DOI:
10.1039/C2RA20228J
(Paper)
RSC Adv., 2012,
2, 5349-5357
Electrochemical, phosphate hydrolysis, DNA binding and DNA cleavage properties of new polyaza macrobicyclic dinickel(II) complexes†
Received
9th February 2012
, Accepted 26th March 2012
First published on 26th March 2012
Abstract
A new class of macrobicyclic dinickel(II) complexes [Ni2L1,2B](ClO4)4 (1–6), where L1,2 are polyaza macrobicyclic binucleating ligands, and B is a N,N-donor heterocyclic base (viz. 2,2′–bipyridine (bipy) and 1,10–phenanthroline (phen)) are synthesized and characterized. The redox, catalytic, DNA binding and DNA cleavage properties were studied. They exhibit two irreversible waves in the cathodic region around Epc = −0.95 V and Epa = −0.85 V vs. Ag/Ag+ in CH3CN–0.1 M TBAP, respectively. The first order rate constants for the hydrolysis of 4-nitrophenylphosphate to 4-nitrophenolate by the dinickel(II) complexes 1–6 are in the range from 3.36 × 10−5 to 10.83 × 10−5 Ms−1. The complexes 3 and 6 show good binding propensity to calf thymus DNA giving binding constant values (Kb) in the range from 3.08 × 105 to 5.37 × 105 M−1. The binding site sizes and viscosity data suggest the DNA intercalative and/or groove binding nature of the complexes. The complexes display significant hydrolytic cleavage of supercoiled pBR322DNA at pH 7.2 and 37 °C. The hydrolytic cleavage of DNA by the complexes is supported by the evidence from free radical quenching and T4 ligase ligation. The pseudo Michaelis–Menten kinetic parameters kcat = 5.44 × 10−2 h−1 and KM = 6.23 × 10−3 M for complex 3 were obtained. Complex 3 also shows an enormous enhancement of the cleavage rate, of 1.5 × 106, in comparison to the uncatalysed hydrolysis rate (k = 3.6 × 10−8 h−1) of ds-DNA.
Introduction
The development of new reagents that can specifically bind and cleave DNA under physiological conditions via oxidative and hydrolytic mechanisms has been attracting great interest in the bioinorganic chemistry field.1 Metal complexes can bind to DNA with non-covalent interactions such as electrostatic binding, groove binding, intercalative binding and partial intercalative binding.2 Many useful applications of these complexes require that the complexes bind to DNA through an intercalative mode. Intercalators are small molecules that contain a planar aromatic heterocyclic functionality which can insert and stack between the base pairs of double helical DNA.3 In past decades, several phenanthroline or bipyridine based nickel(II) complexes were reported as they are avid DNA intercalative binders as well as DNA cleavers.4 Metal complexes that cleave DNA in a hydrolytic manner are increasingly important for a number of reasons.5 They may function as artificial restriction enzymes in molecular biology, offering the possibility of designing systems with different sequence specificities than natural protein endonucleases. This capability may also be useful for targeting DNA sequences to block gene expression at the molecular level in chemotherapy or for studying nucleic acid conformations. Hydrolytic cleavage in the absence of any external additives does not suffer from such drawbacks as the cleaved products can be religated enzymatically.6 A number of proposals have been put forth describing the reactivity of DNA with mononuclear nickel(II) complexes.7 Compared with the number of studies dealing with mononuclear complexes, relatively few studies on dinuclear complexes have been reported to date.8 We have explored the efficient DNA hydrolysis promoted by water coordinated macrocyclic octahedral dinickel(II) complexes under physiological conditions.9 Very recently, we have reported that the heterocyclic bases coordinated polyaza macrobicyclic dicopper(II) complexes10 display better DNA cleavage activities than heterocyclic base free dicopper(II) analogues.11 This significant rate enhancement promoted by the heterocyclic base coordinated dinuclear complexes stimulated us to design and synthesize macrobicyclic dinickel(II) analogues to evaluate and understand the factors on the DNA-binding and cleavage properties. Herein, we report the synthesis, structural characterization, electrochemical, phosphatase-like activity, DNA binding and cleavage properties of the new macrobicyclic dinickel(II) complexes (Scheme 1).
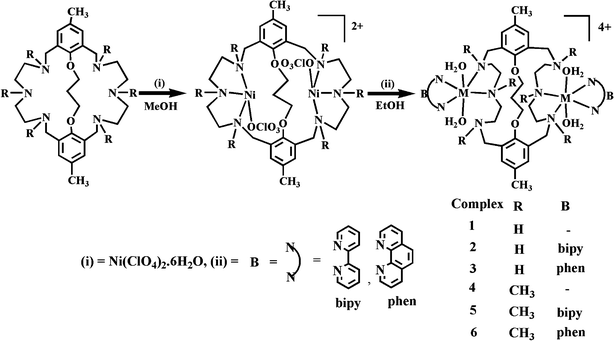 |
| | Scheme 1 Synthesis of macrobicyclic dinuclear nickel(II) complexes. | |
Results and discussion
General properties
A very strong and broad band near 1100 cm−1 and a strong sharp band near 625 cm−1 are observed in the IR spectra of the heterocyclic base coordinated complexes 2, 3, 5 and 6, which is in agreement with the presence of uncoordinated ionic perchlorate in their crystal lattices. Whereas the heterocyclic base free complexes 1 and 4 displayed two peaks around 1105 and 1085 cm−1, indicating that the perchlorate ions present in the complexes are coordinated to the NiII ions. The electronic spectra of the CH3CN solution of complexes 1 and 4 showed only one intense intraligand band in the UV region. In the visible region, complexes 1 and 4 exhibited absorption maxima at 563 and 585 nm, respectively. Whereas, the heterocyclic base containing complexes 2, 3, 5 and 6 showed two intense bands in the UV region and three less intense bands in the visible region. This strongly suggests that the coordination geometry around the metal ion in the heterocyclic base free (1 and 4) and heterocyclic base coordinated complexes (2, 3, 5 and 6) might be distorted square planar and distorted octahedral, respectively.9,12 Interestingly, the absorption maxima (λmax) of complexes 4–6 are higher than those of complexes 1–3. This indicates that the coordination geometry around the NiII ions is more distorted13 in the N-methylated polyaza macrobicyclic ligand (L2) than the N-methyl free polyaza macrobicyclic ligand (L1). The positive ion ESI mass spectra (Fig. S1–S2, ESI†) showed signals for heterocyclic base free dinickel(II) complexes 1 and 4, [Ni2L1,2](ClO4)2 showed major peaks at m/z = 411.72 and 453.45, respectively. The heterocyclic base coordinated complexes 2, 3, 5 and 6 displayed peaks at 603.24, 627.33, 645.65 and 669.83, which have been assigned to the [Ni2L1a–b,2a–b − 2ClO4]2+ ion, respectively. Since all attempts to obtain a suitable single crystal of the as-synthesized complexes for X-ray diffraction analysis resulted in failures, the information about the sizes and shapes of the complexes (3–6) was gleaned from the energy minimized geometry (Fig. 1 and Fig. S3–S4, ESI†) using the molecular mechanics universal force field simulation (MMUFF).14 The analytical and ESI mass spectral data were consistent with the proposed formula of the dinickel(II) complexes.
 |
| | Fig. 1 Energy minimized structure of macrobicyclic dinickel(II) complexes 3 (above) and 5 (below). Color code: Ni = green, O = red, N = blue, C = grey. | |
Electrochemistry
The cyclic voltammograms of the dinickel(II) complexes (1–6) are depicted in Fig. 2 and Fig. S5–S8 (ESI†). Electrochemical data are summarized in Table S1.† All of the dinuclear nickel(II) complexes (1–6) display two irreversible reduction and oxidation waves in the cathodic region around Epc = −0.95 V and Epa = −0.85 V, respectively.
 |
| | Fig. 2 Cyclic voltammograms of dinickel(II) complexes 1 and 4 in CH3CN. | |
Controlled potential electrolysis carried out at 100 mV more negative than the reduction wave ratifies the consumption of two electrons per molecule (n ≅ 1.97) and the experiment showed that the couple corresponded to a two-electron transfer process. This is assigned to the formation of a NiINiI species. An irreversible anodic peak with a small peak current (ia) at cathodic potentials (around −0.85 V) may be due to the deposition of metal on the electrode surface, which causes low electrical conductivity. Hence, the anodic current attributed to the NiINiI oxidation diminishes significantly, leaving only a small anodic peak.15 It is important to note that the complexes 4–6 reduce at less negative potentials than complexes 1–3. This is attributed to the structural differences in the molecules, the amine donor nitrogen of L2 contains a bulky methyl group, whereas L1 has a hydrogen atom. The deviation of the metal from its coordination plane should be larger for L2 complexes in order to destabilize the NiII oxidation state. Thus, the electron density on the nickel ions of the complexes 1–3 is less and the nickel coordination geometry may also be more distorted due to the steric effect of the bulky methyl group substituent.16 It was suggested16b,17 that the reduction in electron density on the nickel ions and distortion in geometry favors the reduction process (NiII → NiI) at a less negative potential, as observed for the complex of ligand L1 relative to ligand L2. The heterocyclic base (such as 2,2′-bipyridine and 1,10-phenanthroline) coordinated complexes (2, 3, 5 and 6) containing nickel(II) ions were reduced at lower potentials than the heterocyclic base free dinickel(II) complexes 1 and 4. This can be attributed to the donor nature, greater planarity and electronic properties that are associated with extended aromatic rings.12 In the positive potential region, the complexes 1–6 showed a two electron transfer quasi-reversible redox wave around E1/2 = +0.60 V vs. Ag/AgCl respectively. Controlled potential electrolysis experiment indicates that the redox peaks at anodic region is associated with the following oxidation process at the metal center.
| | 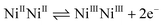 | (1) |
It is noticed that all the dinickel(II) complexes display a two electron redox wave at a single potential in the cathodic and anodic region. This may be due to both the NiII ions being remote to each other in the macrobicyclic ring, as well as them being in identical coordination environments. Thus they reduce/oxidize at a single potential in the cathodic/anodic region.
Phosphatase-like activity
Many hydrolytic processes in enzyme-catalysis involve metal ions that are assumed to activate a water molecule, which easily forms a hydroxyl group as a nucleophilic group in the reaction system.18 Presently, heterocyclic base coordinated dinickel(II) complexes (2, 3, 5 and 6) possess in their structure a potential nucleophile constituted by the metal coordinated water molecule, and their catalytic activities on hydrolysis of 4-nitrophenyl phosphate (4-NPP) was investigated spectrophotometrically by following the absorption increase at 400 nm due to the formation of 4-nitrophenolate ions over time. The effect of pH on the rate of reaction was determined and correlated with the pKa of coordinated water in 5. The pH dependence plot for the dinuclear NiII complexes on the phosphate hydrolysis reaction showed a pH-independent rate above 8.0 and a range below this pH where the initial rate of hydrolysis increases with pH (Fig. 3).
 |
| | Fig. 3 Dependence of the reaction rates on pH hydrolysis of 4-NPP by the macrobicyclic dinuclear NiII complex 5. | |
The derived sigmoidal pH-rate profiles are characteristic of a kinetic process controlled by an acid–base equilibrium and exhibits inflection points corresponding to the pKa value of 7.52 for one of the coordinated water molecules. This indicates that the [Ni2(L1,2)(B)2(H2O)3(OH)] complex is the reactive species. The NiII-bound OH− acts as a nucleophile to attack the phosphate atom of the 4-NPP and hydrolysis takes place. Since the substrate concentration was essentially constant during the measurement, the initial first order rate constant (kobs) was measured at different concentrations of the catalyst at pH 7.2 and 25 ± 0.1 °C. Plots of rate constant (kobs) vs. complex concentration are presented in Fig. 4. As can be seen, for all complexes, the rate of 4-NPP cleavage initially increases linearly with the increase of complex concentration but gradually the reaction order in the catalyst concentration deviates from unity.
![Dependence of the reaction rate on the concentration of 1–6 for the 4-NPP hydrolysis at pH 7.2 and 25 ± 0.1 °C. Conditions: [4-NPP] = 5.0 × 10−5 M, [Ni2 complex] = 5.0 × 10−5 to 5.0 × 10−4, [buffers] = 50 mM, I = 0.1 M NaClO4).](/image/article/2012/RA/c2ra20228j/c2ra20228j-f4.gif) |
| | Fig. 4 Dependence of the reaction rate on the concentration of 1–6 for the 4-NPP hydrolysis at pH 7.2 and 25 ± 0.1 °C. Conditions: [4-NPP] = 5.0 × 10−5 M, [Ni2 complex] = 5.0 × 10−5 to 5.0 × 10−4, [buffers] = 50 mM, I = 0.1 M NaClO4). | |
In other words, the reaction exhibits a first order dependence only at low NiII complex concentrations. The kobs value for phosphate hydrolysis reaction by nickel(II) perchlorate hexahydrate salt was found as 2 × 10−13 s−1. This is negligibly small when compared to the kobs value (108 times faster) for dinuclear NiII complexes. The first order rate constants k were obtained for the NiII complexes, from Lineweaver–Burk plot, i.e., 1/V0vs. 1/[4-NPP] by changing the concentration of the substrate (Fig. 5) and the results of calculation are summarized in Table 1. These values are smaller than our earlier report for the hydrolytic cleavage of 4-NPP by macrobicyclic dicopper(II) analogs.9
 |
| | Fig. 5 Lineweaver–Burk plot for the 4-NPP hydrolysis by dinickel(II) complexes 1–6. Inset shows hydrolysis of 4-NPP by the dinickel(II) complex 1. | |
Table 1 Kinetic data for 4-NPP hydrolysis and DNA binding parameters of dinickel(II) complexes (1–6)
| Complex |
4-NPP hydrolysis k (s−1) |
105Kb [s]a |
105Kappb |
CD Δλmax + Δεc |
Binding constants (M−1) were determined by absorption spectrophotometric titration and [s] is the binding site size.
Apparent binding constants (M−1) were determined by fluorescence spectrophotometric method.
Δλmax is the shift in nm of the positive DNA CD band at 274 nm. Δε (the value in parentheses) is the difference between the maximum ellipticity (in °) observed for the positive CD band in the spectrum of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction mixture, and the ellipticity observed at the same wavelength in the spectrum of free CT DNA. :1 reaction mixture, and the ellipticity observed at the same wavelength in the spectrum of free CT DNA.
|
|
1
|
3.36 × 10−5 |
2.84 [0.65] |
3.28 |
— |
|
2
|
7.15 × 10−5 |
4.55 [1.22] |
4.42 |
— |
|
3
|
6.32 × 10−5 |
5.12 [1.76] |
5.37 |
−4 (8) |
|
4
|
4.18 × 10−5 |
2.78 [0.49] |
3.08 |
−3 (3) |
|
5
|
10.83 × 10−5 |
4.26 [1.08] |
4.74 |
— |
|
6
|
8.32 × 10−5 |
4.64 [1.42] |
4.85 |
−4 (9) |
From Table 1, it can be seen that the rate of the phosphate hydrolysis reaction increased in the following order 1 < 4 < 3 < 2 < 6 < 5. Interestingly, the heterocyclic base coordinated NiII complexes 2, 3, 5 and 6, display higher catalytic hydrolysis activities than the heterocyclic base free NiII analogues 1 and 4. This is due to the NiII complexes 2, 3, 5 and 6 possessing in their structure a potential nucleophile constituted by the metal coordinated water molecule, which is absent in complexes 1 and 4. Interestingly the ligands L2 of complexes (4–6) show better hydrolysis activities than NiII complexes 1–3 of ligand L1. This indicates that the degree of distortion of geometry around the NiII ions in the macrobicyclic ligand L2 is more than that of the NiII ions in ligand L1. It is evident from the literature,12,19 that the rate constants for more distorted complexes are higher than those of less distorted complexes. The rate of phosphate ester hydrolysis enhanced by 1–6 are higher than those reported for mononuclear NiII complexes20a but are less when compared to the reported phenolate bridged dinickel(II) complexes.20b This may be due to the absence of a bridging phenolate or hydroxyl group in complexes 1–6. It is important to note that the phosphate hydrolysis activity of the nickel(II) complexes is due to their labile coordination sites present in the molecule and are fully independent of their redox properties. Thus there is no link between the electrochemical properties and catalytic phosphate hydrolysis activities of dinickel(II) complexes.
DNA binding
Absorption titration technique has been used to monitor the mode of interaction of the dinickel(II) complexes 1–6 with CT DNA (Fig. 6). The intrinsic equilibrium DNA binding constant (Kb) values of the complexes along with the binding site size (s) are given in Table 1. The Kb values of ∼105 M−1 follow the order: 3 > 6 > 2 > 5 > 1 > 2. The binding site size (s) values in the base pairs obtained from the M vs. H fits are in the range of 0.49–1.76, with the phen complexes showing a higher value of s in comparison to their bpy analogs. The better binding of complexes 3 and 6, as compared to complexes 1, 2, 4 and 5, might be due to their extended aromaticity and coplanarity of the phenanthroline ring system. Complexes were expected to be stacked between the base pairs upon interaction of the complex with DNA.21 These spectral characteristics obviously suggest that the two complexes (3 and 6) mentioned here interact with DNA most likely through a mode that involves a stacking interaction between the aromatic choromophore and the base pairs of DNA. Complexes 1, 2, 4 and 5 show a minor bathochromic shift of the spectral band, of ∼3 nm, suggesting a groove-binding preference of the complexes. Structurally, complexes 3 and 6 should provide more a aromatic moiety to overlap with the stacking base pairs of the DNA helix by intercalation, which results in hypochromism and bathochromism. The increasing aromatic moiety in the phenanthroline based ligand of the NiII complex facilitates its potential intercalative DNA binding, while complexes 1, 2, 4 and 5, with or without a bipyridine moiety, may prefer major groove-binding. We have used a fluorescence spectral titration method to obtain the apparent binding constant values (Kapp) of complexes 1–6 (Table 1). Ethidium bromide (EB) has been used as a spectral probe as it exhibits an enhanced emission intensity when it binds to the DNA.
![Absorption spectra of dinickel(ii) complexes 2 and 6 (1 × 10−5 M) in the absence and presence of increasing amounts of CT DNA (0–2.5 × 10−3 M) at 25 °C in 50 mM Tris-HCl/NaCl buffer (pH = 7.5). Arrow shows the absorbance changing upon increasing DNA concentrations. Insets shows the least–squares fit of Δεaf/Δεbfvs. [DNA] for complexes 1–6.](/image/article/2012/RA/c2ra20228j/c2ra20228j-f6.gif) |
| | Fig. 6 Absorption spectra of dinickel(II) complexes 2 and 6 (1 × 10−5 M) in the absence and presence of increasing amounts of CT DNA (0–2.5 × 10−3 M) at 25 °C in 50 mM Tris-HCl/NaCl buffer (pH = 7.5). Arrow shows the absorbance changing upon increasing DNA concentrations. Insets shows the least–squares fit of Δεaf/Δεbfvs. [DNA] for complexes 1–6. | |
The competitive binding of the complexes to DNA could result in the displacement of bound EB and could cause a decrease in the emission intensity because of solvent quenching. The emission spectra of EB bound to DNA in the absence and presence of complexes 3 and 4 are shown in Fig. 7. The Kapp values of the complexes are ∼105 M−1. The binding constant values (Kb) of the dinickel(II) complexes 1–6 are comparable to the reported heterocyclic base coordinated mononuclear NiII complexes,4d but are higher than the heterocyclic base free mononuclear NiII complexes.22 Circular dichorism (CD) is a useful method to assess whether nucleic acids undergo conformational changes as a result of complex formation or changes in the environment.23
![Emission spectrum of EB bound to DNA: (A) in the presence of 3 or 4; (B) ([EB] = 3.3 μM, [DNA] = 40 μM, [complex] = 0–25 μM, λex= 410 nm) in 50 mM Tris-HCl/NaCl buffer (pH = 7.5). Arrow shows the emissions changing upon increasing complex concentrations. Insets show the plots of emission intensity I0/I vs. [DNA]/[NiII complex].](/image/article/2012/RA/c2ra20228j/c2ra20228j-f7.gif) |
| | Fig. 7 Emission spectrum of EB bound to DNA: (A) in the presence of 3 or 4; (B) ([EB] = 3.3 μM, [DNA] = 40 μM, [complex] = 0–25 μM, λex= 410 nm) in 50 mM Tris-HCl/NaCl buffer (pH = 7.5). Arrow shows the emissions changing upon increasing complex concentrations. Insets show the plots of emission intensity I0/I vs. [DNA]/[NiII complex]. | |
At a 2:1 ratio of [Ni2L1,2]:CT DNA, all nickel(II) complexes produced shifts to higher energy for the positive CD signal as well as an enhancement of CD elipticity at 274 nm.
Examination of Fig. 8 shows that the magnitude of the increases in elipticity at 274 nm increases (Table 1) in the following order 4 < 6 < 3. The results reveal that the interaction is through preferentially intercalative binding with CT DNA and induce conformational changes from B–DNA to Z–DNA. However, the changes induced by 3 and 6 are more significant than those by 4. It is reasonable to suggest that the higher affinity of complexes 3 and 6 with CT DNA is due to presence of phenanthroline in both the compartments of the complex, which enhances the interaction.
 |
| | Fig. 8 CD spectra recorded over the wavelength range 230–320 nm for solutions containing a 2:1 ratio of CT DNA (200 μM) and dinuclear NiII complexes 3, 4 and 6 (100 μM) in50 mM Tris-HCl/NaCl buffer (pH = 7.5). (a = CT DNA, b = 3 + DNA, c = 4 + DNA, d = 6 + DNA). | |
The heterocyclic base free NiII complex 4 induced only slight conformational changes in DNA, such as the conversion from a more B-like to C-like structure within the DNA molecule. These CD spectral results are consistent with our early report on dicopper(II) analogs.9
Viscosity measurements have been carried out to examine the effect of the complexes 1, 3, 4 and 5 on the specific relative viscosity of DNA. Since the relative specific viscosity (η/η0) of DNA gives a measure on the increase in contour length associated with the separation of DNA base pairs caused by intercalation, a classical DNA intercalator like EB shows a significant increase in the viscosity of the DNA solutions (η and η0 are the specific viscosities of DNA in the presence and absence of the complexes, respectively).
In contrast, a partial and/or nonintercalation of the complex could result in a less pronounced effect on the viscosity.24 The effects of 1, 3, 4, 5 and EB on the viscosity of rod-like DNA, are shown in Fig. 9. Complexes 1 and 4 are bound by electrostatic interactions only, because they exerted essentially no such effect. After increasing the amounts of 3 and 5 the relative viscosity of DNA increased steadily, similar to EB. The increase in relative viscosity is expected to correlate with the compound's DNA-intercalating potential, and followed the order EB > 3 > 5 > 1 > 4. These results suggest that complexes 3 and 5 can bind to DNA through intercalation, due to the presence of the phenanthroline ring system in both compartments of the ligand.20b
 |
| | Fig. 9 Effect of increasing amounts of EB and dinickel(II) complexes 1, 3, 4 and 5 on the relative viscosity of calf thymus DNA at 25 (±0.1) °C. The total concentration of DNA is 0.5 mM. | |
DNA cleavage activity
The cleavage of supercoiled pBR322 DNA by dinickel(II) complexes 1–6 (30 μM) was studied in a medium of 50 mM Tris-HCl/NaCl buffer (pH = 7.2) at 25 ± 0.2 °C. All the NiII complexes showed remarkable DNA cleavage. The results of the gel electrophoresis separations of plasmid pBR322 DNA treated with complexes 1–6 are depicted in Fig. 10. Control experiments suggest that untreated DNA and DNA incubated with Ni(ClO4)2·6H2O did not show any significant DNA cleavage (lanes 1and 2). At the concentration of 30 μM, complexes 1–6 almost promote the complete conversion of DNA from Form I (supercoiled, SC) to Form II (nicked circular, NC) (lanes 3–8).
 |
| | Fig. 10 Cleavage of SC pBR322 DNA (0.2 μg, 33 μM) by NiII complexes 1–6 (30 μM) in 50 mM Tris–HCl/NaCl buffer (pH 7.2). Lane 1, DNA control; Lane 2, DNA + Ni(ClO4)2·6H2O (30 μM) ;Lanes 3–8, DNA + 1–6 (30 μM), respectively. | |
The cleavage mechanism of pBR322 DNA induced by complexes 3 and 5 was investigated (Fig. 11) and clarified in the presence of singlet oxygen quencher L-histidine25 (lanes 2 and 3), superoxide dismutase SOD (4 units) (lanes 4 and 5), hydroxyl radical scavenger DMSO (0.4 M) (lanes 6 and 7) and EDTA as a chelating agent (lanes 8 and 9) under aerobic conditions.26 As shown in the Fig. 11, the DNA cleavage mechanism by complexes 3 and 5 are as follows: L-histidine, SOD and DMSO (lanes 2–9) do not alter DNA cleavage activity, this rules out the possibility of cleavage by singlet oxygen, superoxide and hydroxyl radical, respectively. EDTA efficiently inhibited the DNA cleavage activity of the NiII complexes in a similar way to that of nuclease.27 Under anaerobic conditions, the complexes display remarkable cleavage (lanes 10 and 11). This fact implies that the DNA cleavage reaction by the dinickel(II) system might be due to a hydrolytic mechanism. To ascertain the hydrolytic nature of the cleavage reaction, the NC form obtained from the cleavage of SC DNA was reacted with a T4 ligase enzyme. We have observed complete conversion of the NC DNA to its original SC form (lanes 12 and 13). Phenanthroline based dinuclear NiII complexes 3 and 6 at lower concentration (30 μM) showed better chemical nuclease activity (>85%) than the bipyridine or heterocyclic base free dinuclear NiII complexes 1, 2, 4 and 5 (<80%). This indicates that the structure of the ligand plays an important role in DNA cleavage.28 The rigid structure of the ligand shows better DNA cleavage ability after combining to the metal ions, because it may involve a synergistic mechanism with the rigid complex.
 |
| | Fig. 11 Lane 1, DNA control; lanes 2–3, DNA + L–histidine (0.21 mM) + 3 and 5 (30 μM) respectively; lanes 4–5, DNA + SOD (4 units) + 3 and 5 respectively; lanes 6–7, DNA + DMSO (0.1 mM) + 3 and 5, respectively; lanes 8–9, DNA + EDTA (50 mM) + 3 and 5 respectively; lanes 10–11, DNA + 3 and 5 (under argon) respectively; lane 12, conversion of NC (obtained from DNA + 3, 30 μM) to SC form on treatment with 4 units of T4 DNA ligase; lane 13, NC form (from DNA + 3) as control without addition of T4 DNA ligase. | |
The kinetic aspect of the hydrolytic DNA cleavage (Fig. 12) by complex 3 is found to vary exponentially with incubation time and it followed pseudo-first order kinetics. Kinetic plots showing the formation of NC DNA and the degradation of SC DNA vs. time followed pseudo-first order kinetics and they fit well to a single exponential curve. Under true Michaelis–Menten conditions in which the complex concentration is kept constant at 30 μM and the DNA concentration is varied from 30–183 μM, we are able to obtain the rate constant of 5.44 ± 0.2 h −1 using 150 μM SC pBR322 DNA. The hydrolytic rate constant was determined from the linear plot of log (% SC DNA) vs. time (Fig. 13). The pseudo Michaelis–Menten kinetic parameters kcat = 5.44 × 10−2 h−1 and KM = 6.23 × 10−3 M for complex 3 was calculated. It is noted that the complex 3 showed higher rate of DNA cleavage than the reported heterocyclic bases free dicopper(II) analogues.10 This rate enhancement in the cleavage process is due to the presence of phenanthroline ring in both the compartments of complex 3. This is well reflected in the cleavage data which showed more than 85% of cleavage with a very low amount of complex concentration (30 μM). The complex 3 also shows an enormous enhancement of the cleavage rate of 1.5 × 106 in comparison to the uncatalysed hydrolysis rate (k = 3.6 × 10−8 h−1) of ds-DNA.29 The rate of DNA hydrolysis enhanced by 3 is higher than those reported for mononuclear nickel(II) complexes30 but less when compared to Mg–EcoRV or Mn–EcoRV (∼1.3 × 109).31
![Cleavage activity of dinickel(ii) complex 3 monitored by 0.8% agarose gel electrophoresis, where [DNA] 0.2 μg, 33 μM, (complex 3) 30 μM. Time course measured in 10 mM Tris buffer, pH 7.4, 37 °C, showing the disappearance of SC DNA at (1) 0 min, (2) 5 min, (3) 10 min, (4) 15 min, (5) 20 min, (6) 25 min, and (7) 30 min, respectively. (Gel image showing SC (Form I) and NC (Form II) DNA).](/image/article/2012/RA/c2ra20228j/c2ra20228j-f12.gif) |
| | Fig. 12 Cleavage activity of dinickel(II) complex 3 monitored by 0.8% agarose gel electrophoresis, where [DNA] 0.2 μg, 33 μM, (complex 3) 30 μM. Time course measured in 10 mM Tris buffer, pH 7.4, 37 °C, showing the disappearance of SC DNA at (1) 0 min, (2) 5 min, (3) 10 min, (4) 15 min, (5) 20 min, (6) 25 min, and (7) 30 min, respectively. (Gel image showing SC (Form I) and NC (Form II) DNA). | |
 |
| | Fig. 13 (A) Saturation kinetics of the cleavage of pBR322 DNA using 30 μM complex 3 with different concentrations of SC DNA (30–183 μM) at 37 °C in 50 mM Tris-HCl/NaCl buffer (pH 7.2). (B) Plot of log (% SC DNA) vs. time for a complex concentration of 30 μM. | |
Conclusions
Based on the electrochemical studies, the heterocyclic bases coordinated Ni(II) ions reduce at less negative potentials than the heterocyclic base free complexes containing Ni(II) ions. Catalytic phosphate hydrolysis studies shows that the N-methyl group containing macrobicyclic (L2) dinickel(II) complexes display better hydrolysis activities than those of the N-methyl group free macrobicyclic (L1) dinickel(II) analogs. This is due to the presence of the bulky N-methyl group of L2, which makes the coordination geometry around the Ni(II) ions more distorted than with L1. The DNA binding experiments results suggests that the interaction of the complexes with DNA is an intercalative mode. The DNA cleavage studies of the dinickel(II) complexes displayed a hydrolytic (O2-independent pathway) mechanism, because the singlet oxygen quencher (L-histidine), hydroxyl radical scavenger (DMSO) and superoxide quencher (SOD) were completely ineffective on the cleavage activity. In conclusion, we have performed a comparative study on the influence of aliphatic and aromatic moieties in macrobicyclic ligands on DNA binding and cleavage activities. The phenanthroline coordinated macrobicyclic dinickel(II) complexes 3 and 6, display better DNA interactions and significant DNA hydrolysis activities than the bipyridine based dinickel(II) analogs.
Experimental
Materials and measurements
Polyaza macrobicyclic binucleating ligands (L1and L2) are synthesized as described in our earlier report.10 Tetra(n-butyl)ammonium perchlorate (TBAP) was purchased from Fluka and recrystallized from hot methanol. (Caution! TBAP is potentially explosive; hence, care should be taken when handling the compound.) Sodium salt of 4-nitrophenyl phosphate (4-NPP) was purchased from Aldrich. CT DNA and pBR322DNA were purchased from Bangalore Genie (India). All other chemicals and solvents were of analytical grade and used as received, without any further purification. FT-IR spectra were obtained on a Perkin Elmer FTIR spectrometer with samples prepared as KBr pellets. UV-visible spectra were recorded using a Perkin Elmer Lambda 35 spectrophotometer operating in the range of 200–1100 nm with quartz cells and ε are given in M−1cm−1. Cyclic voltammetric measurements were made at 25 °C on a CH11008 Electrochemical analyzer using a three-electrode setup comprised of glassy carbon working, platinum wire auxiliary and Ag/Ag+ reference electrodes under oxygen free conditions. The concentration of the complexes was 10−3 M. TBAP (10−1 M) was used as the supporting electrolyte. Electrospray ionization mass spectral measurements were done using a Thermo Finnigan LCQ-6000 Advantage Max-ESI mass spectrometer. Solutions of DNA in the buffer 5 mM Tris-HCl/50 mM NaCl (pH = 7.1) in water gave the ratio of UV absorbance at 260 and 280 nm, A260/A280, of 1.9, indicating that the DNA was sufficiently free of protein.32 Concentrated stock solutions of DNA (10.5 mM) were prepared in a buffer and sonicated for 25 cycles, where each cycle consisted of 30 s with 1 min intervals. The concentration of DNA in nucleotide phosphate (NP) was determined by UV absorbance at 260 nm after 1:100 dilutions. The extinction coefficient, ε260, was taken as 6600 M−1cm−1. Stock solutions were stored at 4 °C and used after no more than 4 d. SC plasmid pBR322 DNA was stored at: −20 °C and the concentration of DNA in base pairs was determined by UV absorbance at 260 nm after appropriate dilutions taking ε260 as 13100 M−1cm−1. Concentrated stock solutions of metal complexes were prepared by dissolving calculated amounts of nickel(II) complexes in respective amounts of solvent and diluted suitably with the corresponding buffer to required concentrations for all experiments.
Synthesis of dinuclear nickel(II) complex [Ni2L1(ClO4)2](ClO4)2 (1)
The dinuclear NiII complex 1 was synthesized by refluxing a methanolic solution of the ligand L1 (1 g, 19 mmol) and a methanolic solution of Ni(ClO4)2·6H2O (1.05 g, 38 mmol). A blue colored solid was separated upon evaporation of the solution at room temperature and the resulting compound was washed with cold methanol and then dried. Yield: 1.62 g (81%). ESI-MS in CH3CN: m/z 411.72 [M − 2ClO4]2+. FT-IR, cm−1(KBr disc): 3231 [w, ν(N–H)], 2879 [s, ν(C–H)], 1086 s, 1102 vs, 623 [s, ν(ClO4−)], 478 [s, ν(Ni–N)] (br, broad; s, strong; m, medium; w, weak). λmax, nm (ε, M−1cm−1) in CH3CN: 563 (380), 268 (84000).
Synthesis of dinuclear nickel(II) complex [Ni2L1(bpy)2(H2O)4] (ClO4)4 (2)
Ethanolic solution of dinuclear Ni(II) complex 1 (0.51 g, 0.5 mmol) was added dropwise to the 2,2′-bipyridine (0.15 g 1 mmol) in ethanol with constant stirring. The reaction mixture was refluxed for 6 h and then the solution concentrated to about 4 mL by evaporation of the solvent under reduced pressure. Upon slow cooling of the resulting solution to room temperature, a pale green solid separated out which was filtered and recrystallized from acetonitrile. Yield: 0.52 g (80%). ESI-MS in CH3CN: m/z 603.24 [M − 2ClO4]2+. FT-IR, cm−1 (KBr disc): 3225 [w, ν(N–H)], 2925 [s, ν(C–H)], 1219 [s, ν(C–O)], 1095 vs, 625 [s, ν(ClO4−)], 481[s, ν(Ni–N)]. λmax, nm (ε, M−1cm−1) in CH3CN: 920 (45), 715 (195), 565 (2400), 272 (89000), 225 (129000).
Synthesis of dinuclear nickel(II) complex [Ni2L1(phen)2(H2O)4](ClO4)4 (3)
The dinuclear complex 3 have been synthesized by following the above procedure using 1,10-phenanthroline (0.18 g, 1 mmol) instead of using 2,2′-bipyridine. Dark green colored solid. Yield: 0.48 g (69%). ESI- MS in CH3CN: m/z 629.21 [M − 2ClO4]2+. FT-IR, cm−1 (KBr disc): 3232 [w, ν(N–H)], 2928 [s, ν(C–H)], 1222 [s, ν(C–O)], 1098 vs,624 [s, ν(ClO4−)], 482 [s, ν(Ni–N)]. λmax, nm (ε, M−1cm−1) in CH3CN : 925 (42), 725 (192), 570 (2300), 272 (84000), 223 (132000).
Synthesis of dinuclear nickel(II) complex [Ni2L2(ClO4)2](ClO4)2 (4)
The dinuclear complex 4 have been synthesized by following the above procedure using ligand L2 (1 g, 1.7 mmol)29 instead of using ligand L1. Blue colored solid. Yield: 1.55 g (81%). ESI-MS in CH3CN: m/z 453.45 [M − 2ClO4]2+. FT-IR, cm−1 (KBr disc): 3221 [w, ν(N–H)], 2928 [s, ν(C–H)], 1224 [s, ν(C–O)], 1085 s, 1105 vs, 624 [s, ν(ClO4−)],482 [s, ν(Ni–N)]. λmax, nm (ε, M−1cm−1) in CH3CN: 585 (380), 258 (89500).
Synthesis of dinuclear nickel(II) complex [Ni2L2(bpy)2(H2O)4] (ClO4)4 (5)
The dinuclear complex 5 have been synthesized by following the procedure of synthesis of complex 2, using complex 4 (0.56 g, 0.5 mmol) and 2,2′-bipyridine (0.16 g 1 mmol) instead of using complex 1. Pale green colored solid. Yield: 0.49 g (68%). ESI-MS in CH3CN: m/z 647.40 [M − 2ClO4]2+. FT-IR, cm−1 (KBr disc): 3228 [w, ν(N–H)], 2927 [s, ν(C–H)], 1222 [s ν(C–O)], 1100 vs, 625 [s, ν(ClO4−)] 484 [s, ν(Ni–N)]. λmax, nm (ε, M−1cm−1) in CH3CN: 935 (39), 745 (180), 558 (2600), 265 (84000), 224 (182000).
Synthesis of dinuclear nickel(II) complex [Ni2L2(phen)2(H2O)4] (ClO4)4 (6)
The dinuclear complex 6 have been synthesized by following the above procedure using 1,10-phenanthroline (0.18 g, 1 mmol) instead of using 2,2′-bipyridine. Dark green colored solid. Yield: 0.50 g (75%). ESI–MS in CH3CN: m/z 669.83 [M − 2ClO4]2+. FT-IR, cm−1(KBr disc): 3225 [w, ν(N–H)], 2925 [s, ν(C–H)], 1224 [s, ν(C–O)], 1096 vs, 625 [s, ν(ClO4−)], 485 [s, ν(Ni–N)]. λmax, nm (ε, M−1 cm−1) in CH3CN: 945 (35), 745, (175), 562 (2650), 268 (88000), 225 (189000).
Acknowledgements
S.A. and S.S are grateful to CSIR (SRF), New Delhi, Government of India, for a fellowship. We thank the Department of Science and Technology (DST-FIST), New Delhi, Government of India, for financial support.
References
-
(a) C. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 2004, 248, 147 CrossRef CAS;
(b) L. K. J. Boerner and J. M. Zaleski, Curr. Opin. Chem. Biol., 2005, 9, 135 CrossRef CAS;
(c) L. Otero, P. Smircich, M. Vieites, M. Ciganda, P. C. Severino, H. Terenzi, H. Cerecetto, D. Gambino and B. Garat, J. Inorg. Biochem., 2007, 101, 74 CrossRef CAS;
(d) E. J. Gao, K. H. Wang and X. F. Gu, J. Inorg. Biochem., 2007, 101, 1404 CrossRef CAS;
(e) H. M. Torshizi, M. Saeidifar, G. R. R. Behbehani, A. Divsalarc and A. A. Saboury, J. Chin. Chem. Soc., 2010, 57, 1299 Search PubMed.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188 CrossRef CAS.
- L. S. Lerman, J. Mol. Biol., 1961, 3, 18 CrossRef CAS.
-
(a) S. Routier, V. Joanny, A. Zaparucha, H. Vezin, J.-P. Catteau, J.-L. Bernier and C. Bailly, J. Chem. Soc., Perkin Trans., 1998, 2, 863 Search PubMed;
(b) N. Megger, L. Welte, F. Zamora and J. Muller, Dalton Trans., 2011, 40, 1802 RSC;
(c) P. K. Bhattacharya, H. J. Lawson and J. K. Barton, Inorg. Chem., 2003, 42, 8811 CrossRef CAS.
-
(a) F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Chem. Commun., 2005, 2540 RSC;
(b) K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777 CrossRef CAS;
(c) W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089 CrossRef CAS;
(d) D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201 CrossRef.
- S. Dhar, A. N. R. Pattubala and A. R. Chakravarty, Dalton Trans., 2004, 697 RSC.
-
(a) S. Karabocek, I. Degirmencioglu and N. Karabocek, Transition Met. Chem., 2003, 28, 529 CrossRef;
(b) M. Frezza, S. S. Hindo, D. Tomco, M. M. Allard, Q.C. Cui, M. J. Heeg, D. Chen, Q. P. Dou and C. N. Verani, Inorg. Chem., 2009, 48, 5928 CrossRef CAS;
(c) S. Arounaguiri, D. Easwaramoorthy, A. Ashokkumar, A. Dattagupta and B. G. Maiyaa, Proc.–Indian Acad. Sci., Chem. Sci., 2000, 112, 1 CrossRef CAS;
(d) N. Tang, J. G. Muller, C. J. Burrows and S. E. Rokita, Biochemistry, 1999, 38, 16648 CrossRef CAS.
-
(a) Z.-Q. Pan, K. Ding, H. Zhou, Q.-R. Cheng, Y.-F. Chen and Q.-M. Huang, Polyhedron, 2011, 30, 2268 CrossRef CAS;
(b) N. Saglam, A. Colak, K. Serbest, S. Karabocek and S. Guner, Monatsh. Chem., 2004, 35, 1023 Search PubMed;
(c) N. Sung and T.-Y. Kim, Agric. Chem. Biotechnol., 2006, 49, 86 CAS;
(d) C. Vichard and T. A. Kaden, Inorg. Chim. Acta, 2002, 337, 173 CrossRef CAS;
(e) Z. Lihong, W. Na and Y. Xiaoqi, Prog. Chem., 2007, 19, 1909 Search PubMed.
- S. Anbu, M. Kandaswamy and B. Varghese, Dalton Trans., 2010, 39, 3823 RSC.
- S. Anbu and M. Kandaswamy, Polyhedron, 2012, 33, 1 CrossRef CAS.
- S. Anbu, M. Kandaswamy, P. Sathya Moorthy, M. Balasubramanian and M. N. Ponnuswamy, Polyhedron, 2009, 28, 49 CrossRef CAS.
- M. Thirumavalavan, P. Akilan and M. Kandaswamy, Supramol. Chem., 2004, 16, 495 CrossRef CAS.
- P. R. Reddy and K. S. Rao, Chem. Biodiversity, 2006, 3, 231 CAS.
-
M. A. Thompson, ArgusLab 4.0; Planaria Software LLC: Seattle, WA; http://www.arguslab.com. Search PubMed.
- A. Malinauskas, M. Bron and R. Holze, Synth. Met., 1998, 92, 127 CrossRef CAS.
-
(a)
J. Zubieta, J. C. Hayes and K. D. Karlin, Nickel coordination chemistry, biochemical and inorganic perspective, Adenine Press, New York, 1983, p. 97 Search PubMed;
(b) H. B. Gray and B. G. Malstrom, Inorg. Chem., 1983, 5, 203 Search PubMed.
- N. A. Rey, A. Neves, A. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS.
- M. Thirumavalavan, P. Akilan and M. Kandaswamy, Polyhedron, 2005, 24, 1781 CrossRef CAS.
-
(a) N. Sengottuvelan, D. Saravanakumar and M. Kandaswamy, Polyhedron, 2007, 26, 3825 CrossRef CAS;
(b) C. T. Yang, M. Vetrichelvan, X. Yang, B. Moubaraki, K. S. Murray and J. J. Vittal, Dalton Trans., 2004, 113 RSC;
(c) K. Yamaguchi, F. Akagi, S. Fujinami, M. Suzuki, M. Shionoya and S. Suzuki, Chem. Commun., 2001, 375 RSC.
- B. Peng, H. Chao, B. Sun, H. Li, F. Gao and L.-N. Ji, J. Inorg. Biochem., 2006, 100, 1487 CrossRef.
- P. K. Sasmal, A. K. Patra, M. Nethaji and A. R. Chakravarty, Inorg. Chem., 2007, 46, 11112 CrossRef CAS.
- V. I. Ivanov, L. E. A. Minchenkova, K. Schyolkina and A. I. Poletayer, Biopolymers, 1973, 12, 89 CrossRef CAS.
- J. Tan, B. Wang and L. Zhu, Bioorg. Med. Chem. Lett., 2007, 17, 1197 CrossRef CAS.
- Q. L. Zhang, J. G. Liu, H. Chao, G. Q. Xue and L. N. Ji, J. Inorg. Biochem., 2001, 83, 49 CrossRef CAS.
- H. Kishikawa, Y. P. Jiang, J. Goodisman and J. C. Dabrowiak, J. Am. Chem. Soc., 1991, 113, 5434 CrossRef CAS.
- J. A. Cowan, Curr. Opin. Chem. Biol., 2001, 5, 634 CrossRef CAS.
- J.-Z. Wu, L. Yuan and J.-F. Wu, J. Inorg. Biochem., 2005, 99, 2211 CrossRef CAS.
- Q. X. Xiang, J. Zhang, P. Y. Liu, C. Q. Xia, Z. Y. Zhou, R. G. Xie and X. Q. Yu, J. Inorg. Biochem., 2005, 99, 1661 CrossRef CAS.
- X. Sheng, X. Guo, X. M. Lu, G. Y. Lu, Y. Shao, F. Liu and Q. Xu, Bioconjugate Chem., 2008, 19, 490 CrossRef CAS.
-
(a) H.-Y. Wang, W.-B. Yuan, Q. Zhang, S.-W. Chen and S.-S. Wu, Transition Met. Chem., 2008, 33, 593 CrossRef CAS;
(b) K. G. Ragunathan and H.-J. Schneider, Angew. Chem., Int. Ed. Engl., 1996, 35, 1219 CrossRef CAS;
(c) M. C. B. Oliveira, M. S. R. Couto, P. C. Severino, T. Foppa, G. T. S. Martins, B. Szpoganicz, R. A. Peralta, A. Neves and H. Terenzi, Polyhedron, 2005, 24, 495 CrossRef CAS.
-
(a) A. Sreedhara, J. D. Freed and J. A. Cowan, J. Am. Chem. Soc., 2000, 122, 8814 CrossRef CAS;
(b) D. J. Wright, W. E. Jack and P. Modrich, J. Biol. Chem., 1999, 274, 31896 CrossRef CAS;
(c) N. P. Stanford, S. E. Halford and G. S. Baldwin, J. Mol. Biol., 1999, 288, 105 CrossRef CAS.
- C. Merill, D. Goldman, S. A. Sedman and M. H. Ebert, Science, 1981, 211, 1437 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.