
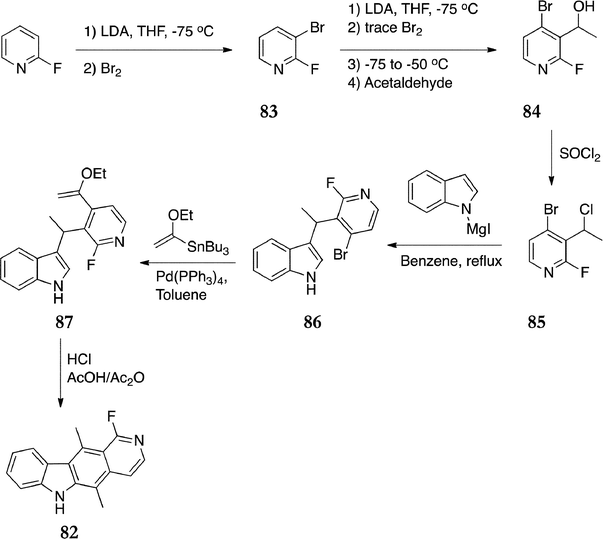
In their synthesis of 1-fluoroellipticine 82, Marsais et al. prepared a tri-functional pyridine for C-3 coupling with an indole (Scheme 17).98 Initially 2-fluoropyridine was brominated to give 3-bromo-2-fluoropyridine 83 in 75% yield. This was lithiated and isomerised by LDA to give 4-bromo-2-fluoro-3-lithiopyridine, followed by treatment with acetaldehyde to give the tri-substituted pyridine 84 (75%). The acetaldehyde had to be added quickly, with efficient reaction cooling in order to avoid forming an isomeric side product. The chloro derivative 85 was prepared by treatment with thionyl chloride (95%) and reacted with 1-indolylmagnesium iodide to give 86 in 65% yield. Next, lithium–bromine exchange followed by treatment with acetaldehyde was attempted but the lithio derivative of 86 did not react with acetaldehyde and gave the debrominated product. However, reaction with a tributylvinylstannane gave the vinyl compound 87 (95%), which readily cyclized under acidic conditions to afford 1-fluoroellipticine 82 in 54% yield.
 | ||
| Scheme 17 | ||
5.2 C-2 coupling of indoles with substituted pyridines
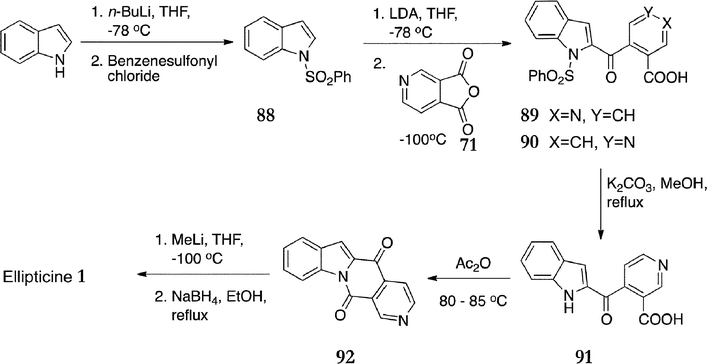
In 1992, Gribble et al. published full detail of their general and versatile C-type route (Scheme 18).99,100 Initially, indole was protected to give N-phenylsulfonylindole 88 in 91% yield. Regiospecific C-2 lithiation with lithium diisopropylamide followed by addition of 3,4-pyridinedicarboxylic anhydride 71 gave a mixture of protected ketoacids 89 & 90 (79%). Ring opening of the anhydride was regioselective for the desired product 89, and the regioisomers were separated by fractional crystallisation from acetone. The minor isomer was insoluble in acetone and the ratio of 89 to 90 was found to be 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8. Deprotection of the indole was achieved with potassium carbonate in methanol to give ketoacid 91 in quantitative yield. At this point, Sauliner and Gribble had envisaged cyclisation to the ellipticine quinone 70, however, after several unsuccessful attempts to cyclise the ketoacid 91, it was found that heating in acetic anhydride affected cyclisation to the ketolactam 92 (100%). This ketolactam was smoothly converted to ellipticine 1 in 82% yield by treatment with two equivalents of methyllithium followed by refluxing with sodium borohydride in ethanol.
:8. Deprotection of the indole was achieved with potassium carbonate in methanol to give ketoacid 91 in quantitative yield. At this point, Sauliner and Gribble had envisaged cyclisation to the ellipticine quinone 70, however, after several unsuccessful attempts to cyclise the ketoacid 91, it was found that heating in acetic anhydride affected cyclisation to the ketolactam 92 (100%). This ketolactam was smoothly converted to ellipticine 1 in 82% yield by treatment with two equivalents of methyllithium followed by refluxing with sodium borohydride in ethanol.
 | ||
| Scheme 18 | ||
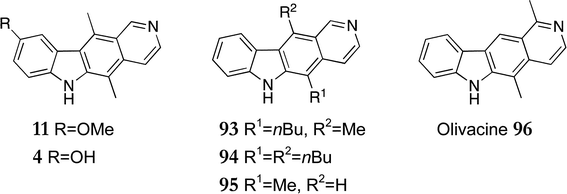
This synthesis has been successfully utilised to produce a wide variety of substituted ellipticines and ellipticine analogues. 9-Methoxyellipticine 11 was synthesised starting from 5-methoxyindole in an overall yield of 47%, subsequent demethylation gave 9-hydroxyellipticine 4 (93%) (Fig. 11).100 Various 5,11-disubstituted ellipticines were also synthesised via sequential regioselective alkylations of ketolactam 92 giving 5-n-butyl-11-methyl-6H-pyrido[4,3-b]carbazole 93 (70%), the 5,11-di-n-butyl derivative 94 (18%) and 11-demethylellipticine 95 (57%). 11-Demethylellipticine 95 had previously been converted to the natural product olivacine 96 by treatment with methyllithium followed by oxidation of a dihydropyridine intermediate with iodine (54%).101
 | ||
| Fig. 11 | ||
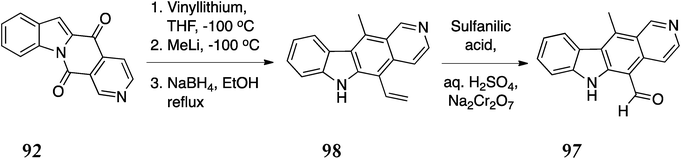
13-Oxoellipticine 97, an alkaloid isolated from the African tree Strychnos dinklagei was also synthesised via this route (Scheme 19).102,103 Ketolactam 92 was treated with vinyllithium followed by methyllithium to give 98, which was oxidatively cleaved with chromic acid to give 13-oxoellipticine 97 (58% from 92).
 | ||
| Scheme 19 | ||
Several ellipticine analogues were accessible via this route by varying the anhydride used (Fig. 12), including isoellipticine104 (5,11-dimethyl-10H-pyrido[3,4-b]carbazole) 51 (85% from the minor isomer of the coupling reaction 90), 5,11-dimethyl-10H-pyrido[2,3-b]carbazole10599 (60% overall) and deazaellipticine106 (5,11-dimethyl-5H-benzo[b]carbazole) 100 (61% overall).
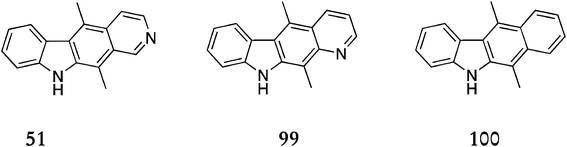 | ||
| Fig. 12 | ||
Modi et al. developed a route for the synthesis of C-11 substituted ellipticines from Gribble's ketolactam 92 (Scheme 20).107,108 Treatment of the ketolactam 92 with one equivalent of methyllithium gave the lactone 101 in 62% yield. This was converted to the bisulfide 102 (45%) and then 12-oxoellipticine 103 (85%). Finally, the formyl group was reduced to the alcohol 5-methyl-11-methanol-6H-pyrido[4,3-b]carbazole 104 in 89% yield.
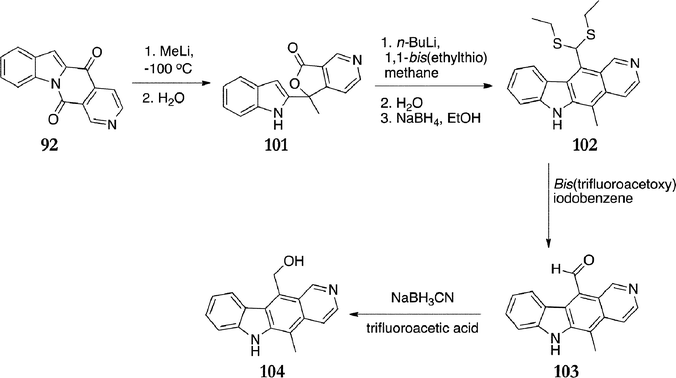 | ||
| Scheme 20 | ||
Overall, Saulnier and Gribble's synthesis is one of the highest yielding and most versatile routes to ellipticine and its analogues. C-5 and C-11 substituents may be introduced via sequential regioselective alkylation of the ketolactam intermediate. A-Ring substituents may be varied using substituted indoles, and D-ring ellipticine analogues (C-1 to C-4) are accessible by variation of the anhydride used.
In 1990, Hibino and Sugino published a route based on thermal electrocyclic reaction of a 2-alkenylindole intermediate (Scheme 21).109 Initially 3-ethylpyridine 105 was converted to the N-oxide 106 in 81% yield. This was converted to 3-ethyl-4-cyanopyridine followed by treatment with methyllithium to give 3-ethyl-4-acetylpyridine 107 (39%). Reaction of 107 with 2-lithio-phenylsulfonylindole afforded the key 2-alkenylinole intermediate 108 in 21% yield. Thermal cyclisation to ellipticine 1 (30%) was only partially successful, as it was accompanied by 11-demethylellipticine 95 (43%).
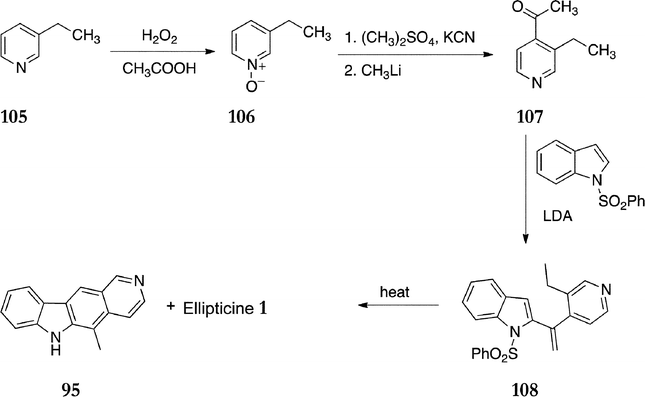 | ||
| Scheme 21 | ||
This route suffered from low yields in both the coupling and cyclisation steps along with the problem of demethylation during thermal cyclisation, resulting in an overall yield of 2% for ellipticine. The thermal cyclisation step may be amenable towards synthesis of 11-demethylellipticine analogues, however, the synthesis of olivacine 96 (Fig. 11) resulted in an overall yield of just 0.3% due to poor yields in the preceding steps.
In parallel with their work on the synthesis of 1-aminoellipticines, Bisagni et al. synthesised a range of 11-aminoellipticines 109via a novel C-2 coupling route (Scheme 22).110 5-Methoxy-1-(phenylsulfonyl)-1H-indole 110 was lithiated at C-2 and reacted with 4-acetyl-N,N-diisopropylnicotinamide 111 to give the coupled product 112 in 62% yield. This was converted to the lactone 113 (80%) and reduced with activated zinc powder in acetic acid followed by hydrochloric acid to give carboxylic acid 114 (62%). Conversion to the amide derivatives 115 was achieved via acylimidazole intermediates in yields of 55–80%. Finally, cyclization in phosphorus oxychloride and removal of the protecting group with RANEY® nickel gave the 11-aminoellipticines 109a–c (23–32%).
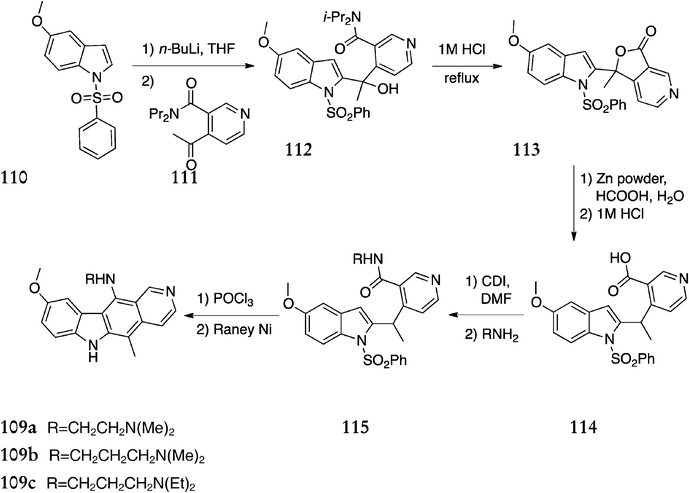 | ||
| Scheme 22 | ||
Bisagni et al. also adapted this route for synthesis of the 9-azaellipticine analogues 116 (Fig. 13).111
 | ||
| Fig. 13 9-Azaellipticine analogues 116. | ||
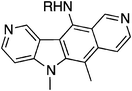
Miki et al. developed a route via coupling of an N-protected indole-2,3-dicarboxylic anhydride with a substituted pyridine.112,113 1-(4-Methoxybenzyl)indole-2,3-dicarboxylic anhydride 117 was reacted with (3-bromo-4-pyridyl)triisopropoxytitanium 118 at low temperature to give the coupled product 119 (61%). Removal of both the para-methoxybenzyl (PMB) and carboxy groups was effected with 20% perchloric acid to give 120 in 81% yield. (In the initial publication,112117 was protected with a benzyl group, however, low yields for deprotection resulted in a change to the para-methoxybenzyl group).113 A Wittig reaction furnished 121 (63%), which was then hydrogenated to 122 in 73% yield. Treatment of 122 with (ethoxyvinyl)tributyltin in the presence of tetrakis(triphenylphosphine)palladium(0) gave the corresponding ethoxyvinyl derivative 123. This was converted to ellipticine by treatment with 10% hydrochloric acid (87%) (Scheme 23).
 | ||
| Scheme 23 | ||
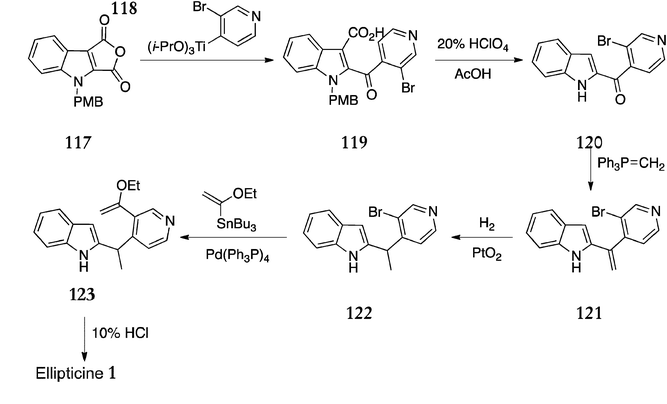
A novel route published in 2000 by Ishikura et al. utilised palladium catalysed tandem cyclisation–cross-coupling of an indolylborate and vinyl bromide (Scheme 24).114 1-(t-Butyloxycarbonyl)indole 124 was converted to the indolylborate 125, which was reacted in situ with vinyl bromide 126 using a 1:4 ratio of Pd2(dba)3·CHCl3 and Ph3P to give the hexatriene 127 (64%). Irradiation of 127 with a high pressure mercury lamp yielded the tetrahydropyridocarbazole 128 in 41% yield. Deprotection of the carboxybenzyl (Cbz) group was achieved by hydrogenation and oxidation with manganese dioxide afforded the Boc-protected ellipticine 129. Finally, the Boc group was removed with trifluoroacetic acid to give ellipticine in 84% yield.
 | ||
| Scheme 24 | ||
This synthesis is a novel strategy towards ellipticine, however, disadvantages include a three step synthesis of the vinyl bromide 126 and a moderate overall yield of 14%. The route may be amenable to synthesis of A-ring substituted ellipticines and variation of the vinyl bromide could be used to produce C or D-ring substituents.
5.3 Cycloaddition reactions
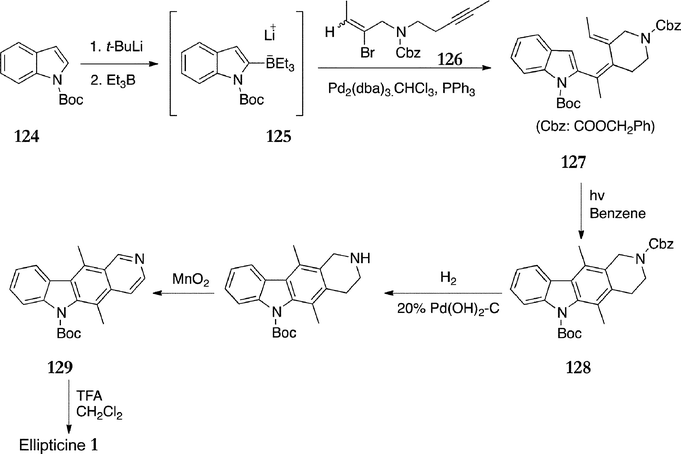
The first route to utilise a cycloaddition as the C-ring forming reaction was published in 1984 by Gribble et al.115 The key step involved a Diels–Alder reaction of furo[3,4-b]indole 130 and 3,4-pyridyne (Scheme 25). 3-Ethylindole 131 was protected with a benzenesulfonyl group to give 132 (74%), followed by C-2 lithiation and quenching with acetaldehyde which furnished the desired alcohol 133 (73%). Oxidation, bromination and hydrolysis gave the hydroxy ketone 134 in 86% yield from 133. This conveniently cyclised to the furo[3,4-b]indole 130 during attempted recrystallisation from dichloromethane in quantitative yield. Furo[3,4-b]indole 130 was also synthesised from indole-3-carboxaldehyde in a yield of 21% over 5 steps in the same publication. | ||
| Scheme 25 | ||
In the cycloaddition reaction, 3,4-pyridyne was generated in situ from reaction of 1-aminotriazolo[4,5-c]pyridine and lead tetraacetate, to which 130 was added to give a mixture of the isomeric cycloadducts 135a+b (38%). Treatment with sodium borohydride and sodium hydroxide effected both desulfonylation and oxygen-bridge extrusion to afford an approximately 1:1 mixture of ellipticine 1 and isoellipticine 51 , which were separated by column chromatography (23% and 29% respectively).
The two main disadvantages of this route were the lengthy synthesis of the furo[3,4-b]indole 130 and the lack of regioselectivity in the cycloaddition step resulting in a 1:1 mixture of ellipticine and isoellipticine.
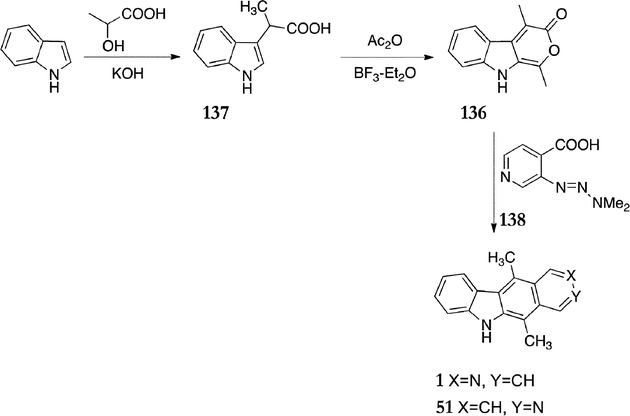
A somewhat similar route was published by May and Moody around the same time as that of Gribble et al. above and was based on Diels–Alder reaction of the pyranoindolone 136 and pyridyne (Scheme 26).116,117 Indole was first converted to 2-(1H-indol-3-yl)propanoic acid 137 (32%) by treatment with lactic acid and base, followed by reaction with boron trifluoride in acetic anhydride to give the pyranoindolone 136 (43%). The pyridyltriazene 138 was synthesised in a one pot procedure by diazotisation of 3-aminopyridine-4-carboxylic acid followed by coupling with dimethylamine (73%). Heating pyranoindolone 136 with excess pyridyltriazene 138 in acetonitrile gave a 1:1 mixture of ellipticine 1 and isoellipticine 51 (40%).
 | ||
| Scheme 26 | ||
While this route was significantly shorter than the previous route and did not require protection of the indole nitrogen, the lack of regioselectivity in the cycloaddition step still remained a major drawback.
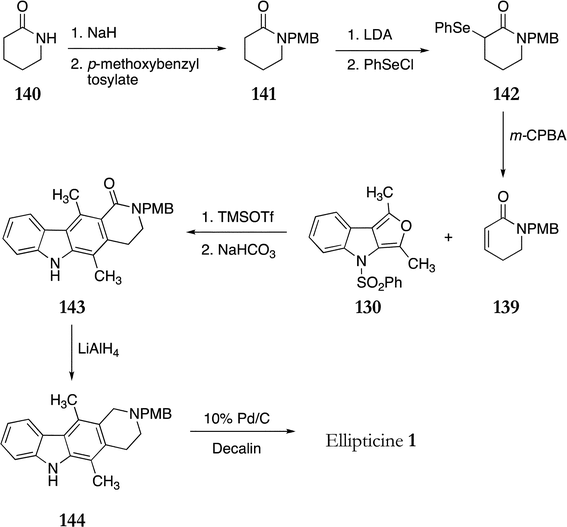
Gribble et al. sought to overcome the problem of regioselectivity by changing the dienophile in their route.118 In preliminary investigations they found that the key intermediate furo[3,4-b]indole 130 reacted completely regioselectively with ethyl acrylate in the presence of aluminium chloride. Thus, they choose 5,6-dihydropyridone 139 as a new dienophile for the route (Scheme 27). Sequential treatment of lactam 140 with sodium hydride and p-methoxybenzyl tosylate gave the N-protected lactam 141 in 68% yield. This was lithiated with lithium diisopropylamide and then quenched with phenylselenenyl chloride to yield 142 (94%), followed by oxidative elimination to give 5,6-dihydropyridone 139 (78%).
 | ||
| Scheme 27 | ||
The Diels–Alder reaction between furo[3,4-b]indole 130 (synthesis in Scheme 25 above) and 5,6-dihydropyridone 139 successfully produced a single regioisomer 143 in 40% yield. This was converted to ellipticine in two steps via reduction of the carbonyl to give 144, followed by N-deprotection and aromatization to give ellipticine (20%). No isoellipticine was detected and, as such this was the first regioselective Diels–Alder synthesis of ellipticine.
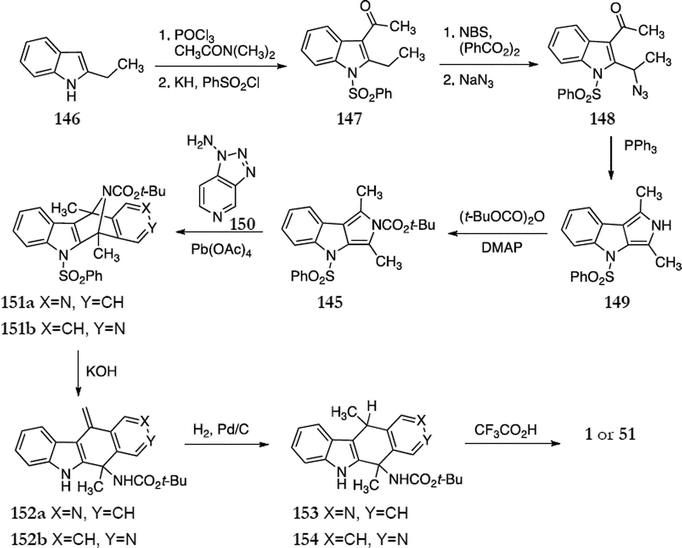
In 1992, Sha and Yang published a synthesis of ellipticine based on a Diels–Alder reaction between 2,4-dihydro-1,3-dimethylpyrrolo[3,4-b]indole 145 and 3,4-pyridyne (Scheme 28).119 This route is a variation on the first Diels–Alder route published by Gribble et al.115 in 1984. Acetylation of 2-ethylindole 146 followed by N-protection of the indole gave 147 in 68% yield. This was brominated and reacted with sodium azide to give the azido derivative 148 (78%). A Staudinger reaction of 148 with triphenylphosphine produced dihydropyrolo[3,4-b]indole 149 (90%) which was reacted with Boc anhydride and 4-dimethylaminopyridine to give the key intermediate 2,4-dihydropyrrolo[3,4-b]indole 145 (90%). 3,4-Pyridyne was generated in situ from 1-aminotriazolo[4,5-c]pyridine 150 and reacted with 145 to give the cycloadducts 151a & b (62% yield, ratio 55:45). Treatment of this mixture with base gave 152a (35%) and 152b (39%) which were separated by column chromatography. Both compounds were separately hydrogenated to give 153 (80%) and 154 (83%) followed by conversion to ellipticine 1 (85%) and isoellipticine 51 (78%).
 | ||
| Scheme 28 | ||
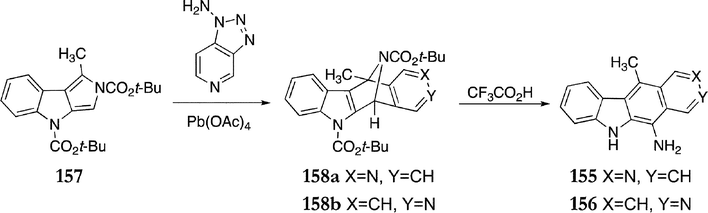
This approach was also used to synthesise new 5-aminoellipticines 155 & 156, with 157 as the key intermediate (Scheme 29). The Diels–Alder reaction gave cycloadducts 158a & b (65%), which were treated with trifluoroacetic acid to give 5-aminoellipticine 155 and 11-aminoisoellipticine 156 (60% combined yield).
 | ||
| Scheme 29 | ||
Diaz et al. investigated the regioselective effect of substituents on the pyridyne in the Diels–Alder reactions, focusing on two previously published routes115,117 (Scheme 25–Gribble & Scheme 26–Moody).120 They synthesised four substituted pyridynes 159a–d (Fig. 14), selected for the ease of introduction and removal of the halogen and to allow modulation of polarisation and steric hindrance.
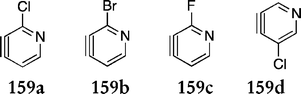 | ||
| Fig. 14 | ||
Reactions of pyridynes 159b–d with Gribble's furo[3,4-b]indole 130 showed no regioselectivity, all gave 1:1 mixtures of cycloadducts. However, pyridyne 159a afforded modest regioselectivity towards the ellipticine cycloadduct 160a over the isoellipticine cycloadduct 160b, with a ratio of 2.4:1 (88%) (Scheme 30). The cycloadducts were separated and 160a was converted to ellipticine in two steps (88%).
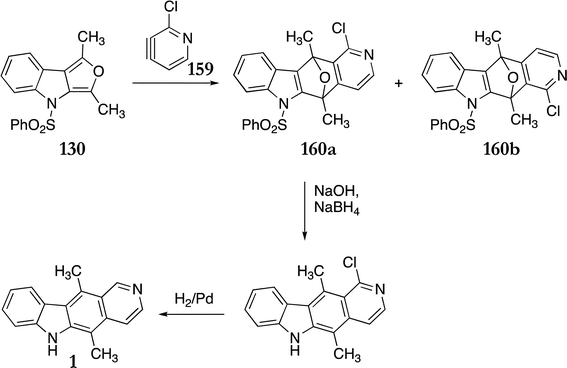 | ||
| Scheme 30 | ||
When Moody's pyranoindole 136 (see Scheme 26 above) was reacted with pyridynes 159a–d no regioselectivity was observed. In an attempt to make Moody's diene more similar to Gribble's, a benzenesulfonyl group was introduced on the indole nitrogen to give 161 in a 78% yield. This pyranoindole reacted regioselectively with pyridyne 159a to give the cycloadduct 162 as the sole product (20%) (Scheme 31). N-Deprotection and hydrogenation gave ellipticine in 46% yield.
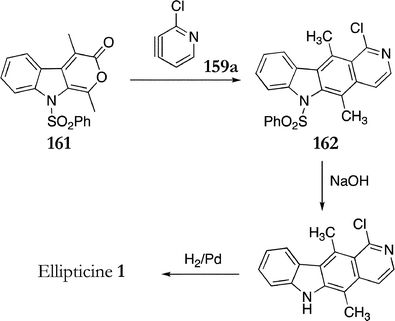 | ||
| Scheme 31 | ||
Mal et al. devised an anionic [4+2] cycloaddition strategy towards the ellipticine quinone and related carbazole quinones.121,122 Indolofuranone 163 was treated with triethylamine and ethylchloroformate to give the N-ethoxycarbonyl derivative 164 in 92% yield (Scheme 32). Reaction of 164 with 3,4-pyridyne (prepared in situ from 3-bromopyridine) in the presence of lithium diisopropylamide gave an inseparable mixture of the ellipticine and isoellipticine quinones 70 & 76 (45%).
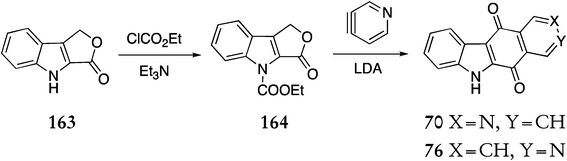 | ||
| Scheme 32 | ||
6. D-type syntheses
To date the vast majority of D-type syntheses have been based on an early route published by Cranwell and Saxton in 1962.123 A wide range of modifications and improvements have been published since, mainly focusing on improvement of carbazole formation or D-ring cyclisation. Two other D-type syntheses, not based on the Cranwell–Saxton route, are discussed under other D-type routes.6.1 Cranwell–Saxton based approaches
Cranwell and Saxton published their D-type route in 1962 and numerous modifications and adaptations have been published since.123 In the original route, 1,4-dimethylcarbazole 165 was prepared from indole and hexane-2,5-dione under acidic conditions in 36% yield (Scheme 33). Vilsmeier–Haack formylation furnished 3-formyl-1,4-dimethylcarbazole 166 along with the expected side product 3,6-diformyl-1,4-dimethylcarbazole and these were separated by chromatography to give 166 in 40% yield. Condensation with 2,2-diethoxyethylamine gave the imine 167 (85%) and attempts to convert this directly to ellipticine via Pomeranz–Fritsch synthesis were unsuccessful, so 167 was hydrogenated to produce the amine 168 (71%). Cyclisation was achieved on treatment with dry hydrogen chloride in ethanol, followed by dehydrogenation with palladium on carbon to furnish ellipticine 1 in a yield of 9.6% over two steps.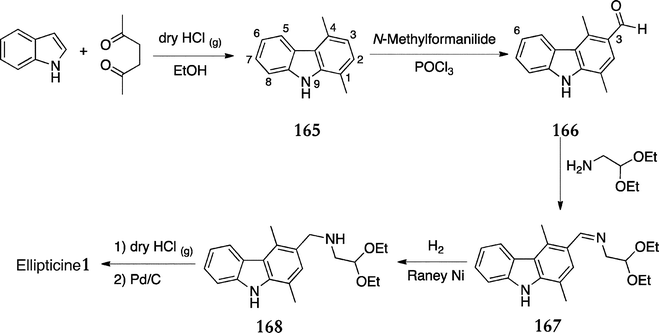 | ||
| Scheme 33 | ||
With an overall yield of just 0.8%, this route was even lower yielding than Woodwards first synthesis in 1959.2 The low yielding carbazole formation, lack of specificity in the Vilsmeier–Haack formylation and the poor yielding cyclisation–aromatisation reaction were the most problematic steps. However, the chemistry was attractively robust and these problematic reactions offered ample opportunity for optimisation, resulting in numerous publications in subsequent years.
Based on their studies of Pomeranz–Fritsch isoquinoline synthesis,124 Jackson et al. published the first and most significant modification of the Cranwell–Saxton route, dramatically improving the yield of the final cyclisation step (Scheme 34).125 Conversion of amine 168 to its corresponding tosylate 169 (93%), followed by cyclisation in 1,4-dioxane and dilute hydrochloric acid, gave ellipticine in 90% yield (84% over the two steps).
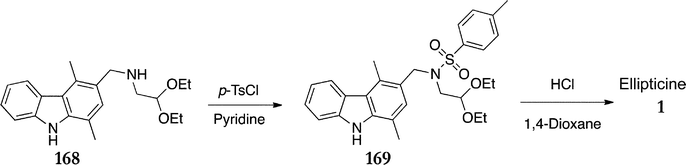 | ||
| Scheme 34 | ||
Later, the same group applied this strategy in their synthesis of 8,9,10-trimethoxyellipticine.126 Gruthrie et al. also utilised this modified Cranwell–Saxton route in their synthesis of 8,9-dimethoxyellipticine 170, starting from the 5,6-dimethoxyindole 171 (Scheme 35).127
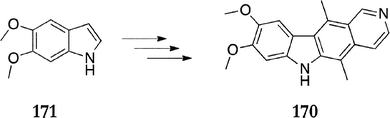 | ||
| Scheme 35 | ||
In 1970, Bruck reported an unusual product of a Vilsmeier–Haack reaction with N-methyl-1,2,3,4-tetrahydrocarbazole.128 Later, Murakami et al. investigated this reaction and subsequently applied it to the synthesis of ellipticine and olivacine (Scheme 36).97,129 When 9-benzyl-1,2,3,4-tetrahydrocarbazoles 172a & b were treated with two equivalents of phosphorous oxychloride in dimethylformamide at 0 °C, 9-benzyl-1-formyl-1,2,3,4-tetrahydrocarbazoles 173 were isolated in good yields. However, when the same reaction was carried out at 120 °C, the unexpected 1-methyl-3-formylcarbazole products 174 & 175 were formed. 9-Benzyl-1,4-dimethyl-9H-carbazole-3-carbaldehyde 174 was converted to ellipticine via the modified Cranwell–Saxton route. The imine intermediate was subjected to Birch reduction to effect simultaneous debenzylation and reduction to the amine. This gave ellipticine in an overall yield of 22% from 9-benzyl-1,2,3,4-tetrahydrocarbazole 172. 9-Benzyl-1-methyl-9H-carbazole-3-carbaldehyde 175 was converted to olivacine 96via treatment with methyllithium and Jones reagent to give the methyl ketone, followed by the usual D-ring steps. This route was less attractive as the final cyclisation gave a 1.2:1 mixture of olivacine 96 and its non-linear isomer 176.
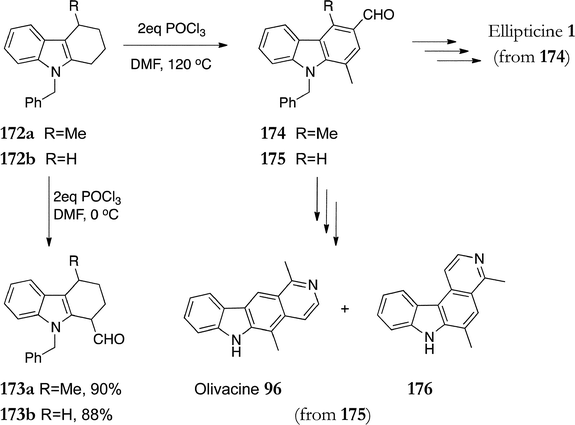 | ||
| Scheme 36 | ||
During the 1990s, Shannon's group focused on the synthesis of A-ring substituted ellipticines via the Cranwell–Saxton route. The main obstacle to overcome was lack of regioselectivity in the Vilsmeier–Haack reaction of A-ring (hydroxy or methoxy) substituted carbazoles. The group published several different strategies, the first of which involved selective bromination of the required carbazole and conversion to a key nitrile intermediate (Scheme 37).130 This strategy was similar to that developed by Hogan et al. in the late 1980s for the synthesis olivacine 96.131,132
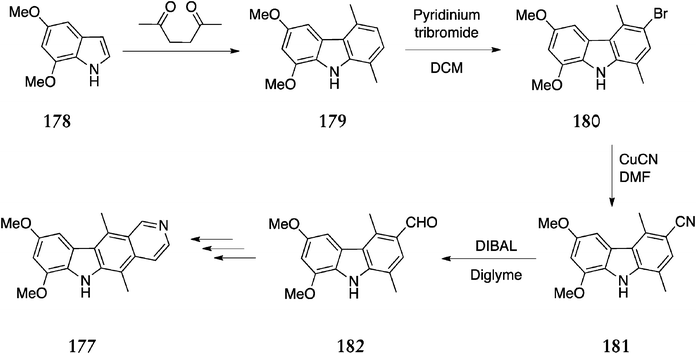 | ||
| Scheme 37 | ||
In Shannon's route to 7,9-dimethoxyellipticine 177, 5,7-dimethoxyindole 178 was condensed with hexane-2,5,-dione to give 6,8-dimethoxy-1,4-dimethyl-9H-carbazole 179 in 72% yield. Vilsmeier–Haack reaction of this compound resulted in formylation at the C-5 position rather than the desired C-3 position. When the solvent was changed to dimethylformamide, formylation occurred at both C-5 and C-3. As a result, selective bromination at C-3 was investigated, and treatment of carbazole 179 with pyridinium tribromide in dichloromethane, gave 3-bromo-6,8-dimethoxy-1,4-dimethyl-9H-carbazole 180 (72%). Conversion to the nitrile 181 was carried out with copper(I)cyanide (52%) and DIBAL reduction gave the desired 3-formyl-6,8-dimethoxy-1,4-dimethylcarbazole 182 (66%). This was converted to 7,9-dimethoxyellipticine 177 in three steps via Jackson's route125 (53%), with an overall yield of 9% from 5,7-dimethoxyindole 178.
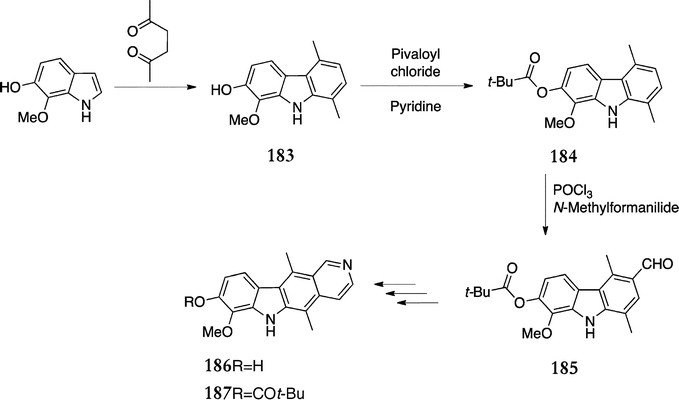
In an alternative route to A-ring substituted ellipticines, Shannon's group also used a t-butylester as protecting group to direct selective Vilsmeier–Haack formylation (Scheme 38).133 6-Hydroxy-7-methoxyindole was converted to the corresponding carbazole 183 and as expected, the Vilsmeier–Haack reaction resulted in formylation at C-6. The group next used an acetate group to protect the alcohol and then successfully formylated at C-3, however the acetate group caused problems later in the synthesis. Finally the t-butylester 184 was formed in 81% yield and subjected to Vilsmeier–Haack conditions to give the 3-formyl product 185 (78%). This was converted to the corresponding ellipticine via Jackson's route,125 with the final cyclisation step yielding a mixture of 8-hydroxy-7-methoxyellipticine 186 (44%) and the t-butylester 187 (27%).
 | ||
| Scheme 38 | ||
Later, the same group published the synthesis of 10-hydroxyellipticine via the above protection strategy.134
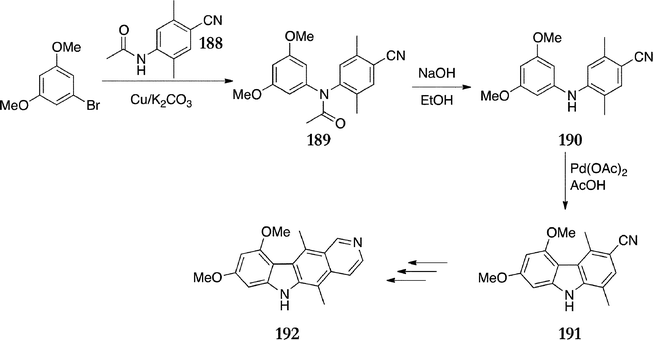
Finally, in a third route to A-ring substituted ellipticines, Shannon et al. circumvented the problematic Vilsmeier–Haack reaction by using substituted diphenylamines as intermediates (Scheme 39).134,135 Goldberg coupling of 1-bromo-3,5-dimethoxybenzene with the amide 188 gave 189 (72%), which was hydrolysed to give the diphenylamine 190 in 98% yield. Cyclisation with palladium acetate afforded the carbazole 191 in a disappointing yield of 32%. DIBAL reduction followed once again by Jackson's modified Cranwell–Saxton route125 gave 8,10-dimethoxyellipticine 192.
 | ||
| Scheme 39 | ||
While overall yields were relatively low, this route was used to synthesise a variety of A-ring mono and disubstituted ellipticines.134,135 Groundwater and Lewis also used a similar strategy in their synthesis of 7-fluoroelliptcine in 1995.136
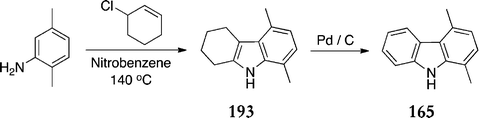
Mustafin et al. also published an alternative route towards carbazoles for use in the Cranwell–Saxton route (Scheme 40).137–139 Reaction of 2,5-dimethylaniline and 3-chlorocyclohexene gave the tetrahydrocarbazole 193via Claisen rearrangement of the initial N-alkenylamine followed by intramolecular cyclisation (61%). Dehydrogenation with palladium on carbon gave 1,4-dimethylcarbazole 165 in 87% yield.
 | ||
| Scheme 40 | ||
While 1,4-dimethylcarbazole 165 was formed in two steps rather than one in the Cranwell–Saxton method, the yield is higher (53% versus 36%). The group also prepared 1-methylcarbazole and 1-methoxycarbazole from the appropriate anilines in yields of 64–78%.
In 2007, Dračínský et al. reported a further improvement of the final cyclisation step.140 Use of a benzenesulfonyl group instead of Jackson's para-toluenesulfonyl group improved both yield and reaction time (Scheme 41). Thus, the benzenesylfonyl amine 194 was refluxed with hydrochloric acid in 1,4-dioxane for just 20 min (previously 6 hours) to give a 97% yield of ellipticine 1 (previously 90%). In addition to this, the product crystallised directly from the reaction mixture and no further purification was required.
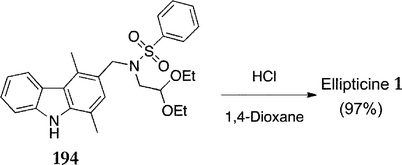 | ||
| Scheme 41 | ||
Recently, Lee et al. investigated the use of microwave reaction conditions on the Cranwell–Saxton route, resulting in improved yields and reaction times (Scheme 42).141 The initial cycloaddition proceeded in 75% yield in the microwave, followed by the conventional Vilsmeier–Haack reaction (55%). Next, a one-pot reaction sequence formed the tosylated amine 169 in 68% yield and the final cyclization step to ellipticine proceeded in 76% yield.
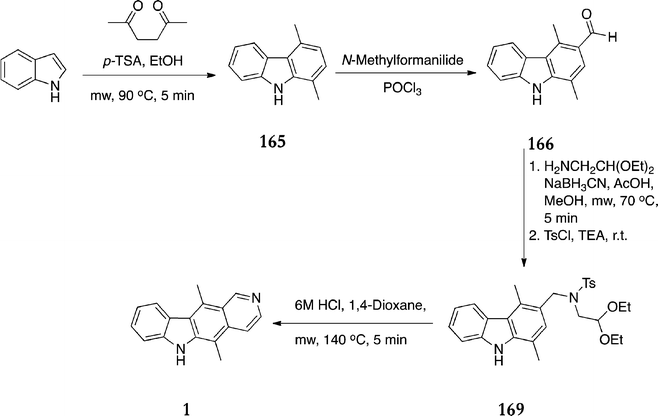 | ||
| Scheme 42 | ||
The incorporation of microwave heating in this route resulted in an overall yield of 21%, which compares very favourably with the original route (0.8%) and also Jackson's modification (7%). Reaction times are also dramatically reduced. Other ellipticines prepared under these conditions include 9-bromoellipticine (17% from 5-bromoindole) and 9-nitroellipticine (7% from 1,4-dimethyl-3-formylcarbazole 166).
Like Shannon130,133–135 and Mustafin,137–139 Konakahara et al. have continued the focus on improved preparation of carbazoles. Their recently published strategy employs Suzuki–Miyaura coupling to give a biphenyl compound, followed by a double N-arylation to form the required carbazole (Scheme 43).142 First, the polyfunctional aryl halide 195 was prepared in two steps from 2,5-dimethylphenol via formylation (70%) and iodination (73%). This was subjected to Suzuki–Miyaura coupling conditions with (2-hydroxyphenyl)boronic acid to give the biphenyl compound 196 in 69% yield. This reaction required optimisation of reaction conditions and solvent and eventually copper acetate was found to significantly decrease the quantities of side products formed. Next, 196 was converted to the ditriflate 197 (92%) and subjected to double N-arylation with O-tert-butyl carbamate to give 1,4-dimethyl-3-formylcarbazole 166 (62%). The final steps were carried out as previously described,125 in yields of 67% and 79%, except that a nosyl group was used instead of tosyl.
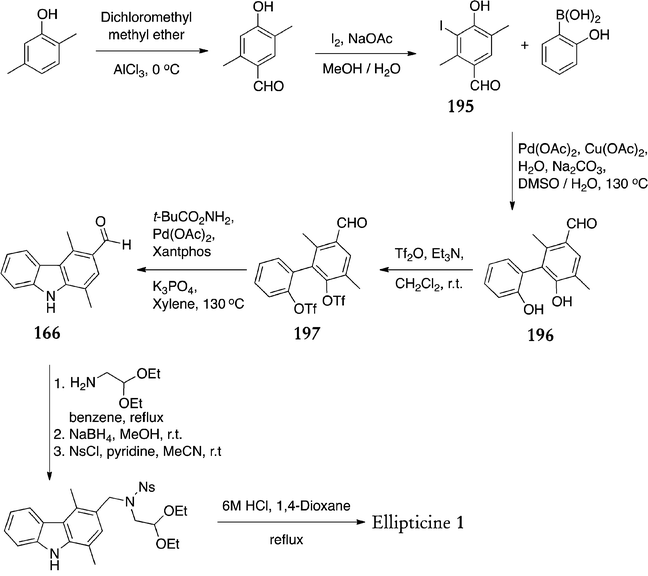 | ||
| Scheme 43 | ||
The overall yield of ellipticine was 10.6% from 2,5-dimethylphenol, thus the improvement in yield is not dramatic, and the number of steps involved is increased compared to earlier routes.
In our own research group, Deane et al. carried out significant optimisation of the Vilsmeier–Haack formylation of 1,4-dimethylcarbazole 165.143 Solvent, reaction time and reagent equivalents were investigated, resulting in improved yield and selectivity of 1,4-dimethyl-3-formylcarbazole 166 (Scheme 44). The optimum conditions were 1.01 equivalents of both phosphorous oxychloride and dimethylformamide refluxing in chlorobenzene for 6.5 h, resulting in a 64% yield of 1,4-dimethyl-3-formylcarbazole 166. The high boiling point solvent and long reaction time were the key factors in the improved yield.
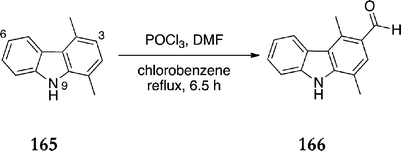 | ||
| Scheme 44 | ||
The Cranwell–Saxton route is by far the most widely investigated and modified synthesis of ellipticine. This has resulted in improved overall yields and preparation of a variety of substituted ellipticines, particularly A-ring substituents.
6.2 Other D-type routes
There have been few D-type syntheses of ellipticine which were not related to the Cranwell–Saxton route. One early route published by Bisagni and co-wokers144 in 1979, towards 1-chloroellipticines was extensively reviewed by Gribble.75In 1990, Bäckvall and Plobeck published a route to ellipticine and olivacine via cycloaddition of 2-phenylsulfonyl-1,3-dienes with indoles (Scheme 45).145 Reaction of indolylmagnesium iodide and 3-(phenylsulfonyl)-2,4-hexandiene gave the tetrahydrocarbazole 198 in 73% yield as a 7:1 mixture of diastereoisomers (major diastereoisomer shown). Michael addition of the lithium salt of acetonitrile, followed by elimination of the sulfone with sodium amalgam gave 199 (92% over two steps). Aromatisation of 199 was attempted by several methods and chloranil in xylene was found to be most successful, affording the carbazole 200 in 64% yield. Reduction and formylation gave the previously known compound 201 (68%), which had been synthesised in 1976 by Oikawa and Yonemitsu146via a different route and converted to ellipticine by a Bischler–Napieralski cyclisation followed by aromatisation (89%). The overall yield of ellipticine was 26%.
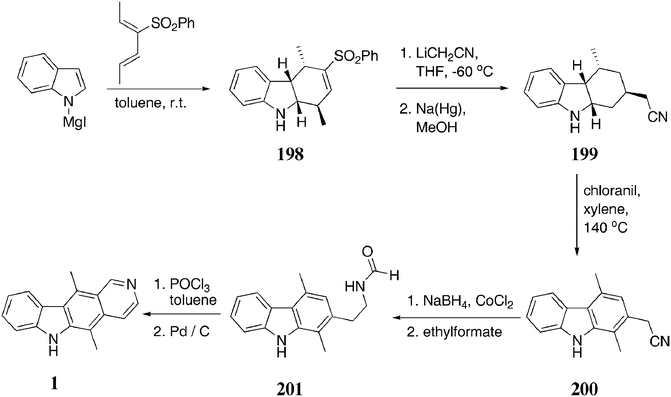 | ||
| Scheme 45 | ||
The route was also used to prepare olivacine 96 in 18% overall yield by reacting with acetic anhydride instead of ethylformate in the penultimate step. Again, the olivacine precursor had previously been prepared by Naito et al. in 1981 via a different route.147
More recently, in 2006, Ho and Hsieh reported a synthesis of ellipticine from 4,7-dimethylindene (Scheme 46).148 Reduction of 4,7-dimethylindene gave the alcohol 202 (99%), which was iodinated to give 203 in 82% yield. Esterification with acetic anhydride gave 204 (99%) and Suzuki coupling of this with (2-nitrophenyl)boronic acid gave the diaryl compound 205 (79%). This was cyclised with triethylphosphite to give 206 in 73% yield. DDQ oxidation of 206 followed by in situ reduction with lithium aluminium hydride gave a mixture of cis and trans diols 207 (68%), which was directly converted to ellipticine in 87% yield.
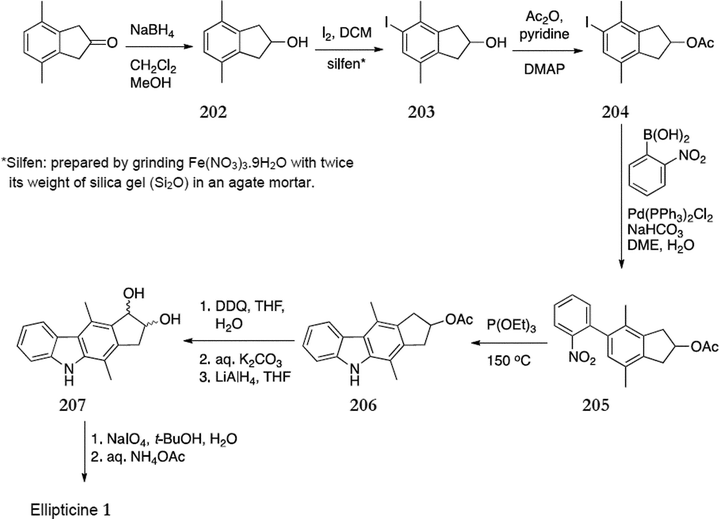 | ||
| Scheme 46 | ||
In 2011 Chaitanya and Nagarajan published a new D-type route via electrophilic cyclisation of 2-alkynyl-3-carbazolylaldimines 208.149 A range of functionalised ellipticinium salts 209 were prepared in > 90% yields by reaction with an amine followed by cyclisation of the resulting imine intermediate with silver trifluoromethanesulfonate (Scheme 47). Unfortunately preparation of the 2-alkynyl-3-carbazolylaldimines 208 was somewhat complicated due to the lack of regioselectivity of the Vilsmeier–Haack formylation previously described in the Cranwell–Saxton series.
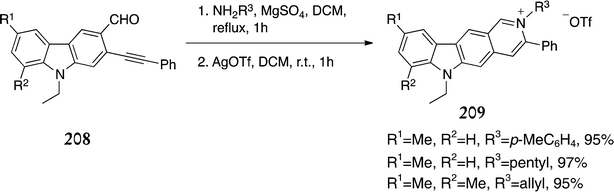 | ||
| Scheme 47 | ||
7. B+C type syntheses
B+C Type syntheses involve simultaneous cyclisation of the B and C rings of ellipticine, this has generally been accomplished by intramolecular cyclisation via radical mechanisms.In the course of their work on intramolecular Diels–Alder cycloaddition reactions for the formation of carbazoles, Differding and Ghosez developed the first B+C type route to ellipticine.150 The key step involved Diels–Alder cycloaddition of acetylenic vinylketenimines 210. N-Methylpiperidone 211 was converted to the conjugated ester 212via a Wittig–Horner reaction (75%). This was treated with lithium diisopropylamide followed by quenching with ammonium chloride to give a β, γ unsaturated ester, which was directly hydrolysed to the acid 213 (75%). Carboxylic acid 213 was converted to an acid chloride with hydrogen chloride and Ghosez's reagent and then coupled with the aniline derivative 214 to give anilide 215 (75%). The vinylketenimine 210 was successfully generated and underwent intramolecular cycloaddition in situ to give 216 (50%). Finally, reduction gave N-methyl-tetrahydroellipticine 217 (71%) (Scheme 48) which had previously been converted to ellipticine.151
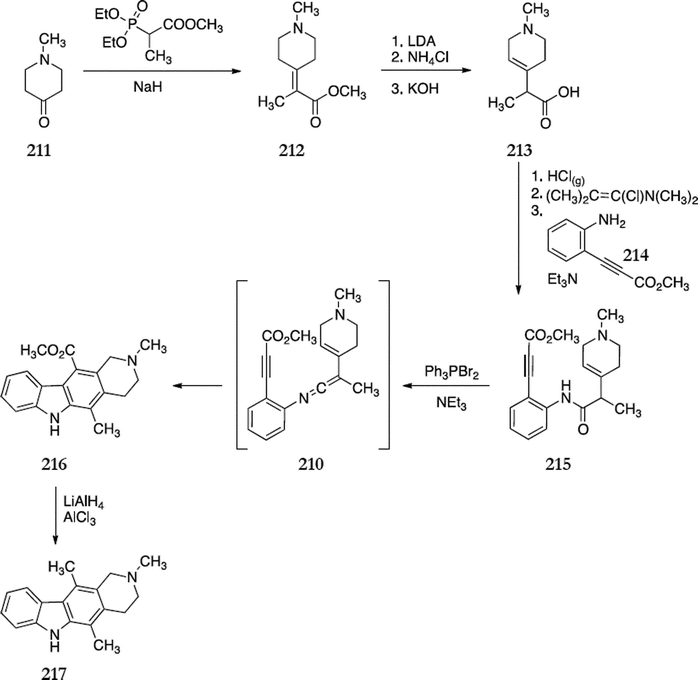 | ||
| Scheme 48 | ||
In 2000, a new B+C type route was published by Zhang et al. towards the synthesis of 5-aza analogues of ellipticine (Scheme 49).152 The key step involved generation of benzoenynyl carbodiimides 218 and thermolysis to give the 6H-indolo[2,3-b][1,6]naphthyridines. Initially, methyl-2-iodobenzoate 219 underwent Pd-catalysed cross-coupling to give the alkyne derivative 220 in 98% yield. This was hydrolysed with base to give the corresponding acid 221 (61%), followed by treatment with diphenyl phosphorazidate to give 222via a modified Curtius rearrangement. The aza-Wittig reaction of 222 and iminophosphorane 223 produced the benzoenynyl carbodiimides 218in situ which were submitted to thermolysis to give the 6H-indolo[2,3-b][1,6]naphthyridine 224 in 49% yield.
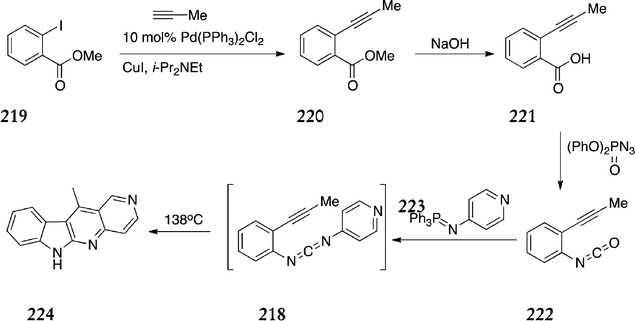 | ||
| Scheme 49 | ||
This route was used to synthesise a range of substituted 6H-indolo[2,3-b][1,6]naphthyridines 225a–c, D-ring analogues 226a & b, and a dimer with two linearly fused indoloquinoline units 227 (Fig. 15).
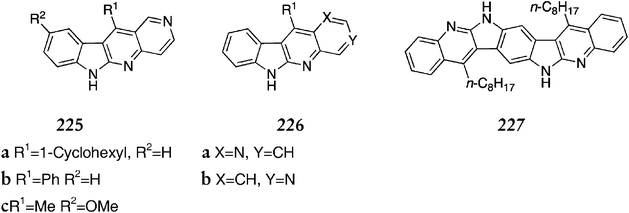 | ||
| Fig. 15 | ||
Subsequently, the same group published work on A-ring analogues via the same route, starting from a substituted pyridine in place of methyl-2-iodobenzoate 219.153 This gave compounds 228, 229 & 230 in good to moderate yields (Fig. 16).
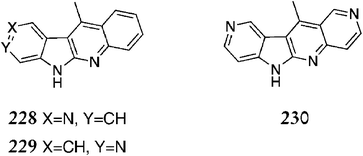 | ||
| Fig. 16 | ||
Moody et al. modified this route to produce 5-aza-1-chloro-9-methoxyellipticine 231 (Scheme 50).154 2-Iodo-4-methoxyaniline 232 was subjected to Sonogashira coupling to give the alkyne derivative 233 (86%), which was converted to the isocyanate 234 in 96% yield using triphosgene. The aza-Wittig reaction and radical cycloaddition was accomplished in one pot by heating 234 with the phosphorane 235 to give 5-aza-1-chloro-9-methoxyellipticine 231 in 17% yield. The chlorine at position 1 was then readily replaced with amine side chains to give 236a–c.
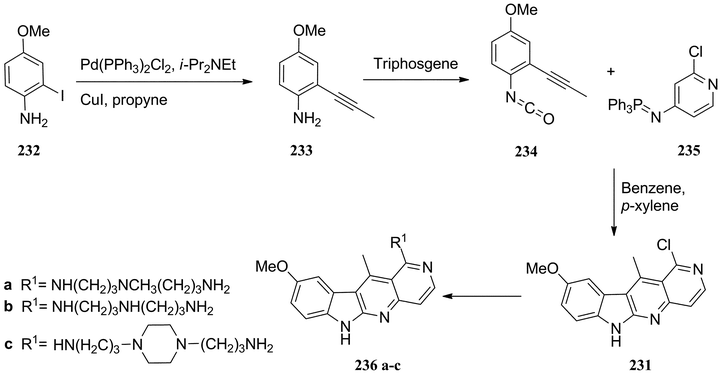 | ||
| Scheme 50 | ||
Pederson et al. developed a radical cascade route to ellipticine from ethyl-2-(4-pyridyl)acetate 237 (Scheme 51).155 Methylation was effected with lithium diisopropylamide followed by methyliodide to give 238 in 93% yield. Amide coupling with trimethylaluminium and 2-iodoaniline yielded the amide 239 (81%). Sonogashira coupling conditions were utilised to introduce the alkyne 240 (88%), which was then converted to the imidoyl selanide 241 (48%). The final radical cyclisation step was accomplished with triethylboron, oxygen gas and tributyltin hydride to give ellipticine in 61% yield.
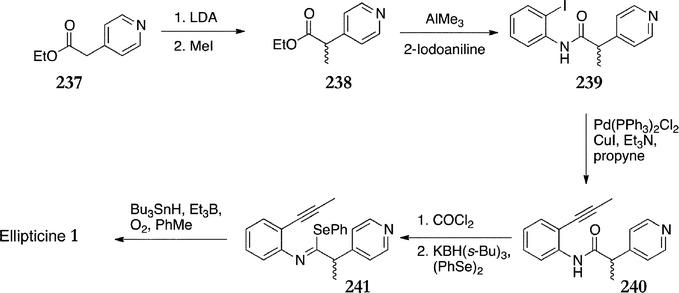 | ||
| Scheme 51 | ||
The utility of this route was demonstrated by synthesis of a range of substituted benzo[b]carbazoles (Scheme 52 & Table 2). Variation of the alkyne used in the Sonogashira coupling reaction gave a range of imidoyl selanides 242a–f, which underwent radical cyclisation to benzo[b]carbazoles 243a–f. In the case of benzo[b]carbazoles 243e & f, indole side products 244e & f were also isolated.
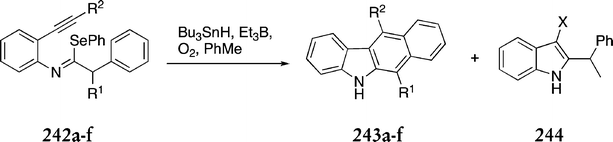 | ||
| Scheme 52 | ||
| R1 | R2 | % Yield 243 | 244 | |
|---|---|---|---|---|
| a | H | Ph | 33 | — |
| b | CH3 | Ph | 55 | — |
| c | CH3 | CH2OAc | 40 | — |
| d | CH3 | H | 18 | — |
| e | CH3 | SiMe3 | 0 | X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHO, 33% CHO, 33% |
| f | CH3 | CO2Me | 19 | XCH2CO2Me, 29% |
The overall yield of ellipticine from this route was 19%. In an effort to make the route more attractive on a large scale, the toxic tributyltin hydride in the final step was replaced with tributylgermanium hydride (Bu3GeH), which resulted in a 49% yield (versus 61% with tributyltin hydride). The main advantage of this route lies in the scope for variation at C-5 and C-11. The initial alkylation could be carried out with a variety of alkylating agents and, along with use of different alkynes in the Sonogashira step, there is scope for a wide range of substituted ellipticines.
In the course of their work on intramolecular dehydro Diels–Alder reactions of N-(o-ethynyl)arylamides, Martínez–Esperón et al. developed a new route to the demethylated ellipticine derivative 245 (Scheme 53).156 Sonogashira coupling of o-iodoaniline 246 and the alkyne 247, followed by N-tosylation gave 248 in 78% yield. This was treated with the (trimethylsilyl)ethynyliodonium salt 249 to give 250 in 85% yield. A second Sonogashira coupling with 4-iodopyridine gave the arylamide 251, which was not isolated but treated in situ with tributylammonium fluoride, followed by heating, to give the demethylated ellipticine derivative 245 (21%).
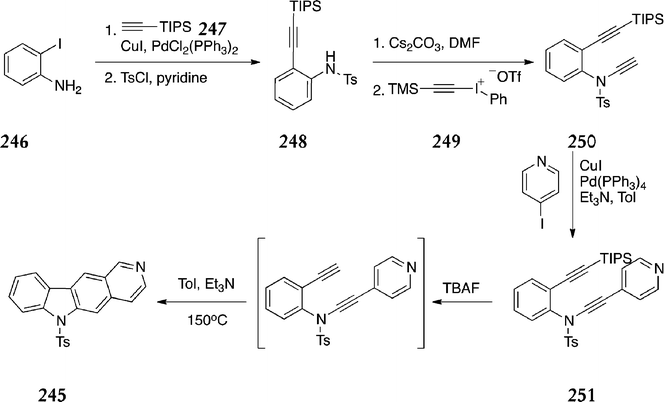 | ||
| Scheme 53 | ||
A variety of substituted benzo[b]carbazoles 252 were prepared in a similar fashion in moderate to good yields from the corresponding arylamides (Fig. 17).
![Substituted benzo[b]carbazoles.](/image/article/2012/RA/c2ra20584j/c2ra20584j-f17.gif) | ||
| Fig. 17 Substituted benzo[b]carbazoles. | ||
8. Summary
While the Cranwell–Saxton route is clearly the most widely investigated and modified synthesis of ellipticine, it is not the highest yielding or most versatile route.123 Jackson's modification improved the overall yield from 0.8% to 8% and Lee's incorporation of microwave heating gave further improvement to 21%.125,141 Of the B-type routes, Miller's nitrene insertion gave a 40% yield of ellipticine over seven steps and was applied to the synthesis of the analogue, indazolo[2,3-b]isoquinoline.88 In a similar vein, Liu and Knochel's nitrene route offered yields of 31–40% over five steps, but with greater variation of substituents.90 In the C-type category, the early routes employed harsh conditions and were low yielding.2,93 Gribble's ketolactam route was the most important progression in this area and easily stands out as the highest yielding synthesis of ellipticine overall (59% over five steps).100 It has also been shown to be versatile for preparation of both substituted ellipticines and analogues.102–106,108 Several of the C-type cycloaddition routes may be useful if both ellipticine and isoellipticine are required, however the issue of regioselectivity still remains problematic, and those routes which are regioselective suffer from much lower yields.118,120 Finally, B+C type routes have primarily been used for the synthesis of ellipticine analogues, in particular, Zhang's route offers an attractive preparation of 5-azaellipticines and was also modified by Moody.152,154 Overall, the interest in ellipticines and related pyridocarbazoles is set to expand given the diversity of structure and emerging bioactivity inherent in this compound class.Acknowledgments
The authors would like to acknowledge the Higher Education Authority of Ireland under the auspices of PRTLI4 for funding this research.References
- S. Goodwin, A. F. Smith and E. C. Horning, J. Am. Chem. Soc., 1959, 81, 1903–8 CrossRef CAS.
- R. B. Woodward, G. A. Iacobucci and F. A. Hochstein, J. Am. Chem. Soc., 1959, 81, 4434–5 CrossRef CAS.
- J. Gruneton and A. Cave, Phytochemistry, 1972, 11, 846–7 CrossRef CAS.
- A. A. Salim, M. J. Garson and D. J. Craik, J. Nat. Prod., 2004, 67, 1719–21 CrossRef CAS.
- A. R. Carroll, R. Addepalli, G. Fechner, J. Smith, G. P. Guymer, P. I. Forster and R. J. Quinn, J. Nat. Prod., 2008, 71, 1063–1065 CrossRef CAS.
- S. Michel, F. Tillequin, M. Koch and A. Ake, J. Nat. Prod., 1982, 45, 489–94 CrossRef CAS.
- S. Michel, F. Tillequin and M. Koch, J. Chem. Soc., Chem. Commun., 1987, 229 RSC.
- K. W. Kohn, M. J. Waring, D. Glaubiger and C. A. Friedman, Cancer Res., 1975, 35, 71–6 CAS.
- G. Dodin, M. A. Schwaller, J. Aubard and C. Paoletti, Eur. J. Biochem., 1988, 176, 371–6 CrossRef CAS.
- M. A. Schwaller, J. Aubard, C. Auclair, C. Paoletti and G. Dodin, Eur. J. Biochem., 1989, 181, 129–34 CrossRef CAS.
- M. Monnot, O. Mauffret, V. Simon, E. Lescot, B. Psaume, J. M. Saucier, J. Belehradek and S. Fermandjian, FEBS Lett., 1990, 273, 71–4 CrossRef CAS.
- M. Monnot, O. Mauffret, V. Simon, E. Lescot, B. Psaume, J. M. Saucier, M. Charra, J. Belehradek and S. Fermandjian, J. Biol. Chem., 1991, 266, 1820–9 CAS.
- A. Canals, M. Purciolas, J. Aymamí and M. Coll, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 1009–1012 CrossRef.
- S. C. Jain, K. K. Bhandary and H. M. Sobell, J. Mol. Biol., 1979, 135, 813–40 CrossRef CAS.
- W. Ross, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1981, 654, 129–134 CrossRef CAS.
- S. J. Froelich-Ammon, M. W. Patchan, N. Osheroff and R. Thompson, J. Biol. Chem., 1995, 270, 14988–15004 Search PubMed.
- C. Auclair and C. Paoletti, J. Med. Chem., 1981, 24, 289 CrossRef CAS.
- M. Stiborová, J. Sejbal, L. Bořek-Dohalská, D. Aimová, J. Poljaková, K. Forsterová, M. Rupertová, J. Wiesner, J. Hudeček, M. Wiessler and E. Frei, Cancer Res., 2004, 64, 8374–8380 CrossRef.
- M. Stiborova, J. Poljakova, H. Ryslava, M. Dracinsky, T. Eckschlager and E. Frei, Int. J. Cancer, 2006, 120, 243–251 CrossRef.
- J. Poljakova, E. Frei, J. Gomez, D. Aimova, T. Eckschlager, J. Hrabeta and M. Stiborova, Cancer Lett., 2007, 252, 270–279 CrossRef CAS.
- M. Stiborová, J. Poljaková, E. Martínková, L. Bořek-Dohalská, T. Eckschlager, R. Kizek and E. Frei, Interdiscip. Toxicol., 2011, 4, 98–105 CrossRef.
- T. Tsujimura, K. Hashimoto, H. Kitayama, H. Ikeda, H. Sugahara, I. Matsumura, T. Kaisho, N. Terada, Y. Kitamura and Y. Kanakura, Blood, 1999, 93, 1319–1329 CAS.
- J. D. Huizinga, L. Thuneberg, M. Kluppel, J. Malysz, H. B. Mikkelsen and A. Bernstein, Nature, 1995, 373, 347–9 CrossRef CAS.
- C. Akin, J. Mol. Diagn., 2006, 8, 412–419 CrossRef CAS.
- A. T. Liao, M. B. Chien, N. Shenoy, D. B. Mendel, G. McMahon, J. M. Cherrington and C. A. London, Blood, 2002, 100, 585–593 CrossRef CAS.
- T. Tsujimura, T. Furitsu, M. Morimoto, K. Isozaki, S. Nomura, Y. Matsuzawa, Y. Kitamura and Y. Kanakura, Blood, 1994, 83, 2619–2626 CAS.
- J. Vendome, S. Letard, F. Martin, F. Svinarchuk, P. Dubreuil, C. Auclair and M. Le Bret, J. Med. Chem., 2005, 48, 6194–6201 CrossRef CAS.
- D. Thompson, C. Miller and F. O. McCarthy, Biochemistry, 2008, 47, 10333–10344 CrossRef CAS.
- X. Jin, D. R. Gossett, S. Wang, D. Yang, Y. Cao, J. Chen, R. Guo, R. K. Reynolds and J. Lin, Br. J. Cancer, 2004, 91, 1808–1812 CrossRef CAS.
- T. F. Franke, D. R. Kaplan and L. C. Cantley, Cell, 1997, 88, 435–437 CrossRef CAS.
- G. Kulik, A. Klippel and M. J. Weber, Mol. Cell. Biol., 1997, 17, 1595–1606 CAS.
- H.-J. Tang, X. Jin, S. Wang, D. Yang, Y. Cao, J. Chen, D. R. Gossett and J. Lin, Gynecol. Oncol., 2006, 100, 308–317 CrossRef CAS.
- K. Fang, S.-P. Chen, C.-W. Lin, W.-C. Cheng and H.-T. Huang, Lung Cancer, 2009, 63, 227–34 CrossRef.
- R. King, Cancer Biol., Longman, Harlow Essex England, 1996 Search PubMed.
- M. Hollstein, K. Rice, M. S. Greenblatt, T. Soussi, R. Fuchs, T. Sorlie, E. Hovig, B. Smith-Sorensen, R. Montesano and C. C. Harris, Nucleic Acids Res., 1994, 22, 3551–5 CAS.
- H. B. Newton, Expert Rev. Anti-Infect. Ther., 2005, 5, 177–191 CrossRef CAS.
- L. M. Shi, T. G. Myers, Y. Fan, P. M. O'connor, K. D. Paull, S. H. Friend and J. N. Weinstein, Mol. Pharmacol., 1998, 53, 241–251 CAS.
- E. Sugikawa, T. Hosoi, N. Yazaki, M. Gamanuma, N. Nakanishi and M. Ohashi, Anticancer Res., 1999, 19, 3099–3108 CAS.
- K. Mizumoto, N. Sato, M. Kusumoto, H. Niiyama, N. Maehara, S. Nishio, Z. Li, T. Ogawa and M. Tanaka, Cancer Lett., 2000, 149, 85–94 CrossRef CAS.
- Y. Peng, C. Li, L. Chen, S. Sebti and J. Chen, Oncogene, 2003, 22, 4478–4487 CrossRef CAS.
- P.-L. Kuo, Y.-L. Hsu, C.-H. Chang and C.-C. Lin, Cancer Lett., 2005, 223, 293–301 CrossRef CAS.
- Y.-C. Kuo, P.-L. Kuo, Y.-L. Hsu, C.-Y. Cho and C.-C. Lin, Life Sci., 2006, 78, 2550–2557 CrossRef CAS.
- G. W. Xu, I. A. Mawji, C. J. Macrae, C. A. Koch, A. Datti, J. L. Wrana, J. W. Dennis and A. D. Schimmer, Apoptosis, 2008, 13, 413–422 CrossRef CAS.
- C. Lu, W. Wang and W. S. El-Deiry, Cancer Biol. Ther., 2008, 7, 2039–2046 CrossRef CAS.
- C. Huang, X. M. Zhang, R. T. Tavaluc, L. S. Hart, D. T. Dicker, W. Wang and W. S. El-Deiry, Cancer Biol. Ther., 2009, 8, 2185–2192 CrossRef CAS.
- F. Wang, J. Liu, D. Robbins, K. Morris, A. Sit, Y.-Y. Liu and Y. Zhao, Apoptosis, 2010, 16, 301–310 CrossRef.
- C. Paoletti, P. Le, N. Dat-Xuong, P. Juret, H. Garnier, J. L. Amiel and J. Rouesse, Recent Results Cancer Res., 1980, 74, 107–23 CrossRef CAS.
- M. Ohashi and T. Oki, Expert Opin. Ther. Pat., 1996, 6, 1285–1294 CrossRef CAS.
- J. G. Rouesse, C. T. Le, P. Caille, J. M. Mondesir, H. Sancho-Garnier, F. May-Levin, M. Spielmann, J. R. De and J. L. Amiel, Cancer Treat. Rep., 1985, 69, 707–8 CAS.
- D. T. Reardan, L. J. Smith and R. E. Klem, US. Pat., US 2008/0167263 Search PubMed.
- R. Jain, K. C. Jindal and S. K. Devarajan, Int. Pat., WO 2007/110878 Search PubMed.
- J. Shaughnessy and E. Tian, US Pat., US 2007/0027175 Search PubMed.
- F. Searle, S. Gac-Breton, R. Duncan, S. Brocchini and D. Newman, Int. Pat., WO 2001/036002 Search PubMed.
- J. Tazi, J. Soret and P. Jeanteur, Fr. Pat., 2005, FR2859475 Search PubMed.
- D. Ellies and W. Rosenberg, Int. Pat., WO 2010/107704 Search PubMed.
- C. Auclair, Arch. Biochem. Biophys., 1987, 259, 1–14 CrossRef CAS.
- E. M. Acton, V. L. Narayanan, P. A. Risbood, R. H. Shoemaker, D. T. Vistica and M. R. Boyd, J. Med. Chem., 1994, 37, 2185–2189 CrossRef CAS.
- W. K. Anderson, A. Gopalsamy and P. S. Reddy, J. Med. Chem., 1994, 37, 1955–63 CrossRef CAS.
- J. Jurayj, R. D. Haugwitz, R. K. Varma, K. D. Paull, J. F. Barrett and M. Cushman, J. Med. Chem., 1994, 37, 2190–2197 CrossRef CAS.
- P. Fossé, B. René, M. Charra, C. Paoletti and J. M. Saucier, Mol. Pharmacol., 1992, 42, 590–595 Search PubMed.
- T. Honda, M. Inoue, M. Kato, K. Shima and T. Shimamoto, Chem. Pharm. Bull., 1987, 35, 3975–8 CrossRef CAS.
- T. Honda, M. Kato, M. Inoue, T. Shimamoto, K. Shima, T. Nakanishi, T. Yoshida and T. Noguchi, J. Med. Chem., 1988, 31, 1295–1305 CrossRef CAS.
- A. R. Grummitt, M. M. Harding, P. I. Anderberg and A. Rodger, Eur. J. Org. Chem., 2003, 63–71 CrossRef CAS.
- A. J. Ratcliffe, M. Sainsbury, A. D. Smith and D. I. C. Scopes, J. Chem. Soc., Perkin Trans. 1, 1988, 2933 RSC.
- L. Ding, G. Etemad-Moghadam, S. Cros, C. Auclair and B. Meunier, J. Med. Chem., 1991, 34, 900–906 CrossRef CAS.
- C. Bourdouxhe, P. Colson, C. Houssier, J. S. Sun, T. Montenay-Garestier, C. Helene, C. Rivalle, E. Bisagni and M. J. Waring, Biochemistry, 1992, 31, 12385–12396 CrossRef CAS.
- C. Bailly, C. Michaux, P. Colson, C. Houssier, J.-S. Sun, T. Garestier, C. Helene, J.-P. Henichart and C. Rivalle, Biochemistry, 1994, 33, 15348–15364 CrossRef CAS.
- S. Routier, J.-L. Bernier, J.-P. Catteau, P. Colson, C. Houssier, C. Rivalle, E. Bisagni and C. Bailly, Bioconjugate Chem., 1997, 8, 789–792 CrossRef CAS.
- R. Devraj, J. F. Barrett, J. A. Fernandez, J. A. Katzenellenbogen and M. Cushman, J. Med. Chem., 1996, 39, 3367–3374 CrossRef CAS.
- G. Czerwinski, N. I. Tarasova and C. J. Michejda, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11520–11525 CrossRef CAS.
- T. W. Moody, G. Czerwinski, N. I. Tarasova, D. L. Moody and C. J. Michejda, Regul. Pept., 2004, 123, 187–192 CrossRef CAS.
- O. Sedlacek, M. Hruby, M. Studenovsky, J. Kucka, D. Vetvicka, L. Kovar, B. Rihova and K. Ulbrich, Bioconjugate Chem., 2011, 22, 1194–1201 CrossRef CAS.
- M. Sainsbury, Synthesis, 1977, 437–448 CrossRef CAS.
- M. J. Hewlins, A. M. Oliveira-Campos and P. V. Shannon, Synthesis, 1984, 289–302 CrossRef CAS.
- G. W. Gribble and M. G. Saulnier, Heterocycles, 1985, 23, 1277–315 CrossRef CAS.
- V. K. Kansal and P. Potier, Tetrahedron, 1986, 42, 2389–2408 CrossRef CAS.
- M. Alvarez and J. A. Joule, Alkaloids: Chem. Biol., 2001, 57, 235–273 CrossRef CAS.
- H.-J. Knölker and K. R. Reddy, Chem. Rev., 2002, 102, 4303–4428 CrossRef.
- R. N. Stillwell, Thesis, Harvard University, 1964 Search PubMed.
- S. Archer, B. S. Ross, L. Pica-Mattoccia and D. Cioli, J. Med. Chem., 1987, 30, 1204–10 CrossRef CAS.
- C. Rivalle, C. Ducrocq and E. Bisagni, J. Chem. Soc., Perkin Trans. 1, 1979, 138 RSC.
- C. Ducrocq, E. Bisagni, C. Rivalle and J.-M. Lhoste, J. Chem. Soc., Perkin Trans. 1, 1979, 142 RSC.
- C. Rivalle, C. Ducrocq, J. M. Lhoste and E. Bisagni, J. Org. Chem., 1980, 45, 2176–2180 CrossRef CAS.
- C. Ducrocq, F. Wendling, J. C. Chermann, M. Tourbez-Perrin, C. Rivalle, P. Tambourin, F. Pochon and E. Bisagni, J. Med. Chem., 1980, 23, 1212–1216 CrossRef CAS.
- R. B. Miller and T. Moock, Tetrahedron Lett., 1980, 21, 3319–3322 CrossRef CAS.
- R. B. Miller and J. G. Stowell, J. Org. Chem., 1983, 48, 886–888 CrossRef CAS.
- P. Vera-Luque, R. Alajarín, J. Alvarez-Builla and J. J. Vaquero, Org. Lett., 2006, 8, 415–418 CrossRef CAS.
- R. B. Miller and S. Dugar, Tetrahedron Lett., 1989, 30, 297–300 CrossRef CAS.
- G. Timári, T. Soós, G. Hajós, A. Messmer, J. Nacsa and J. Molnár, Bioorg. Med. Chem. Lett., 1996, 6, 2831–2836 CrossRef.
- C.-Y. Liu and P. Knochel, J. Org. Chem., 2007, 72, 7106–7115 CrossRef CAS.
- C. Asche, F. Walker, A. Albert and U. Kucklaender, Bioorg. Med. Chem., 2005, 13, 819–837 CrossRef CAS.
- S. Bouclé and J. Guillard, Synthesis, 2011, 2011, 1616–1620 CrossRef.
- S. H. Zee and H. P. Su, J. Chinese Chem. Soc., 1987, 34, 135–139 CAS.
- D. M. Ketcha and G. W. Gribble, J. Org. Chem., 1985, 50, 5451–7 CrossRef CAS.
- D. A. Taylor, M. M. Baradarani, S. J. Martinez and J. A. Joule, J. Chem. Res., 1979, 387 Search PubMed.
- M. L. Bennasar, T. Roca and F. Ferrando, J. Org. Chem., 2005, 70, 9077–9080 CrossRef CAS.
- Y. Yokoyama, N. Okuyama, S. Iwadate, T. Momoi and Y. Murakami, J. Chem. Soc., Perkin Trans. 1, 1990, 1319–1329 RSC.
- F. Marsais, P. Pineau, F. Nivolliers, M. Mallet, A. Turck, A. Godard and G. Queguiner, J. Org. Chem., 1992, 57, 565–573 CrossRef CAS.
- M. G. Saulnier and G. W. Gribble, J. Org. Chem., 1982, 47, 2810–12 CrossRef CAS.
- G. W. Gribble, M. G. Saulnier, J. A. Obaza-Nutaitis and D. M. Ketcha, J. Org. Chem., 1992, 57, 5891–9 CrossRef CAS.
- J. P. Kutney, M. Noda, N. G. Lewis, B. Monteiro, D. Mostowicz and B. R. Worth, Can. J. Chem., 1982, 60, 2426–2430 CrossRef CAS.
- M. G. Saulnier and G. W. Gribble, Tetrahedron Lett., 1983, 24, 3831–3834 CrossRef CAS.
- J. A. Obaza-Nutaitis and G. W. Gribble, J. Nat. Prod., 1986, 49, 449–51 CrossRef CAS.
- M. G. Saulnier and G. W. Gribble, Abstracts of Papers Am. Chem. Soc., 1983, 185, 17 Search PubMed.
- G. W. Gribble, G. L. Fletcher, D. M. Ketcha and M. Rajopadhye, J. Org. Chem., 1989, 54, 3264–9 CrossRef CAS.
- H. L. Fraser and G. W. Gribble, Can. J. Chem., 2001, 79, 1515–1521 CrossRef CAS.
- S. P. Modi, J. J. Carey and S. Archer, Tetrahedron Lett., 1990, 31, 5845–8 CrossRef CAS.
- S. P. Modi, M. A. Michael, S. Archer and J. J. Carey, Tetrahedron, 1991, 47, 6539–48 CrossRef CAS.
- S. Hibino and E. Sugino, J. Heterocycl. Chem., 1990, 27, 1751–1755 CrossRef CAS.
- I. Praly-Deprez, C. Rivalle, J. Belehradek, C. Huel and E. Bisagni, J. Chem. Soc., Perkin Trans. 1, 1991, 3173 RSC.
- I. Praly-Deprez, C. Rivalle, C. Huel, J. Belehradek, C. Paoletti and E. Bisagni, J. Chem. Soc., Perkin Trans. 1, 1991, 3165 RSC.
- Y. Miki, Y. Tada, N. Yanase, H. Hachiken and K. Matsushita, Tetrahedron Lett., 1996, 37, 7753–7754 CrossRef CAS.
- Y. Miki, H. Hachiken and N. Yanase, J. Chem. Soc., Perkin Trans. 1, 2001, 2213–2216 RSC.
- M. Ishikura, A. Hino, T. Yaginuma, I. Agata and N. Katagiri, Tetrahedron, 2000, 56, 193–207 CrossRef CAS.
- G. W. Gribble, M. G. Saulnier, M. P. Sibi and J. A. Obazanutaitis, J. Org. Chem., 1984, 49, 4518–4523 CrossRef CAS.
- C. May and C. J. Moody, J. Chem. Soc., Chem. Commun., 1984, 926–927 RSC.
- C. May and C. J. Moody, J. Chem. Soc., Perkin Trans. 1, 1988, 247–250 RSC.
- D. A. Davis and G. W. Gribble, Tetrahedron Lett., 1990, 31, 1081–1084 CrossRef CAS.
- C.-K. Sha and J.-F. Yang, Tetrahedron, 1992, 48, 10645–10654 CrossRef CAS.
- M. Diaz, A. Cobas, E. Guitian and L. Castedo, Eur. J. Org. Chem., 2001, 4543–4549 CrossRef CAS.
- D. Mal, B. K. Senapati and P. Pahari, Synlett, 2005, 994–996 CrossRef CAS.
- D. Mal, B. K. Senapati and P. Pahari, Tetrahedron, 2007, 63, 3768–3781 CrossRef CAS.
- P. A. Cranwell and J. E. Saxton, J. Am. Chem. Soc., 1962, 3482–3487 CAS.
- A. J. Birch, A. H. Jackson and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1974, 2185 RSC.
- A. H. Jackson, P. R. Jenkins and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1977, 1698 RSC.
- M. J. E. Hewlins, A. H. Jackson, A.-M. Oliveira-Campos and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1981, 2906 RSC.
- R. W. Guthrie, A. Brossi, F. A. Mennona, J. G. Mullin and R. W. Kierstead, J. Med. Chem., 1975, 18, 755 CrossRef CAS.
- P. Bruck, Chem. Comm, 1970, 1690 RSC.
- Y. Murakami, Y. Yokoyama and N. Okuyama, Tetrahedron Lett., 1983, 24, 2189–2192 CrossRef CAS.
- R. J. Hall, P. V. R. Shannon, A. M. F. Oliveira-Campos and M. J. R. P. Queiroz, J. Chem. Res., Synop., 1992, 2–3 CAS.
- I. Hogan, P. Jenkins and M. Sainsbury, Tetrahedron Lett., 1988, 29, 6505–6508 CrossRef CAS.
- I. Hogan, P. D. Jenkins and M. Sainsbury, Tetrahedron, 1990, 46, 2943–2964 CrossRef CAS.
- P. M. Dharmasena and P. V. R. Shannon, Tetrahedron Lett., 1994, 35, 7119–7122 CrossRef CAS.
- L. Chunchatprasert, P. Dharmasena, A. OliveiraCampos, M. Queiroz, M. Raposo and P. Shannon, J. Chem. Res., Synopses, 1996, 84–85 CAS.
- R. J. Hall, J. Marchant, A. M. F. Oliveira-Campos, M.-J. R. P. Queiroz and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1992, 3439 RSC.
- P. Groundwater and R. Lewis, J. Chem. Res., Synopses, 1995, 215 CAS.
- A. G. Mustafin, I. N. Khalilov, E. V. Tal'vinskii, I. B. Abdrakhmanov, L. V. Spirikhin and G. A. Tolstikov, Khim. Prir. Soedin., 1992, 549–554 CAS.
- A. G. Mustafin, I. N. Khalilov, V. M. Sharafutdinov, D. I. D'yachenko, I. B. Abdrakhmanov and G. A. Tolstikov, Russ. Chem. Bull., 1997, 46, 608–609 CrossRef CAS.
- A. G. Mustafin, I. N. Khalilov, R. R. Ismagilov, Z. M. Baimetov, L. V. Spirikhin, I. B. Abdrakhmanov and G. A. Tolstikov, Russ. Chem. Bull., 1999, 48, 2121–2126 CrossRef CAS.
- M. Dracinsky, J. Sejbal, B. Rygerova and M. Stiborova, Tetrahedron Lett., 2007, 48, 6893–6895 CrossRef CAS.
- H.-Y. Lee, G. S. Chen, C.-S. Chen and J.-W. Chern, J. Het. Chem., 2010, 47, 454–458 CAS.
- T. Konakahara, Y. B. Kiran, Y. Okuno, R. Ikeda and N. Sakai, Tetrahedron Lett., 2010, 51, 2335–2338 CrossRef CAS.
- F. M. Deane, C. M. Miller, A. R. Maguire and F. O. McCarthy, J. Heterocycl. Chem., 2011, 48, 814–823 CrossRef CAS.
- E. Bisagni, C. Ducrocq, J. M. Lhoste, C. Rivalle and A. Civier, J. Chem. Soc., Perkin Trans. 1, 1979, 1706–1711 RSC.
- J. E. Baeckvall and N. A. Plobeck, J. Org. Chem., 1990, 55, 4528–4531 CrossRef CAS.
- Y. Oikawa and O. Yonemitsu, J. Chem. Soc., Perkin Trans. 1, 1976, 1479 RSC.
- T. Naito, N. Iida and I. Ninomiya, J. Chem. Soc., Chem. Commun., 1981, 44 RSC.
- T.-L. Ho and S.-Y. Hsieh, Helv. Chim. Acta, 2006, 89, 111–116 CrossRef CAS.
- T. K. Chaitanya and R. Nagarajan, Org. Biomol. Chem., 2011, 9, 4662 CAS.
- E. Differding and L. Ghosez, Tetrahedron Lett., 1985, 26, 1647–1650 CrossRef CAS.
- R. Besselievre and H. P. Husson, Tetrahedron, 1981, 37, 241–246 CrossRef.
- Q. Zhang, C. S. Shi, H. R. Zhang and K. K. Wang, J. Org. Chem., 2000, 65, 7977–7983 CrossRef CAS.
- X. L. Lu, J. L. Petersen and K. K. Wang, J. Org. Chem., 2002, 67, 5412–5415 CrossRef CAS.
- D. L. Moody, M. Dyba, T. Kosakowska-Cholody, N. I. Tarasova and C. J. Michejda, Bioorg. Med. Chem. Lett., 2007, 17, 2380–2384 CrossRef CAS.
- J. M. Pedersen, W. R. Bowman, M. R. J. Elsegood, A. J. Fletcher and P. J. Lovell, J. Org. Chem., 2005, 70, 10615–10618 CrossRef CAS.
- A. F. Martinez-Esperon, D. Rodriguez, L. Castedo and C. Saa, Tetrahedron, 2008, 64, 3674–3686 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |