Sustainable formation of fatty acid alkyl esters by transesterification of triglycerides with chlorotrimethylsilane
Antonella
Salvini
,
Donatella
Giomi
,
Giacomo
Cipriani
,
Giovanni
Bartolozzi
,
Renzo
Alfini
and
Alberto
Brandi
*
Department of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 13, 50019 Sesto Fiorentino (FI), Italy. E-mail: alberto.brandi@unifi.it; Fax: +39 055 457 3572; Tel: +39 055 457 3485
First published on 29th March 2012
Abstract
A new efficient and sustainable method for the transesterification of triglycerides and production of fatty acid alkyl esters in acidic conditions is disclosed. The transesterification process can be extended, with the same efficiency, to methyl, ethyl and n-butyl esters. The method utilizes chlorotrimethylsilane as the mediator for the transesterification process, with a slight excess of the alcohol reagent and a low temperature (60 °C), without the use of water in any step of the process. The reaction provides a perfect separation of the glyceric phase and the fatty acid alkyl esters, which can be recovered, starting from pure vegetable oil, with high conversion and high purity. The method was positively extended to animal fats, exhausted vegetable oils and highly acidic oils without any pretreatment.
1. Introduction
The production of BioDiesel (BD) from vegetable oils is a well known process utilized in many production sites of different sizes. The interest in BD has grown due to the possibility of utilizing the product in already available combustion plants without any particular variation in the combustion system. Nevertheless, processes employed up to date reveal several limitations and bothersome problems. The production of fuel from vegetable oils is interesting because it is an alternative to the production of fuel from mineral oils. On the other hand, its intensive industrial development is criticized nowadays, particularly in underdeveloped countries and in international organizations (Food and Agriculture Organization, FAO), because it utilizes feed stock materials that belong to the food supply. Of particular interest, therefore, is the production of BD from exhausted vegetable oils, which constitute an environmental problem. These oils, in fact, deriving from a diffuse family production, are generally inappropriately disposed of. In this sense, their use as an energy supply represents an additional double benefit.BD is mainly produced by transesterification reactions of lipids,1–5 particularly triglycerides, vegetable oils and/or animal fats, with low molecular weight alcohols, like methanol, ethanol, or n-butanol. The most common process employs methanol to synthesize methyl esters (fatty acid methyl esters, FAMEs). As a byproduct of the transesterification process glycerol is obtained. The transesterification reaction requires a catalyst to occur, and an alkaline catalyst, like sodium or potassium methoxide or hydroxide, has been predominantly used. Notwithstanding the large industrial use of base catalysts, the catalyst system brings about several problems. In fact, the total free fatty acid content in vegetable oils must be below 0.5% by weight to avoid the formation of soaps (Na or K salts of fatty acids) and their emulsions with water, which make the separation of pure BD difficult, and increase the waste water disposal issue. Also the byproduct glycerol requires further purification and separation from water before further transformation or utilization. The base catalyzed BD production process suffers from higher costs due to the issue of purification and the need for refined vegetable oils as starting materials.
Acid catalysts (sulfuric acid, organic sulfonic acids, hydrochloric acid, phosphoric acid and boron trifluoride) have been largely studied6–10 as alternatives to the basic ones, but their industrial promotion has been limited, because the conversion rate of the oil is lower than in the alkaline process. However, they have the clear advantage that they allow the direct use of starting materials rich in free fatty acids. Homogeneous acid catalysts studied up-to-date bring about separation and purification problems that can lead to an increase in production costs. Heterogeneous acid catalysts have been developed to avoid the separation of the catalyst, to be used in continuous flow reactors. However, their efficiency is lower than the homogeneous ones requiring higher reaction temperatures (200–250 °C) and pressures (50 atm).11–14
We now report a new sustainable process15 for the transesterification of triglycerides 1 which is able to convert triglycerides in fatty acid alkyl esters by employing an efficient reagent that can operate in the homogeneous phase, but at the same time can be easily separated from the products. As a consequence the process is efficient also with raw starting materials, like exhausted vegetable oils, strongly acidic vegetable oils and animal fats. At the same time it is operationally simple and allows an easy separation of the fatty acid alkyl esters from other byproducts of the transesterification process.
2. Experimental
2.1 Materials
The oil employed in this work was ‘high-oleic sunflower oil’ of Type c2, as reported in the study by Guillén and Ruiz16 The proportions of the different acyl groups in the starting material, as well as in the FAMEs, were determined via1H NMR analysis through application of the equations reported by the same authors. Methanol (VWR, 99.8%), chlorotrimethylsilane (Aldrich, 98%), chlorotriethylsilane (Aldrich, 99%), calcium carbonate (Carlo Erba, 99.5%), oleic acid (Aldrich, 65–88%), deuterated chloroform (Aldrich, 99.8%), deuterated methanol-d4 (Merck, 99.8%), were utilized as obtained from the producer without further purification.2.2 Analytical methods
1H NMR and 13C NMR spectra were recorded with Varian VXR 200 and Varian Mercuryplus 400 instruments, operating at 200 and 50 MHz and 400 and 100 MHz, respectively. The chemical shifts are reported in ppm and referenced to tetramethylsilane (TMS) as the internal standard. Spectra elaboration was carried out by means of iNMR 3.0.1 software. The determination of the fatty acid methyl ester (FAME) content (%) by 1H NMR was carried out by the comparison of the integral of the methylene hydrogens –CH2–COO– signal with that of the CH3O– signal of the methyl esters produced in the transesterification process.17The determination of the fatty acid methyl ester (FAME) content (%) by gas chromatography (GC) was carried out using methyl heptadecanoate as the internal standard, according to DIN EN 14103. The determination of polymeric materials in the exhausted oil was carried out by comparing the FAME content (%) of two different samples obtained by the transesterification of raw sunflower oil and exhausted sunflower oil, following a literature procedure.18 GC analysis was performed using a Shimadzu GC-2010 gas chromatograph equipped with a programmable temperature vaporizing (PTV) injector, a flame ionization detector (FID) and a Zebron ZB-5 HT INFERNO capillary column (5% phenyl–95% polydimethylsiloxane, 15 m × 0.32 mm ID × 0.1 μm df). GC conditions: isotherm at 50 °C (2 min) ramp at 15 °C min−1 to 180 °C, isotherm at 180 °C (10 min), ramp at 25 °C min−1 to 380 °C, isotherm at 380 °C (1 min). The carrier gas was helium with a flow rate of 1.83 mL min−1. PTV injector conditions: ramp at 200 °C min−1 from 50 °C to 300 °C, isotherm at 300 °C (10 min). The temperature of the detector was set at 385 °C. A sample volume of 1 μL was injected using a split mode, with a split ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.
:10.
2.3 Synthesis of esters of fatty acids from natural refined sunflower oil
1H NMR (CDCl3, 400 MHz) δ (ppm): 0.86 (m, CH3), 1.23–1.32 (m, CH2), 1.60 (m, CH2CH2CO2CH3), 1.98–2.04 (m, CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 2.28 (t, J = 7.5 Hz, CH2CO2CH3), 2.75 (t, J = 6.5 Hz, CHCHCH2CHCH), 3.64 (s, OCH3), 5.30–5.36 (m, CHCH); 13C NMR (CDCl3, 50 MHz) δ (ppm): 14.3 (q), 14.4 (q), 22.8 (t), 22.9 (t), 25.2 (t), 25.9 (t), 27.4 (t), 29.1 (t), 29.3 (t), 29.4 (t), 29.5 (t), 29.6 (t), 29.7 (t), 29.8 (t, 2C), 29.9 (t), 30.0 (t), 31.8 (t), 32.2 (t), 34.3 (t), 51.6 (q), 127.9 (d), 128.0 (d), 128.1 (d), 129.7 (d), 130.0 (d), 130.1 (d), 130.2 (d), 174.2 (s), 174.3 (s).
CH), 2.28 (t, J = 7.5 Hz, CH2CO2CH3), 2.75 (t, J = 6.5 Hz, CHCHCH2CHCH), 3.64 (s, OCH3), 5.30–5.36 (m, CHCH); 13C NMR (CDCl3, 50 MHz) δ (ppm): 14.3 (q), 14.4 (q), 22.8 (t), 22.9 (t), 25.2 (t), 25.9 (t), 27.4 (t), 29.1 (t), 29.3 (t), 29.4 (t), 29.5 (t), 29.6 (t), 29.7 (t), 29.8 (t, 2C), 29.9 (t), 30.0 (t), 31.8 (t), 32.2 (t), 34.3 (t), 51.6 (q), 127.9 (d), 128.0 (d), 128.1 (d), 129.7 (d), 130.0 (d), 130.1 (d), 130.2 (d), 174.2 (s), 174.3 (s).
The volatile byproducts collected in the cool trap consisted of a mixture of hexamethyldisiloxane (TMS2O), MeOH, and traces of methyltrimethylsilylether (TMSOMe) and dimethylether. The mixture was distilled at 95 °C to obtain pure TMS2O (1.6 g, 99% by GC analysis).
The lower phase was dissolved in methanol (5 mL) and neutralized with calcium carbonate (1.0 g). After filtration, the solution was concentrated to dryness under reduced pressure to obtain a yellow oil (1.1 g) containing glycerol and α-monochlorohydrin in 52:48 ratio (evaluated by 13C NMR).
2.4 Synthesis of FAMEs from a 50% mixture of animal fat and sunflower oil
The animal fat was dissolved in the vegetable oil (50% of fat and 50% of oil by weight) by heating under stirring at 120 °C for 2 h in an open vessel. After filtration, a mass reduction of about 10% was observed. The oily mixture (10 g), placed in a 25 mL screw-cap Sovirel® tube, was added to methanol (1.64 g, 2.08 mL, 51.3 mmol) and TMSCl (2.78 g, 25.6 mmol) and heated under continuous stirring at 60 °C for 8 h. After cooling to room temperature, the mixture separated into two phases. The upper phase was heated under reduced pressure (2 mmHg) at 65 °C to distill off volatiles and a pale yellow oil containing the FAMEs (10 g) was obtained. 1H NMR and GC analyses showed, respectively, 94% and 96.1% conversions of the oil to FAMEs.2.5 Synthesis of FAMEs from exhausted oil
Exhausted oil was obtained by heating the same previously used sunflower oil at 120 °C for 72 h under stirring in air. GC analysis (see Section 2.2) showed the presence of roughly 12% of polymeric material.In a 25 mL screw-cap Sovirel® tube, methanol (1.64 g, 2.08 mL, 51.3 mmol) and TMSCl (2.78 g, 25.6 mmol) were added to the exhausted oil (10 g) under a nitrogen atmosphere and the mixture was allowed to react under stirring at 60 °C for 8 h. The oil, by addition of reagents, became light brown, and the color darkened during heating. After cooling to room temperature, the mixture separated into two phases. The upper phase was heated under reduced pressure (2 mmHg) at 65 °C for 3 h to obtain a dark brown liquid containing the FAMEs (10.19 g). 1H NMR analysis gave a 94% conversion of the oil to FAMEs, whereas a 79.5% conversion was obtained by GC analysis, because of the presence of polymeric materials. The upper phase was distilled at 110–125 °C under reduced pressure (10−2 mmHg) to obtain pure FAMEs (6.28 g, 96.1% by GC).
The lower phase was dissolved in methanol (5 mL) and pH-neutralized with calcium carbonate (1.0 g). After filtration, the solution was concentrated under reduced pressure to obtain a pink oil (0.69 g) containing glycerol (58.5%) and α-monochlorohydrin (41.5%) (evaluated by 13C NMR).
2.6 Synthesis of esters of fatty acids from a 50% mixture of sunflower oil and oleic acid
In a 25 mL screw-cap Sovirel® tube, sunflower oil [5 g, 5.7 mmol (MWcalc = 878 g mol−1)] and oleic acid (5 g, 17.7 mmol) were added under nitrogen to methanol (1.64 g, 2.08 mL, 1.5 equiv., 51.3 mmol) and TMSCl (2.78 g, 25.6 mmol). The resulting emulsion was allowed to react under continuous stirring at 60 °C for 8 h and then cooled to room temperature. Two phases separated immediately after ceasing the stirring. The upper phase, containing the methyl esters, was separated and heated under reduced pressure (2 mmHg) at 65 °C to distill off volatiles and to obtain a pale yellow liquid (10.2 g). 1H NMR and GC analyses showed, respectively, 96% and 89% conversions of the starting material to FAMEs. The upper phase was distilled at 110–130 °C under reduced pressure (10−2 mmHg) to obtain pure FAMEs (7.7 g, 99% by GC).2.7 Synthesis of esters of fatty acids from natural refined sunflower oil using triethylchlorosilane
In a 25 mL screw-cap Sovirel® tube, chlorotriethylsilane (TESCl, 3.86 g, 25.6 mmol) was added to methanol (1.64 g, 2.08 mL, 1.5 equiv., 51.3 mmol) and sunflower oil [10 g, 11.4 mmol (MWcalc = 878 g mol−1)] under a nitrogen atmosphere. The resulting emulsion was allowed to react under continuous stirring at 60 °C for 8 h, then the two phases were readily separated and the upper phase was heated under reduced pressure (2 mmHg) at 65 °C to distill off volatiles and to obtain a pale yellow liquid (11.6 g). 1H NMR analysis gave a 83% conversion of the oil to FAMEs. The spectrum also showed the presence of hexaethyldisiloxane (TES2O) in the mixture, not removed by distillation because of its low volatility. Volatile byproducts collected in the cool trap consisted of a mixture of triethylsilanol (TESOH), MeOH, and traces of methyltriethylsilylether (TESOMe) and dimethylether. The lower phase was dissolved in methanol (5 mL) and neutralized with calcium carbonate (1.0 g). After filtration, the solution was concentrated to dryness under reduced pressure to obtain a yellow oil (0.57 g) containing glycerol and α-monochlorohydrin in 68:32 ratio (evaluated by 13C NMR).
3. Results and discussion
In a standard procedure (see Section 2.3.1), the transesterification process was carried out by mixing the oil 1 (commercial sunflower oil used invariantly in the study), an alcohol, and chlorotrimethylsilane (2) in a Sovirel® vial, and heating the emulsion at 60 °C under stirring for 8 h. The alcohol was used with a slight excess (1.5 mol equiv.) with respect to the triglycerides. Chlorotrimethylsilane (2) was used in 50 mol% with respect to alcohol to reach a full conversion of the oil to fatty acid alkyl esters 5 (Scheme 1). By stopping the stirring two phases immediately formed (Fig. 1). | ||
| Fig. 1 Photos of reaction vessels at the beginning and at the end of the reaction, showing the perfect separation of the final two phases. | ||
 | ||
| Scheme 1 Transesterification of triglycerides with alcohols catalyzed by chlorotrimethylsilane (TMSCl). | ||
The two phases were perfectly separated by decantation. The pale yellow upper phase contained fatty acid alkyl esters (BD) 5 together with hexamethyldisiloxane, the product of the transformation of TMSCl, and traces of alcohol and trimethylsilylethers of the alcohol used. It is remarkable that the upper phase did not contain any traces of glycerol. A distillation of the volatile products of this phase at reduced pressure by gentle heating (see Section 2.3.1) left a pure mixture of fatty acid alkyl esters in a quantitative yield. The lower viscous glyceric phase that resulted was very acidic. It contained, besides glycerol (3), 3-Cl-1,2-propanediol (α-monochlorohydrin) (4), likely obtained by the chlorination of glycerol with TMSCl (2) and HCl. The different conversions of oil to fatty acid methyl esters (FAMEs) 5 on varying the amount of TMSCl used, and the consequent variation of the composition of the glyceric phase (glycerol:α-monochlorohydrin ratio) are reported in Table 1. The conversions were determined by 1H NMR analysis, by comparison of the integral of the methylene hydrogens –CH2–COO– signal with that of the CH3O– signal of the methyl esters produced in the transesterification process.17 The results were consistent with parallel GC analyses.
| Entry | TMSCla (2) | TMSCl–CH3OH ratio | FAME 5 (%)b | Glycerol–3-Cl-1,2-propandiolc |
|---|---|---|---|---|
| a Amount of TMSCl added to 10 g of pure oil. b Evaluated by 1H-NMR analyses. c Evaluated by 13C-NMR analyses. | ||||
| 1 | 1.11 g, 10.2 mmol | 20:100 |
85 | 77.5:22.5 |
| 2 | 1.67 g, 15.4 mmol | 30:100 |
89 | 64.5:35.5 |
| 3 | 2.23 g, 20.5 mmol | 40:100 |
92 | 58.5:41.5 |
| 4 | 2.78 g, 25.6 mmol | 50:100 |
97 | 52.0:48.0 |
The effect of the temperature was also verified (Table 2). Conversion to FAME 5 under the best conditions (entry 4, Table 1) was optimal only at 60 °C.
| Temperature / °C | FAME 5 Conversion (%) |
|---|---|
| 28–30 | 77 |
| 40 | 90 |
| 60 | 97 |
The lower glyceric phase was diluted with alcohol, neutralized with calcium carbonate, and concentrated to give a pure mixture of α-monochlorohydrin (4) and glycerol (3) which was subjected to fractional distillation.19 Different alcohols, i.e. EtOH or n-BuOH, were used for the transesterification with similar efficiency (97% by 1H NMR for both alcohols).
Animal fat can also be transformed with similar efficiency with a slight modification of the procedure. The animal fat was first dissolved in pure vegetable oil (1:1 w/w) by heating in an open vessel at 120 °C for 2 h. This dissolving pretreatment was also useful to remove the water contained in the fat. The resulting mixture was filtered to remove proteic materials and residues, then cooled and subjected to the standard conditions to give the fatty acid esters with similar efficiency (Fig. 1) (94% by 1H NMR, 96.1% by GC analysis).
Exhausted vegetable oils were also subjected to the same process with similar efficiency (94% by 1H NMR, 79.5% by GC analysis). A reference exhausted vegetable oil was prepared by heating the oil in air at 120 °C for 72 h, leading to a deeper colored and more viscous material. GC control (see Section 2.2) of the exhausted oil showed that roughly 12% polymeric material was produced with the treatment. By submitting this oil to the standard transesterification conditions a very similar result was obtained, the only difference being the darkening of both the phases formed at the end of the reaction and the presence of polymeric materials. After separation of the two phases, pure fatty acid methyl esters (96.1% by GC analysis) were obtained by distillation at 110–125 °C under reduced pressure (10−2 mmHg).
Another common source of raw materials is represented by highly acidic oils obtained from refinery processes. In order to reproduce this material as a model, a mixture of raw sunflower oil and oleic acid (1:1 w/w) was treated in the standard conditions to give a mixture of fatty acid methyl esters (89% by GC analysis). Also in this case, after separation of the two phases, pure fatty acid methyl esters (99% by GC analysis) were obtained by distillation at 110–130 °C under reduced pressure (10−2 mmHg).
Other chlorosilanes, like triethylchlorosilane (see Section 2.7), dimethyldichlorosilane, and tetrachlorosilane were tested in the same process giving comparable results with regards to the transesterification process, but proved less practical in the work up and recovery of the final products because of the low volatility of some of the silane byproducts.
The role of chlorotrimethylsilane in this transesterification process is unequivocal. It is known that TMSCl (2) can be commonly used as a substitute for HCl in esterification processes,20,21 albeit it has been utilized very rarely in transesterification reactions.22 A comparative test was carried out using for the transesterification a methanolic solution of HCl (3N), with the same experimental conditions (60 °C, 8 h). A much lower conversion of the oil (75% by 1H NMR) was obtained, in agreement with literature data.6–10 The role of chlorotrimethylsilane can be well evidenced in Scheme 2.
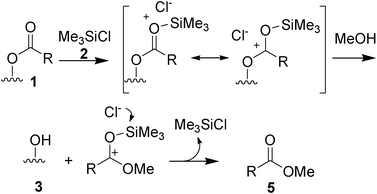 | ||
| Scheme 2 Catalytic activity of chlorotrimethylsilane in the transesterification. | ||
The somewhat high amount of reagent necessary to carry out the process is justified by its hydrolysis under several pathways, described in Scheme 3, including the process that leads to 3-Cl-1,2-propanediol (α-monochlorohydrin), in which chlorotrimethylsilane might play a role together with HCl. All the hydrolytic pathways lead to the formation of hexamethyldisiloxane (7), the stable ether that forms from two molecules of trimethylsilanol (6) that, in fact, can be recovered quantitatively from the reaction mixture.
 | ||
| Scheme 3 Rationale for formation of byproducts. | ||
The volatile trimethylsilylethers (8) formed by reaction of 2 with the alcohol reagent, observed in traces in the crude mixtures, could be involved as well in the process reported in Scheme 2, acting as the effective mediator that delivers the alkoxy group intramolecularly to the carbenium ion.
4. Conclusions
In conclusion, a new efficient method for the transesterification of triglycerides and the production of fatty acid alkyl esters (BioDiesel) in acidic conditions was disclosed. Due to the simplicity and efficiency of the method it can be extended to animal fats and exhausted vegetable oils without the need for pretreatment, as well as highly acidic oils. In particular it allows the easy separation of fatty acid esters and the glyceric phase. The process occurs at a moderate temperature and can be extended to alcohols other than MeOH, in particular EtOH and n-BuOH. Glycerol, during the process, is transformed into the more valuable 3-Cl-1,2-propanediol (α-monochlorohydrin),25 a useful intermediate for the synthesis of glycidol, as well as the 1,3-dichloropropanol precursor of epichlorohydrin, a commodity with numerous industrial applications. The acid mediator, chlorotrimethylsilane, is a low cost commercially available reagent with low toxicity and environmental impact. Its use in rather large quantities in the process represents the only drawback for the described procedure, however, it can be easily overcome. In fact, its transformation product, hexamethyldisiloxane, an inert product recovered quantitatively from the reaction, could be recycled by the transformation back to chlorotrimethylsilane by known procedures,23,24 by utilizing the formed HCl side product. In this way, this new method for BD production shows its low environmental impact, because only a few non toxic and/or recyclable byproducts are formed in the process.Acknowledgements
The authors thank Gatti S.r.l Castelnuovo Rangone (MO)-Italy, Adriatica Oli S.r.l Montecosaro (MC)- Italy, and ALTEC SAM Monaco for financial and technical support.References
- H. J. Wright, J. B. Segur, H. W. Clark, S. K. Cobum, E. E. Langdon and R. N. DuPuis, J. Am. Oil Chem. Soc., 1944, 21, 145–148 CrossRef CAS.
- U. Schuchardt, R. Sercheli and R. M. Vargas, J. Braz. Chem. Soc., 1998, 9, 199–210 CrossRef CAS.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- G. Vicente, M. Martínez and J. Aracil, Bioresour. Technol., 2004, 92, 297–305 CrossRef CAS.
- M. Mittelbach and C. Remschmidt, Biodiesel, the comprehensive handbook, Boersedruck GmbH, 1st edn, 2004 Search PubMed.
- K. J. Harrington and C. D'Arcy-Evans, Ind. Eng. Chem. Prod. Res. Dev., 1985, 24, 314–318 CrossRef CAS.
- B. Freedman, R. O. Butterfield and E. H. Pryde, J. Am. Oil Chem. Soc., 1986, 63, 1375–1380 CrossRef CAS.
- K. S. Liu, J. Am. Oil Chem. Soc., 1994, 71, 1179–1187 CrossRef CAS.
- H. Fukuda, A. Kondo and H. Noda, J. Biosci. Bioeng., 2001, 92, 405–416 CAS.
- T. Kocsisová, J. Cvengros and J. Lutisan, Eur. J. Lipid Sci. Technol., 2005, 107, 87–92 CrossRef.
- E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- D. E. López, J. G. Goodwin Jr, D. A. Bruce and E. Lotero, Appl. Catal., A, 2005, 295, 97–105 CrossRef.
- F. Zaccheria, Chim Ind, 2009, 91, 126–131 CAS.
- S. Yan, C. DiMaggio, S. Mohan, M. Kim, S. O. Salley and K. Y. S. Ng, Top. Catal., 2010, 53, 721–736 CrossRef CAS.
- A. Brandi, A. Salvini, G. Cipriani, D. Giomi and G. Bartolozzi, WO 2011/023712 A1, 2011.
- M. D. Guillén and A. Ruiz, Eur. J. Lipid Sci. Technol., 2003, 105, 688–696 CrossRef.
- G. Gelbard, O. Brès, R. M. Vargas, F. Vielfaure and U. F. Schuchardt, J. Am. Oil Chem. Soc., 1995, 72, 1239–1341 CrossRef CAS.
- J. Eras, F. Montañes, J. Ferran and R. Canela, J. Chromatogr., A, 2001, 918, 227–232 CrossRef CAS.
- J. B. Conant and O. R. Quayle, Org. Synth., 1941, 1, 292 Search PubMed; J. B. Conant and O. R. Quayle, Org. Synth., 1922, 2, 29 Search PubMed.
- R. Kumareswaran, A. Gupta and Y. D. Vankar, Synth. Commun., 1997, 27, 277–282 CrossRef CAS.
- F. Pisaneschi, C. Della Monica, F. M. Cordero and A. Brandi, Tetrahedron Lett., 2002, 43, 5711–5714 CrossRef CAS.
- G. A. Grasa, R. Singh and S. P. Nolan, Synthesis, 2004, 7, 971–985 Search PubMed.
- B.-h. Zhang and L.-x. Shi, Hebei Gongye Keji, 2007, 24, 63–65 Search PubMed.
- X. Cheng and Q. Zhang, Huaxue Shijie, 1988, 29, 65–67 CAS.
- (a) E. Santacesaria, M. Di Serio and R. Tesser, Process for monochlorohydrins production from glycerol and hydrochloric acid, WO 2008/132770 A1, 2008 Search PubMed; (b) M. Pagliaro and M. Rossi, The Future of Glycerol, RSC Green Chemistry No. 8, RSC Publishing, Cambridge UK, 2nd edn., 2010 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |