Insights on Ag doped porous TiO2 nanostructures: a comprehensive study of their structural and morphological characteristics†
D.
Georgescu
ab,
L.
Roiban
ac,
O.
Ersen
*a,
D.
Ihiawakrim
a,
L.
Baia
b and
S.
Simon
b
aInstitut de Physique et Chimie des Matériaux de Strasbourg, 23, rue du Loess, 67034, Strasbourg, France. E-mail: Ovidiu.Ersen@ipcms.u-strasbg.fr
bFaculty of Physics & Institute for Interdisciplinary Research on Bio-Nano-Science Cluj-Napoca, “Babes-Bolyai” University, M. Kogalniceanu 1, 400084, Romania
cInorganic Chemistry and Catalysis, Universiteitweg 99, 3584 CG Utrecht, Netherlands
First published on 29th March 2012
Abstract
A comprehensive analysis of heterogeneous nanostructures based on TiO2 aerogels and Ag nanoparticles (NPs) is reported herein. The experimental techniques engaged along this study enable a multi-scale approach for the characterization of this composite system. The results clearly indicate that an increase of the Ag amount within the porous structure of TiO2 nanograins leads to a lower dispersion of Ag on the support. For a higher Ag global amount, the proportion of encapsulated NPs increases considerably, reducing in this manner the amount of accessible Ag. The effect of the thermal treatment on such nanostructures has been evaluated as well. The specimen annealing appears to induce a considerable diminution of the microporosity, finding that is in good agreement with the results obtained by measurement of the specific surface area of the TiO2 grains. The annealing also changes the microstructural characteristics of these nanostructures, as expected. More specific, it leads to an enhanced degree of encapsulation of Ag NPs, which as consequence become considerably less accessible. Being focused on the analysis of porous nanocomposites with specific properties, the current investigation aims as well at emphasizing the remarkable potential of electron tomography, a technique that can be successfully employed for precise analysis of three dimensional nanostructures.
1. Introduction
During the last few years, TiO2 based porous materials have received particular attention owing to their special properties that make them appropriate candidates for various applications, like for heterogeneous (photo) catalysis, solar cells, integrated wave guides, pigments and electronic devices etc.1 TiO2 has also been extensively used in more complex structures, in particular, as supports for different types of nanoparticles (NPs) in nanocomposites. In this context, it has been recently demonstrated that the presence of silver species, i.e. Ag+ and Ag0, as well-defined NPs or atomic clusters inside a TiO2 porous structure improves and/or extends the composite properties highly demanded in applications.2–4 For example, these nanocomposites based on TiO2 and Ag can simultaneously exhibit excellent photocatalytic performances,5,6 great ability for sensing minute concentrations of chemical pollutants from contaminated water7,8 and antimicrobial effects as they minimize efficiently the risk of microbial contamination.9–12 By taking into account their enormous potential for a variety of devices, the development of such multifunctional structures requires the precise control of their structural and morphological characteristics.13 For a more efficient use in applications, the microstructural characteristics have to be closely correlated with the synthesis parameters, in order to finely tune the properties of interest by adjusting the preparation conditions. Indeed, an important quantity of work has been devoted to the study of porous materials. It has been systematically shown that, depending on the preparation conditions, a porous network can be opened or closed, organized or random, and can exhibit narrow or broad pore size distribution.Besides the particular properties of the nanostructures constituted by Ag and TiO2 as compared to pure TiO2, it has been previously reported that the addition of Ag, even in small amounts, has a considerable impact on the microstructure of the TiO2 porous material.14–17 As such, certain microstructural parameters can be modified by the presence of Ag: the type of crystalline structure, the mean size of TiO2 aggregates, their specific surface area, the type and the size distribution of the pores, their connectivity etc. In addition, the total amount of Ag inserted in the TiO2 matrix clearly governs the Ag NP–TiO2 support interface properties, as well as the fraction of accessible (non-encapsulated) Ag particles. It is worth mentioning that all these parameters are of extreme importance, as they control most of the specific properties exhibited by these porous nanocomposites.
From a general perspective, for characterizing the structure and morphology of nanomaterials and nano-objects at an atomic scale, transmission electron microscopy (TEM) techniques are nowadays extensively employed, especially when applied in traditional 2D modes (acquisition of a unique image, diffraction pattern or spectrum). In the case of porous materials used as supports for the insertion of a second phase, the crucial parameters are three-dimensional. The 3D characteristics comprise the spatial organization of the components, the system morphology and the organization of the porous network. Such parameters are very difficult to get by analyzing simple 2D images, where all the details of the investigated object are superimposed on the same plane and the result is integrated along the thickness on a 2D projection. In these circumstances, the use of the 3D-TEM (or electron tomography, ET) technique was found to be the only good alternative to precisely solve the three dimensional characteristics mentioned above.18–21 It consists of the reconstruction of the object's volume from a tilt series recorded in one of the various modes within the transmission electron microscopy: bright field (BF) conventional TEM,22,23 energy filtered TEM (EFTEM) imaging24,25 or high angle annular dark field (HAADF) in the scanning TEM (STEM) mode.26 The choice of the mode used for the recording depends on the specimen characteristics and the type of information required. The traditional TEM mode is generally not appropriate for the study of crystalline materials, due to the presence of diffraction contrast in the TEM images. However, due to its easier implementation as compared with the other modes, it is strongly recommended for the analysis of amorphous or weakly crystallized materials. One of the main benefits of the tomographic techniques as employed for the study of porous materials relates to their ability of providing direct information in the nanoscale range from the 3D porous structure (pore shape and accessibility, their size distribution). In addition, the 3D packing of aggregates composing the macroscopic materials can be successfully probed as well.27 As for the case of multi-component materials, such as nanostructures made of NPs deposited onto 3D supports or located within the material, the ET allows one to get precise information on the spatial distribution of the different materials and/or phases.28 However, it is worth pointing out here one of the main drawbacks of ET: similarly to others nanoscale imaging techniques, the ET investigations are performed at specific locations and consequently the information achieved is not unconditionally representative for the whole specimen. It is therefore strongly recommended to combine the tomographic analysis with some other more macroscopic techniques, such as IR or Raman spectroscopy, X-ray diffraction or N2 or Hg sorption measurements. In the case of porous materials containing different types of porosity, the gas sorption measurements can bring complementary information which in combination with the 3D-TEM results provides a complete and reliable microstructural characterization. However, as compared to 3D-TEM, where the analysis is performed directly in the real space, the information attained from sorption measurements is strongly model-dependent.29
In spite of the benefits for subsequent applications to obtain precise information on the spatial distribution of nanoparticles within the porous network, 3D-analysis by electron tomography was rarely applied to the study of Ag–TiO2 nanocomposites. In this framework, one can mention a 2D HAADF-STEM study applied to an Ag–TiO2 system, in order to obtain primary information on the spatial distribution of Ag particles into the TiO2 matrix as a function of the annealing conditions.30 One should emphasize that this study deals with Ag–TiO2 composites prepared by a combined sputtering/sol–gel approach, where the Ag particles were deposited by sputtering onto the sol–gel titania xerogels. The microstructure of the as-prepared samples was found to be dominated by the presence of Ag-rich nanoaggregates distributed apparently over the whole matrix surface. A careful analysis of the 2D images has also revealed the presence of smaller Ag particles inside the TiO2 matrix, considered responsible for the partial Ag oxidation and the inhibition of the TiO2 crystallization process. More recently, a 3D study in which ET was applied to determine the specific surface area of mesopores and to investigate shapes and 3D distributions of pores and Ag particles in Ag–TiO2 porous samples was reported.31 In this case, the nanocomposites were prepared by using a Pluronic P-123 self-assembly process on which Ag NPs were precipitated on the surface by UV irradiation. It was found that the porous TiO2 structure produced via the block copolymer method presents a high number of effective reaction sites for the degradation of methylene blue. In addition to the mentioned investigations devoted precisely to the study of Ag–TiO2 systems, one can also mention a work reporting the quantitative 3D-TEM analysis of Au nanoparticles supported on oxides of high atomic number (Ce, Tb and Zr).32 In this last study, quantitative data about the surface crystallography of the support and the distribution of the nanoparticles have been determined. More precisely, it has been shown that the Au nanoparticles have preferential locations at the boundaries and stepped sites of the oxide support.
In this context, the general goal of the present work is to analyze the relation between the Ag loading and the microstructure and 3D organization of the TiO2 matrix. More precisely, with respect to the previous studies, the aim here is to determine via a multi-scale approach the role played by the Ag loading on the structural and morphological characteristics of a highly porous aerogel TiO2 network. One of the particularities of this study is that the Ag–TiO2 nanocomposites were prepared by a supercritical drying process. For this purpose, a pure TiO2 and two samples with different Ag loadings have been considered. Afterwards, the samples were subjected to a thermal treatment to investigate the microstructural changes induced by the annealing process. For a complete analysis at different scales, several complementary techniques have been used: on one hand, the traditional selected area electron diffraction (SAED), high resolution transmission electron microscopy (HRTEM) and X-ray diffraction (XRD) techniques were able to furnish local or global structural information on the analysed specimens; on the other hand, the more specific 3D-TEM and N2-sorption analysis tools, which give access to the porous characteristics of the TiO2 support. Particular attention has been paid to a quantitative application of the electron tomography. The final goal is to provide combined information from the porous network of TiO2 aerogel, size and spatial localization of the Ag NPs on the support, as well as the interface between the two components. Obtaining comparative information on the raw (as-prepared) and annealed samples is the key-issue here, as the antimicrobial investigations performed on these specimens have revealed the presence in high amounts of inhibition zones only for the raw specimens.
2. Experimental
2.1. Sample preparation
The TiO2 gels were prepared by the sol–gel method using tetraisopropoxide (TIP), HNO3, Et–OH and H2O (9.65/32.55/0.25/2.18 g) and afterwards put down to age four weeks. For obtaining the Ag–TiO2 nanocomposites, the gels were then immersed for one day in AgNO3 solutions with concentration of 5.10−3 and 5.10−2 mol dm−3, by using 100 ml Et–OH, and then were supercritically dried with LCO2 (T > 38 °C and p = 1350 psi) using Tousimis Autosamdri 815 equipment. The samples will be further denoted as I1 (pure TiO2), A1 (prepared from the less concentrated solution) and B1 (prepared from the highly concentrated solution). The three TiO2 aerogels were submitted to a thermal treatment at 500 °C for 1 h and will be further denoted as I2, A2 and B2.2.2. Sample characterization
The elemental analyses of the porous samples containing silver were performed by using a SCIEX Perkin Elmer Elan DRC II inductively coupled plasma mass spectrometer.The XRD patterns of the six samples were recorded by using CuK (λCuK = 1.5406 Å) radiation of a standard DRON-3M powder diffractometer working at 45 kV and 30 mA. The XRD patterns of the annealed samples were analyzed by using a Ritveld refinement.
The specific surface area, the porous volume and the mean diameters of the pores within the studied samples were estimated from N2-adsorption–desorption isotherms measured by a Sorptomatic 1990 equipment. The values of the specific surface areas were obtained by the Brunauer–Emmett–Teller (BET) method,33 whereas the pore volume and the pore size distribution were determined by the Barrett-Joyner-Halenda (BJH) method.34 Before the experiment, the masses of the analyzed powders were accurately measured subsequent to a pretreatment at 120 °C during two hours; this thermal treatment has been carried out in order to remove the contaminants from the specimen surface.
The high resolution TEM images, selected area diffraction patterns and tomographic measurements were carried out on a JEOL 2100F electron microscope operating at 200 kV, with a field emission gun and a point-to-point resolution in TEM mode of 0.21 nm (high resolution polar piece), equipped with an EELS spectrometer, EDX detector and a spherical aberration probe corrector. Before observation, the samples were ultrasonically dispersed in ethanol for 5 min and a drop of suspension was deposited on a carbon membrane grid. The HRTEM images were analyzed using the Digital Micrograph software, whereas the JEMS software was used for indexing the diffraction patterns. For the tomography analysis, a drop of a solution containing fiducial markers (gold NPs with a diameter of 5 nm) was deposited on the grid to make the alignment step of the tomographic data treatment easier. The acquisition of the tilt series used to calculate the reconstructed volume was performed in bright field TEM using the Digital Micrograph software, with images recorded on a CCD Ultrascan array detector. The angular interval was generally between −72° and 72°, with an angular step of 2° at 0° tilt which varies at higher angles according to the Saxton algorithm.35 Basically, the number of images within each tilt series was about 100. After acquisition, the individual images were aligned in the IMOD software,36 using a rough alignment based on the cross-correlation procedure followed by a finer alignment using the geometrical positions of the fiducial markers. The reconstructed volumes were calculated using the iterative methods of the TomoJ plugin37,38 implemented in the ImageJ software. The 3D representation, analysis and modeling of the reconstructed volumes were performed using the 3D Slicer39 and Chimera40 software. To obtain 3D models of the analyzed aggregates, a segmentation procedure based on the grey-level intensity of the voxels was systematically performed. Using the analytical formula proposed by Frank,41 the spatial resolution in the reconstructions was estimated to be about 0.2 nm, with an elongation factor in the direction of the electron beam lower than 1.1.
3. Results and discussion
3.1. Structural characterization
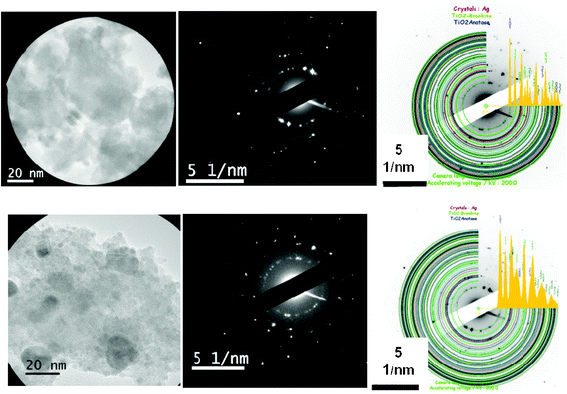 | ||
| Fig. 1 Left: TEM images taken on some typical areas of A1 (top) and B1 (bottom) specimens selected for the recording of the diffraction patterns. Middle: the corresponding SAED patterns. Right: the indexing procedure of the SAED patterns, performed by using the JEMS software. | ||
The thermal treatment leads to a considerable increase of the fraction of the crystalline phase, as expected. Indeed, compared to the as-prepared samples, the analysis of the diffraction patterns of A2 and B2 reveals a strong augmentation of the number of individual crystallites randomly oriented. A classical indexing procedure indicates the presence of both anatase and brookite TiO2 polycrystalline phases, with the anatase in the major proportion. Once again, the structural homogeneity within the specimens has been checked by comparing the diffraction patterns recorded on selected circular areas with diameters of 100, 200 and 500 nm and no significant difference was observed.
The structural characteristics as evaluated from electron diffraction were confirmed by the analysis performed on the high resolution images (Fig. 2 and SI 2 from the ESI†). It is worth noting the more pronounced local character of the HRTEM analysis as compared to SAED. The HRTEM observations are performed generally on the regions marked by typical contrasts associated with the presence of crystalline planes. On the image of the as-prepared sample, a crystalline grain with a size of about 10 nm has been observed in the center of a bigger amorphous aggregate; by measuring the corresponding distances, we were able to assign this grain to the anatase phase. On the same image, we also observe some dark areas, with sizes between 2 and 5 nm, which can be clearly assigned to the Ag NPs. Note that smaller Ag particles can be present as well within the analyzed area, but their observation in the HRTEM mode is difficult, particularly due to the specimen thickness.
 | ||
| Fig. 2 HRTEM images for B1 (left) and B2 (right) samples. The yellow arrows indicate the locations of several Ag particles within the chosen aggregate of the B1 sample. The Fourier transform as calculated on the square area shows the presence of an Ag NP for the second specimen. | ||
By inspecting the HRTEM images of the annealed samples, one can identify several dark areas. In a similar way as for the as-prepared specimens, the direct measurement of the interreticular distances allowed us to associate these regions to Ag NPs. Their size seems to be larger compared to the as-prepared samples, an observation further confirmed by the tomographic measurements from the next section. As expected, the bright areas from the TiO2 support represent the major part of the analyzed aggregates. A typical contrast associated with the presence of a crystallographic network can be seen in most of these areas. It confirms the previous results obtained by SAED, i.e. the high crystallization degree and the small amount of the amorphous TiO2 phase for the annealed samples. The derived crystallographic distances are summarized in Table 1 together with the corresponding distances as measured by SAED. Both indicate that the anatase is present in major proportion, but do not exclude the occurrence of the brookite phase. It is illustrated in Table 1 that some distances measured by HRTEM and SAED can be assigned to the brookite phase. It is worth noting that this second phase is generally less interesting for potential applications of these nanocomposites. As a consequence, the conditions for the thermal treatment were chosen for promoting the formation of the anatase phase. However, to estimate the relative amount of the two phases, a more global and quantitative structural analysis is required and therefore an XRD analysis has been further carried out.
| Sample | HRTEM | SAED | ||||
|---|---|---|---|---|---|---|
| d (Å) | Crystalline phase | Miller indices (h k l) | d (Å) | Crystalline phase | Miller indices (h k l) | |
| A1 | 3.52 | Anatase | (1 0 1) | 3.52 | Anatase | (1 0 1) |
| 3.50 | Brookite | (1 2 0) | 3.46 | Brookite | (1 1 1) | |
| 2.36 | Anatase | (0 0 4) | 1.66 | Anatase | (2 1 1) | |
| B1 | 2.20 | Ag | (1 1 1) | 3.52 | Anatase | (1 0 1) |
| 2.47 | Brookite | (0 1 2) | 3.46 | Brookite | (1 1 1) | |
| 3.53 | Anatase | (1 0 1) | 1.66 | Anatase | (2 1 1) | |
| A2 | 2.00 | Ag | (2 0 0) | 3.52 | Anatase | (1 0 1) |
| 1.86 | Anatase | (2 0 0) | 3.46 | Brookite | (1 1 1) | |
| 1.88 | Anatase | (2 0 0) | 1.66 | Anatase | (2 1 1) | |
| B2 | 3.53 | Anatase | (1 0 1) | 3.52 | Anatase | (1 0 1) |
| 2.41 | Brookite | (2 0 1) | 3.46 | Brookite | (1 1 1) | |
| 2.35 | Ag | (1 1 1) | 1.66 | Anatase | (2 1 1) | |
 | ||
| Fig. 3 XRD patterns for the four Ag–TiO2 nanocomposites, the as-prepared A1 and B1 (left), and the annealed A2 and B2 (right). Rietveld refinements were systematically applied in order to allow quantitative analyses of the four XRD patterns. | ||
Analysis of the porous network within the TiO2 matrix and 3D localization of Ag NPs
As pointed out in the introduction, for obtaining valuable information on the morphology and porous network of the TiO2 aerogel grains as well as on the 3D distribution of Ag NPs therein, ET experiments have been performed for all the specimens investigated here, i.e. I1, I2, A1, A2, B1 and B2. The typical procedure for the data analysis is shown in Fig. 4 for an aggregate belonging to the sample B1. In a first step, once the reconstructed volume is calculated, it can be analyzed slice by slice along any chosen direction. A typical example of the three orthogonal slices through the reconstruction is presented in Fig. 4b, c and d. In a second step, the reconstructed volume of the analyzed region can be used to calculate the 3D model of the aggregate. In such 3D representation, the different components (here titania and silver, represented in white and blue in Fig. 4e and f) are spatially separated and can be analyzed individually. Fig. 5 shows the equivalent 3D models for all other specimens, achieved in a similar manner by data segmentation of the corresponding reconstructed volumes. They provide a real 3D visualization of the architecture of the analyzed Ag–TiO2 nanocomposites grains. For a better visualization of the 3D relative distributions of the Ag NPs, one can choose to represent only their sub-volumes from the whole 3D model (bottom part on Fig. 5). Besides the realism achieved by direct 3D visualization of the grains, the main benefit of the 3D modeling procedure is that it can be subsequently used for performing quantitative analyses. Such an analysis allows one to precisely assess the specific parameters, such as the volume and specific area of each component as well as the characteristics of the interface between the components. | ||
| Fig. 4 (a) Typical TEM image from the tilt series used for volume reconstruction of a representative aggregate from the B1 sample. (b), (c) and (d) Three orthogonal slices through the reconstruction; the z-axis corresponds to the electron beam direction at 0° tilt; the yellow arrows indicate the localization of some Ag NPs. (e) 3D tomographic model of the analyzed aggregate, with TiO2 in white and Ag in blue. (f) Cross-section view through the 3D model, illustrating the internal architecture of the analyzed grain. | ||
 | ||
| Fig. 5 Top: 3D tomographic models of the analyzed grains of the samples A1, B1, A2 and B2. Bottom: 3D representations of the same aggregates, where only the Ag NPs are represented. | ||
The visualization of the Ag 3D model shows primary information on the localization of Ag NPs with respect to the external surface of the TiO2 grains. Corresponding to samples B1 and B2 (with a high amount of Ag), one can observe a homogenous distribution of the Ag NPs within the TiO2 grains. In contrast, in the case of A1 and A2 (with a low Ag amount) the density of NPs increases from the aggregate’s core to its external surface, yielding thus a rather heterogeneous distribution. This discrepancy in Ag distribution is certainly due to the difference in the Ag amount (between A1 and A2, and B1 and B2, respectively) and could be explained by a preferential growth of Ag NPs at the surface of the grains in the first stage of the supercritical drying process. More specifically, for the samples B1 and B2 the Ag amount is large enough to assure its complete penetration within the individual aggregates, whereas in the case of A1 and A2 the Ag quantity is ten times lower leading to its selective deposition rather at the vicinity of the surface. This behavior is in high agreement with the results of Krylova et al.15 who reported a preferential localization of Ag NPs on the outer surface of sol–gel Ag–TiO2 composites if the Ag loading is low and a more homogenous distribution of NPs at higher Ag loading, especially for high calcination temperatures.
From a quantitative point of view, the porous characteristics of the TiO2 aerogel supports obtained by analyzing the 3D model are summarized in Table 2. Similarly to the most recent analyses devoted to the porosity characterization at nanometer scale, these tomographic results must be compared to the values obtained by nitrogen sorption. For illustration, Fig. 6 shows the nitrogen sorption isotherms for three samples (the three others are shown in ESI,† Fig. SI 3). All these isotherms are of type IV and denote the presence of a mesostructured material. More precisely, the hysteresis loops are characteristics of porous materials with a well-defined pore structure and a relatively uniform mesopore distribution (shown in Fig. 7 and SI 4 from the ESI†). A close analysis of the hysteresis loops reveals the appearance, for all annealed samples, of a flatness of the volume for 0 < P/P0 < 0.2. This behavior indicates a decrease of the amount of micropores as a consequence of the applied thermal treatment and is confirmed by the diminution of the cumulative volume of micropores as derived from the Horvath–Kawazoe (HK) model (see Table 2). The annealing induces as well a significant diminution of the mesopores dimension for both the Ag-free samples and the composites with the highest Ag content, i.e. I1, I2, B1 and B2 (Table 1). These dissimilarities will be exhaustively discussed in the next paragraphs.
 | ||
| Fig. 6 Nitrogen adsorption isotherms for the as-prepared pure TiO2 I1 (a), Ag-rich as-prepared B1 (b), annealed pure TiO2 I2 (c), Ag-rich annealed B2 (d) specimens. | ||
 | ||
| Fig. 7 Size distribution of mesopores for the as-prepared pure TiO2 I1 (a), Ag-rich as-prepared B1 (b), annealed pure TiO2 I2 (c), Ag-rich annealed B2 (d) specimens. | ||
| Sample | Ag concentration (from elemental analysis) (%) | Specific surface area (from tomography) (m2 g−1) | Specific surface area (from BET) (m2 g−1) | Surface of Ag (from the total aggregate surface, from tomography) (%) | Accessible Ag surface (from the total Ag surface, from tomography) (%) | Mean diameter of mesopores (from BJH) (nm) | Cumulative micropore volume (from HK) (cm3 g−1) |
|---|---|---|---|---|---|---|---|
| I1 | — | 80 | 614 | 15.6 | 0.3 | ||
| I2 | — | 100 | 120 | 12.6 | 0.07 | ||
| A1 | 0.23 | 48 | 474.8 | 3 | 15 | 10 | 0.25 |
| B1 | 4.48 | 51 | 550.3 | 7 | 15 | 13 | 0.27 |
| A2 | 0.28 | 79 | 129.7 | 6 | 30 | 10.5 | 0.07 |
| B2 | 4.48 | 72 | 58.6 | 0.4 | 1 | 11.5 | 0.03 |
A significant discrepancy can be observed between the specific surface area values as deduced by electron tomography and nitrogen sorption, especially for the as-prepared samples. To explain this difference, several issues need to be accounted for. It is well known that the electron tomography technique has generally limited spatial resolution (of the order of nanometers) and therefore is not able to identify the micropores and some of mesopores (lower than 2 nm). In this context, several previous studies clearly indicate that the specific surface areas deduced from ET experiments are one order of magnitude smaller than the values obtained from nitrogen sorption measurements.16,22,42 In our case, a similar difference for the as-prepared samples appears, which is certainly due to the high contribution of the micropores to the total surface area. By contrast, the difference is considerably reduced in the case of the annealed samples. This can be unambiguously attributed to the vanishing of a high number of micropores during the thermal treatment, leading globally to a significant reduction of the total porosity within the annealed nanocomposites. From a quantitative perspective, the comparative analysis of the specific surface area values as deduced from BET and tomography for the annealed samples A2 and B2 suggests that the microporosity is minor in B2, whereas in the case of A2 it remains still significant. One can however remark that the cumulative micropore volume is the same for the annealed sample (I2) without silver and with the smallest silver content (A2) and it decreases to less than half for the annealed composite with the highest silver amount (see Table 2). Moreover, by analyzing the mesopores surface as deduced from ET, one can observe a relevant augmentation of the corresponding values for the three pairs of samples. This behavior could be tentatively attributed to the drastic diminution of the microporosity during annealing, assuming that the formation of new mesopores can be partially caused by the coalescence of several micropores. The results obtained from the HK model on the cumulative micropore volume (Table 2) confirm this hypothesis. Regarding the size of mesopores, it is important to mention that we have analyzed preferentially their mean values as deduced from nitrogen sorption, which are less affected by the local character as compared with the ET results. We have obtained thus 10 nm for the sample A1, while the corresponding value for the B1 (with higher Ag amount) is slightly higher, about 13 nm (Fig. 7a and SI 4a from the ESI†). The thermal treatment induces a different and unexpected behavior: the median pore diameter remains similar for the sample with lower Ag amount (10.5 nm for A2), while for the sample with high Ag amount a significant diminution has been observed (11.5 nm for B2, compared to the initial value of B1, i.e. 13 nm).
Once the 3D models of the aggregates are calculated, cross-sectional views at different depths and orientations can be redrawn in order to visualize better the interface between Ag and TiO2. Such typical 3D views for the four specimens are shown on Fig. 8. In a first step, their simple analysis allows one to get direct information about the TiO2 pore characteristics. In this sense, though the presence of some closed pores can be evidenced, most of them appear as open and directly connect with the outer surface of the grain. Afterwards, by analyzing the cross-sectional 3D representations, precise information on the spatial localization of Ag NPs within the inner porous network has been obtained. Surprisingly, apart from some NPs located on the pores surface the great majority of NPs appear to be encapsulated by the TiO2 porous matrix. It is worth mentioning here that this result concerns only the NPs visible in the tomographic reconstruction e.g. with diameter higher than 1.5 nm. In other words, the Ag atomic clusters most probably located inside the nanocomposite are excluded from the actual discussion.
 | ||
| Fig. 8 Cross-sectional views through the 3D models corresponding to the aggregates chosen for tomographic studies of specimens: (a) A1, (b) B1, (c) A2, (d) B2. Such 3D representations allow us to directly visualize the internal porous network and the localization of Ag NPs within this porosity, on the external surface of the grains or inside the TiO2 pores. | ||
A statistical analysis of Ag NPs size has been also performed, by considering the 3D modeling alone. Since a high number of NPs has been analyzed (about 600), one considers that the results are relevant for the whole specimen. The size histograms are represented in Fig. 9 for the samples with high Ag amounts (B1 and B2), whereas the histograms for the two other specimens (A1 and A2) are given in the ESI† (Fig. SI 5). As expected, the mean diameter of Ag NPs increases after annealing, while their number decreases, as the effect of NPs diffusion and coalescence, and/or to the Ostwald ripening process. A rather unexpected finding concerns the higher value of the Ag NPs’ mean size in the specimen A1 (small amount of Ag) as compared to the other as-prepared sample, B1. A possible explanation of this variation can be found by taking into account the smaller diameter of the mesopores in A1 as compared to B1. This microstructural difference can as well be at the origin of different diffusion rates for the two specimens along the supercritical drying process. The diffusion of silver inside the aged gel certainly depends on the initial TiO2 pore diameter, whilst the nucleation centers were found to be preferentially located in the vicinity of the grain surfaces as evidenced for the specimens doped with a low Ag amount (A1 and A2). As a consequence, in the case of A1, where the final diameter of mesopore was found to be smaller than for B1, the first Ag NPs, formed at the beginning of the growth process, block the pores, thus limiting the diffusion rate of the unconsumed reactants. This is not the case for the sample B1, where higher pores diameters permit a considerable higher diffusion rate and for a longer time. Thus, smaller size NPs form with a more uniform distribution within the TiO2 grains.
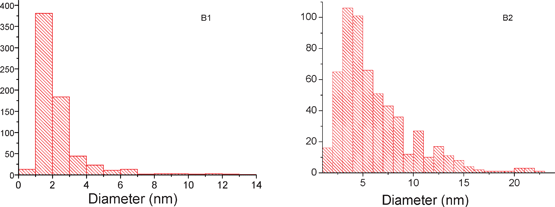 | ||
| Fig. 9 Histograms illustrating the size distribution of Ag NPs for the Ag-rich as-prepared B1 (left) and annealed B2 (right) samples. | ||
It is reminded here that all the hypotheses advanced do not take into account the eventual presence of Ag atomic clusters which cannot be resolved by our reconstruction. It is still worth mentioning the significant difference between the mean pore diameters for the annealed samples A2 and B2. It most probably originates from the initial difference between the mean pore diameters of the corresponding as-prepared samples.
For the applications involving the antimicrobial effect of the silver, one of the key parameters is the Ag surface which should be easily accessible. As observed before, some Ag NPs are encapsulated in the TiO2 porous network and consequently their surfaces became inaccessible. To quantify this effect, two types of parameters have been calculated from the 3D tomographic model: (i) the Ag contribution to the total grain surface, referred to as the outside Ag surface which depends on the number of Ag NPs located at the surface of the TiO2 grains; (ii) the Ag surface in contact with the vacuum relative to the total surface area proposed by the Ag NPs, referred to as the accessible Ag surface, a parameter that accounts for the NPs encapsulation. To assess the outside Ag surface (the specific surface area of Ag), the specific surface area of the whole porous grain (including Ag and TiO2) was calculated in the first place from the 3D model and afterwards the contribution of Ag to this surface was estimated. Further, the accessible Ag surface has been determined by comparing the 3D modeling of Ag and TiO2 and subtracting the interface between the two components from the total Ag surface. By comparing the corresponding data displayed in Table 2, one can observe that the outside Ag surface of B1 is only two times larger than for A1, whereas the relative amount of Ag is ten times higher. Since the mean diameter of NPs is smaller in B1 as compared to A1 (and that generally induces an increase of the surface area), one can unambiguously conclude that for the Ag-rich sample the number of encapsulated Ag NPs is higher. This result is in good agreement with some previous works which have shown that, if the antimicrobial activity increases at low Ag loadings with respect to pure TiO2, it decreases for loadings higher than an optimum value.43 Additionally, note that for both samples the accessible Ag surfaces are very similar, in the order of 15%.
The effects of the thermal treatment on the Ag accessibility are contradictory for the two Ag–TiO2 nanocomposites studied here. For the sample with smaller Ag amount, the outside Ag surface and the accessible Ag surface are two times higher as compared to the corresponding values of the not-treated specimens. That is certainly a combined effect of an increased number of NPs located on the pore surface and a diminishing of the encapsulation degree of Ag NPs. Contrary, for the Ag-rich sample, the outside Ag surface and the accessible Ag surface become insignificant after annealing. The strong intensification of the encapsulation degree of Ag can be at the origin of this finding. A possible hypothesis that can be advanced for explaining the different behavior of the two samples is the presence of micropores in A2 and their complete lacking in B2. In such a case, the pore surface from the grains of B2 has became relatively flat, favoring in this way the diffusion of Ag species towards the surface and leading to the augmentation of the encapsulation degree of Ag NPs. The first conclusion that can be redrawn from this comparative analysis denotes the specimen A2 as the most interesting sample for the applications making use of the Ag effective surface area. Indeed, though the total Ag amount is ten times lower as compared to the sample B2, it can be characterized by a relatively high surface specific area of the TiO2 aerogel as well as by a high effective Ag surface.
For a clear assessment of the impact of Ag on the porous TiO2 microstructural characteristics, we have analyzed by ET and nitrogen sorption two pure TiO2 aerogels prepared in the same conditions as the Ag–TiO2 nanocomposites. These specimens are denoted as I1 and I2, for the as-prepared and annealed samples, respectively. The tomographic analyses schematized in Fig. 10 and the information obtained (e.g. the surface specific areas of the grains) is summarized in Table 2. By analyzing some typical slices extracted from the reconstructions, one observes the high degree of porosity for the two pure TiO2 samples. From a quantitative point of view, the values of the surface specific areas are about two times higher than in the case of Ag–TiO2 nanocomposites. For both as-prepared and annealed specimens, this observation is in agreement with the nitrogen sorption measurements. A possible explanation is that, in the case of Ag doped samples, the Ag NPs and TiO2 growths happen simultaneously blocking the further development of pores created at the beginning of the supercritical drying process leading in this way to a diminution of the final porosity. Moreover, the thermal treatment leads to an increase of the mesoporosity for the undoped specimens, similar to the Ag-doped ones. This behavior is rather unexpected for a homogenous sample, where the thermal treatment induces most likely the build up of a new population of bigger grains as an effect of coalescence. Consequently, the main finding of this analysis is that the simultaneous synthesis of Ag NPs and TiO2 porous structures has a considerable impact on the porous characteristics of the final aerogel nanocomposites, at variance to the similar ones in which the Ag NPs are subsequently deposited by impregnation of the TiO2 matrix.
 | ||
| Fig. 10 Steps of the tomographic analyses performed on the pure TiO2 aerogels, the as-prepared (top) and annealed (bottom) specimens. Left: typical 2D-images from the tilt series used to calculate the 3D reconstructions of the chosen grains. Middle: examples of slices extracted from the reconstructions for the two specimens. Right: 3D models obtained by using a data segmentation procedure. The small NPs visible on the carbon membrane are Au fiducial markers used in the fine alignment process. | ||
Conclusions
We have reported here a complete analysis of heterogeneous nanostructures made of a porous TiO2 aerogel and Ag NPs, a very interesting composite that can be successfully employed for various applications. The experimental method proposed combines several characterization techniques that allowed us to precisely determine the structural and morphological characteristics of the studied specimens via a multi-scale approach. We have shown that increasing the Ag amount does not necessarily improve the accessible surface of the Ag species: for a relatively high amount of Ag in the grains of nanocomposite with a homogenous spatial distribution of Ag NPs, the accessibility to the Ag surface diminishes, leading thus to a moderation of the antimicrobial activity. Alternatively, we have demonstrated that the effect of the thermal treatment is relatively complex: it leads, on one hand, to an increase of the mean diameters of NPs and therefore to the reduction of the surface contribution as against the bulk, but on the other hand, it modifies the fraction of encapsulated NPs as well. This latter effect seems to depend strongly on the morphological 3D characteristics: for the samples where the thermal treatment induces a diminution of the microporosity, the surface diffusion of the different species is higher leading to more encapsulated Ag NPs and consequently lower accessibility.From a general point of view, this work underlines once again the impressive potential of electron tomography when applied to the analysis of multicomposite specimens with 3D nanostructured architectures. It is however worth noting that for a complete structural, morphological and chemical analysis of such heterogeneous nanostructures, the use of several characterization tools with different selectivity is a crucial requirement.
Acknowledgements
This research was accomplished in the framework of PNII Idei PCCE-129/2008 project granted by the Romanian National University Research Council—CNCSIS. D. G. author wishes to thank for the financial support provided from programs co-financed by The Sectoral Operational Programme Human Resources Development, Contract POSDRU 6/1.5/S/3—“Doctoral studies: through science towards society”. The authors thank Dr Simona Moldovan for her careful and critical reading of the manuscript.References
- N. Hüsing and U. Schubert, Angew. Chem., Int. Ed., 1998, 37, 22–45 CrossRef.
- A. B. Jarzebski and J. Lorenc, Chem. Eng. Sci., 1995, 50, 357–60 CrossRef CAS.
- M. S. Lee, S. S. Hong and M. Mohseni, J. Mol. Catal. A: Chem., 2005, 242, 135–40 CrossRef CAS.
- O. Akhavan and E. Ghaderi, Surf & Coat Technology, 204(210), 3676–3683 CAS.
- B. Tian, Z. Shao, Y. Ma, J. Zhang and F. Chen, J. Phys. Chem. Solids, 2011, 72, 1290–1295 CrossRef CAS.
- Y. Ao, J. Xu, D. Fu and C. Yuan, J. Phys. Chem. Solids, 2008, 69, 2660–2664 CrossRef CAS.
- L. Baia, L. Diamandescu, L. Barbu-Tudoran, A. Peter, G. Melinte, V. Danciu and M. Baia, J. Alloys Compd., 2011, 509, 2672–2678 CrossRef CAS.
- M. Baia, V. Danciu, V. Cosoveanu and L. Baia, Vib. Spectrosc., 2008, 48, 206–209 CrossRef CAS.
- L. Zhao, H. Wang, K. Huonext, L. Cuia, W. Zhangb, H. Nib, Y. Zhanga, Z. Wua and P. K. Chuc, Biomaterials, 2011, 32, 5706–5716 CrossRef CAS.
- B. Yu, K. M. Leung, Q. Guo, W. M. Lau and J. Yang, Nanotechnology, 2011, 22, 115603 CrossRef.
- X. Pana, I. Medina-Ramirezb, R. Mernaughc and J. Liu, Colloids Surf., B, 2010, 77, 82–89 CrossRef.
- Y. Liu, W. Wang, F. Yang and X. Yang, Microporous Mesoporous Mater., 2008, 114, 431–439 CrossRef CAS.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, S. Gross, C. Maragno and E. Tondello, Coord. Chem. Rev., 2006, 250, 1294–1314 CrossRef CAS.
- K. Kaneko, W-J. Moon, K. Inoke, Z. Horita, S. Ohara, T. Adschiri, H. Abe and M. Naito, Mater. Sci. Eng., A, 2005, 403, 32–36 CrossRef.
- G. V. Krylova, Yu. I. Gnatyuk, N. P. Smirnova, A. M. Eremenko and V. M. Gun'ko, J. Sol-Gel Sci. Technol., 2009, 50, 216–228 CrossRef CAS.
- N. Bahadur, K. Jain, R. Pasricha, S. Govind and S. Chand, Sens. Actuators, B, 2011, 159, 112–120 CrossRef CAS.
- X. S. Li, G. E. Fryxell, C. Wang and M. H. Engelhard, Microporous Mesoporous Mater., 2008, 111, 639–642 CrossRef CAS.
- H. Fredrich, J. R. A. Sietsma, P. E. de Jongh, A. J. Verkleij and K. P. de Jong, J. Am. Chem. Soc., 2007, 129, 10249–10254 CrossRef.
- M. Weyland, Top. Catal., 2002, 21, 175–183 CrossRef CAS.
- S. Bals, K. J. Batenburg, D. Liang, O. Lebedev, G. Van Tendeloo, A. Aerts, J. A. Martens and C. E. A. Kirschhock, J. Am. Chem. Soc., 2009, 131, 4769–4773 CrossRef CAS.
- I. Florea, M. Houlle, O. Ersen, L. Roiban, A. Deneuve, I. Janowska, P. Nguyen, C. Pham and C. Pham-Huu, J. Phys. Chem. C, 2009, 113, 17711–17719 CAS.
- H. Friedrich, P. E. de Jongh, A. J. Verkleij and K. P. de Jong, Chem. Rev., 2009, 109, 1613–1629 CrossRef CAS.
- O. Ersen, C. Hirlimann, M. Drillon, J. Werckmann, F. Tihay, C. Pham-Huu, C. Crucifix and P. Schultz, Solid State Sci., 2007, 9, 1088–1098 CrossRef CAS.
- P. A. Midgley and M. Weyland, Ultramicroscopy, 2003, 96, 413–431 CrossRef CAS.
- I. Florea, O. Ersen, C. Hirlimann, L. Roiban, A. Deneuve, M. Houlle, I. Janowska, P. Nguyen, C. Pham and C. Pham-Huu, Nanoscale, 2010, 2, 2668–2678 RSC.
- S. Bals, G. Van Tendeloo and C. Kisielowski, Adv. Mater., 2006, 18, 892–895 CrossRef CAS.
- I. Florea, L. Roiban, C. Hirlimann, F. Tihay, C. Pham-Huu, J. Werckmann, C. Pham, P. Nguyen, M. Drillon and O. Ersen, Adv. Eng. Mater., 2011, 13, 122–127 CrossRef CAS.
- J. C. González, J. C. Hernandez, M. López-Haro, E. del Río, J. J. Delgado, A. B. Hungría, S. Trasobares, S. Bernal, P. A. Midgley and J. Juan Calvino, Angew. Chem., Int. Ed., 2009, 48, 5313–5315 CrossRef.
- O. Ersen, J. Parmentier, L. A. Solovyov, M. Drillon, C. Pham-Huu, J. Werckmann and P. Schultz, J. Am. Chem. Soc., 2008, 130, 16800–16806 CrossRef CAS.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maccato, E. Tondello, O. I. Lebedev, S. Turner, G. Van Tendeloo, C. Sada and U. L. Stangar, ChemPhysChem, 2009, 10, 3249–3259 CrossRef CAS.
- K. Yoshida, M. Makihara, N. Tanaka, S. Aoyagi, E. Nishibori, M. Sakata, E. D. Boyes and P. L. Gai, Microsc. Microanal., 2011, 17, 264–273 CrossRef CAS.
- J. C. Gonzalez, J. C. Hernandez, M. Lopez-Haro, E. del Rio, J. J. Delgado, A. B. Hungria, S. Trasobares, S. Bernal, P. A. Midgley and Jose Juan Calvino, Angew. Chem., Int. Ed., 2009, 48, 5313–5315 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- W. Saxton, W. Baumeister and M. Hahn, Ultramicroscopy, 1984, 13, 57–70 CrossRef CAS.
- J. R. Kremer, D. N. Mastronarde and J. R. McIntos, J. Struct. Biol., 1996, 116, 71–76 CrossRef CAS.
- C. Messaoudi, T. Boudier, C. O. S. Sorzano and S. Marco, BMC Bioinformatics, 2008, 8, 288 CrossRef.
- C. O. S. Sorzano, C. Messaoudi, M. Eibauer, J. R. Bilbao-Castro, R. Hegerl, S. Nickell, S. Marco and J. M. Carazo, BMC Bioinformatics, 2009, 10, 124 CrossRef.
- http://www.slicer.org/ .
- http://www.cgl.ucsf.edu/chimera/ .
- J. Frank, Electron Tomography, Plenum, New York, 1992 Search PubMed.
- J. Frank, Three dimensional Electron Microscopy of macromolecular assemblies, Academic Press, San Diego, 1996 Search PubMed.
- Z. Xiong, J. Ma, W. J. Ng, T. D. Waite and X.S. Zhao, Water Res., 2011, 45, 2095 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20568h/ |
| This journal is © The Royal Society of Chemistry 2012 |