Rehydratable gel for rapid loading of nanoliter solution and its application in protein crystallization†
Yuefang
Li
,
Dameng
Guo
and
Bo
Zheng
*
Department of Chemistry, The Chinese University of Hong Kong, Shatin, Hong Kong. E-mail: bozheng@cuhk.edu.hk; Fax: +852 26035057; Tel: +852 39436261
First published on 23rd April 2012
Abstract
In the work presented, rehydratable polyacrylamide gel is introduced as a medium to uptake and store nanoliter protein solutions in microwells for multiplex bioanalysis. The polyacrylamide gel, produced and stored in the microwells, shrank by 97% upon dehydration and could be reversibly rehydrated to 95% of the initial volume by absorbing aqueous solution. We employed the rehydratable gel to load aqueous solutions of different proteins with molecular weights in the range of 14.7–250 kDa. The protein loading occurred simultaneously with the gel rehydration and reached saturated state in 5 min. The relative protein concentrations in the gel ranged from 92% to 53%, depending on the molecular weight of the proteins. Particularly, the rehydratable gel had a much higher protein loading efficiency than the fresh gel. We applied the protein-carrying gel to the crystallization of four model proteins in the microwells and produced diffraction-quality protein crystals. The rehydratable gel is simple to fabricate, efficient to load with protein, and has good capacity for storing the protein solutions in microwells with minimal dilution effects on the protein solution. The rehydratable gel incorporated microwell chip should be useful in multiplex analysis that requires small sample consumption and high throughput.
1. Introduction
Microwell arrays are a useful tool that enables high throughput experiments of biology and chemistry with the volume of reagents downscaled to the nanoliter level. Microwell arrays of high density and uniform sizes could be easily fabricated from various materials, including thermoplastics, polysiloxane rubbers and glass, all of which are cost effective and vary in physical properties and surface functionalities to meet the requirements of the diverse applications. The delivery of liquid into the microwells in an accurate and fast manner, however, becomes more difficult as the volume of the microwells descreases to nanoliter or smaller. Liquid deposition techniques have been developed to deliver nanoliter liquid efficiently, such as piezodispensing1–3 and pin tools.4 The performance of these techniques depends on the liquid compositions, viscosities, and surface tensions, and could be affected by the clogging of nozzles or needles. Microfluidic methods, such as pneumatic valve controlled multichannels5 and serial droplets,6 provide an alternative to deliver liquid to nanoliter microwells. The sophisticated control systems and complicated operations on these microfluidic chips, however, make these methods less accessible.Previously we developed a liquid-dispensing method using a degassed poly(dimethylsiloxane) (PDMS) slab engraved with microchannels as an internal vacuum source to aspirate liquids into the nanoliter microwells. The PDMS slab was removed afterwards to allow further mixing and reaction within the microwells.7 The dispensing method is simple as it does not require any apparatus in manipulating the liquids but relies on the reduced pressure in the microchannels. However, in this method the removal of the PDMS slab tends to remove the liquids in the microwells due to surface tension. The same problem exists in other microwell-based platforms.6,8,9 A strategy to solve this problem is to produce high contrast in hydrophobicity between the microwell wall and the chip surface by surface patterning and modification,6 which complicates the fabrication of the microwells. In addition, the nanoliter liquids in the microwells are susceptible to evaporation and could dry out within seconds if the environmental humidity is not well-controlled. The problem severely limits the application of the microwell-based platform, especially when the liquids contain proteins which demand a hydrating environment to maintain their activities. In this work we present a simple approach to tackle the problem of liquid loss during the dispensing and mitigate the influence of water evaporation by incorporating the liquid into rehydratable polyacrylamide (PA) hydrogel inside the microwells.
Hydrogels are often used to carry proteins and nucleic acids to provide precise control over the environment surrounding the biomolecules.10–13 The hydrated and biocompatible environment created by the hydrogel facilitates long-term preservation and manipulation of the biomolecules, while the flexible network of the hydrogel provides commodious space so that the biological functions of the loading biomolecules will not be affected. In the field of proteomics, for example, hydrogels served as hydrophilic substrates with high water contents for protein microarrays to maintain the biological functions of proteins.14,15 Hydrogels were also used to transfer and concentrate membrane proteins by contact printing.16
To incorporate biomolecules into a hydrogel network, a common method is to mix the biomolecules with the gel precursor prior to gelation, and the biomolecules are entrapped in the gel bulk by crosslinking the gel precursor. The application of this method is limited by the specific gelation reactions that could potentially denature the biomolecules. Another method is to immerse the gel into a concentrated stock solution of the biomolecules which then gradually diffuse into the gel. This method has low efficiency in getting biomolecules at high concentration in the gel and is time-consuming. In the current work we solved the problems of solution loading in gels by PA gel rehydration. PA gel could be stored in a dehydrated state after gelation and rehydrated before use to simplify the gel handling in experiments of electrophoresis-based protein separation.17–20 The dehydrated PA gel possesses a strong affinity to water and has been used as a dehydrating reagent.21 In our study, PA gel was produced in the microwells and dehydrated for long-term storage. Nanoliter solutions were dispensed into the microwells along the microchannels engraved in the degassed PDMS slab and were simultaneously absorbed by the gel during gel rehydration. As the gel showed high efficiency in loading protein solutions, we applied the method to protein crystallization by dispensing protein solutions in the microwells, simultaneously rehydrating the gel, and generating an array of gel storing the protein solutions for subsequent crystallization (Fig. 1).
 | ||
| Fig. 1 Frontal (left) and cross-sectional (right) illustrations of using the rehydratable gel array for protein loading and crystallization: a) prepare the gel particles in the microwells and dehydrate the gel; b) dispense protein solutions into the microwells using degassed PDMS microchannels and rehydrate the gel simultaneously; c) remove the PDMS microchannel; d) bind the microwells containing gel particles loaded with proteins with microwells containing precipitants to allow the mixing. | ||
2. Experimental
2.1. Preparing PA gel in microwells
The microwell chip was fabricated from a soda-lime glass slide (Shaoguang) coated with chromium and photoresistant using photolithography and wet etching techniques.22 64 square microwells were engraved on the chip with an interval of 400 μm between two adjacent microwells. The width of the microwell was 400 μm and the depth was 100 μm. The chip was cleaned by oxygen plasma (Plasma-Prep II, SPI), and then the surface between the microwells was treated by octadecyltrichlorosilane (Acros) via contact printing. After the treatment, the chip surface became hydrophobic while the walls of the microwells were still hydrophilic.To synthesize the PA gel in the microwells, a reaction mixture was prepared by mixing 1.7 mL 30% (w/v) acrylamide/bis-acrylamide aqueous solution (Beyotime) with 100 μL 20% VA-086 (Wako) solution and 8.2 mL DI water. The concentrations of monomer and initiator in the final mixture were 5% and 0.2% (w/v), respectively. 100 μL deoxygenated mixture was dripped in the microwells, then the chip was immediately covered by a piece of cover slide and placed under UV illumination (1 mW cm−2) for 10 min. The glass cover was then removed carefully, leaving an array of PA gel embedded in the microwells, which was rinsed thoroughly with DI water. To fully dehydrate the gel, the chip carrying the gel was incubated in an oven at 50 °C for 10 min.
2.2. Loading protein in the rehydratable gel
To load protein into the dehydrated gel, a liquid dispensing method with degassed PDMS was used.7 A PDMS slab engraved with microchannels was aligned and reversibly bound to the microwells. Then the whole chip was degassed at 6 kPa for 15 min to remove dissolved air out of the PDMS. 5 μL of the protein solutions were pipetted on the inlets of the microchannels, which flowed into the microchannels and the microwells, and the PA gel was rehydrated. Finally, when the gel was fully rehydrated, the PDMS slab was carefully peeled off, leaving the gel carrying proteins in the microwell array, which were ready for further assays. Fluorinated oil FC3283 (3 M) was applied to help peel off the PDMS slab. It took around 5 min for the sample liquid to fill all the microwells and rehydrate the gel completely.2.3. Evaluating protein loading capacity and efficiency
∼10 ng ml−1 enhanced green fluorescence protein (eGFP) was derived from in vitro expression.23 Rhodamine B labeled PA gel was synthesized following the procedure mentioned in 2.1 except that DI water was replaced by 0.1 mg mL−1 aqueous solution of methacryloxyethyl thiocarbamoyl rhodamine B (Polysciences). The loading of eGFP into the PA gel was traced by a confocal laser scanning microscope (C1si, Nikon).Five protein solutions were used to rehydrate the gel: 20 mg mL−1 thaumatin (Wako) in 0.1 M N-(2-acetamido)-iminodiacetic acid buffer, pH 6.5; 40 mg mL−1 trypsin (Sigma) in 10 mM CaCl2, 10 mg mL−1 benzamidine hydrochloride, 25 mM HEPES buffer, pH 7.0; 10 mg mL−1 catalase (Sigma) in 50 mM phosphate buffer, pH 6.8; 50 mg mL−1 chicken egg-white lysozyme (Sigma) in 50 mM sodium acetate buffer, pH 4.5; and 10 mg mL−1 bovine serium albumin (BSA, Sigma) in 50 mM phophate buffer, pH 7.4. All reagents were filtered by 0.45 micron filter membrane (DISMIC-3cp, Advantec) prior to use.
To estimate the amount of protein absorbed by gel rehydration, 5.0 mg dehydrated PA gel was mixed with 190 μL protein solution and rehydrated to ∼95 mg after incubation for 5 min. The volume of the leftover protein solution was measured afterwards. The absorption at 280 nm of the original protein solution and the leftover solution was determined by a UV-Vis spectrometer (Nanodrop 1000, Thermo). Control experiments were carried out by immersing 95.0 mg fresh PA gel in 95 μL protein solutions for 5 min and measuring the volume and 280-nm absorbance of the leftover solution. The initial protein concentration in the control experiment was twice as high as the initial protein concentration in experiment using dehydrated PA gel.
2.4. Protein crystallization
Four model proteins were used for the crystallization experiments. The crystallization reagents were 2.0 M sodium potassium tartrate in 0.1 M HEPES buffer pH 7.5 for thaumatin, 0.2 M (NH4)2SO4 and 30% PEG 8000 in 0.1 M sodium cacodylate buffer pH 6.5 for trypsin, 0.2 M MgCl2 and 30% iso-propanol in 0.1 M HEPES buffer pH 7.5 for catalase, and 2 M sodium formate in 0.1 M acetate buffer pH 4.6 for lysozyme. All protein solutions were prepared as in 2.3 and filtered by 0.45 micron filter membrane prior to use.5 μL crystallization reagents were pipetted on the inlets of microchannels and were dispensed into the microwells using degassed PDMS microchannels,7 and the chip was immersed in FC3283 oil afterwards. The chip containing protein-loading gel and the other chip containing precipitants were aligned and bound to allow reagents mixing in the resulting microchambers. The two chips were clamped together and stored in paraffin oil. For the control experiments that grew crystals in solution phase, both the protein solutions and the precipitants were directly dispensed into the microwells without the gel.7 The growth of protein crystals was monitored by a polarized light stereomicroscope (MZ 16, Leica) equipped with a CCD camera (SPOT Insight, Diagnostic Instruments).
To harvest a protein crystal, the bound microwell slabs were carefully separated apart, and 5 μL of cryoprotectant (10% glycol in the corresponding crystallization reagent) was pipetted on the microwells containing crystals. The crystal was picked up using a cryoloop (Hampton Research) and then flash-frozen by liquid nitrogen. The X-ray diffraction data was collected at 100 K by an X-ray generator (MicroMax-007, Rigaku) and recorded by an IP detector (R-AXIS IV++, Rigaku).
3. Results and discussion
3.1. Rehydration of the PA gel in the microwells
In our study, the PA gel was synthesized directly in the microwells (Fig. 2a), which served both as the template for molding the gel and as the container for the gel and the loading liquid. The PA gel was derived from 5% acrylamide solution and the molar ratio of the monomer to the crosslinker was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The monomer and crosslinker concentration was optimal to produce a gel with good mechanical strength and a maximum pore size. The resulting pore size of PA gel was ∼300 nm, as determined from the SEM image of the freeze-dried gel (Fig. s1†), which was larger than the reported pore size of ∼150 nm in bulk PA gel.24 Compared to the gel of higher solid content and with smaller pore size of tens of nanometers,24,25 the 300 nm pore size in the current gel facilitated a faster protein loading and releasing rate. Before usage, the PA gels were rinsed with DI water thoroughly to wash away soluble substances inside the gel, including residue monomers, initiators, and short-chain oligomers. Then the chip carrying the gel was incubated at 50 °C to dehydrate the gel.
:1. The monomer and crosslinker concentration was optimal to produce a gel with good mechanical strength and a maximum pore size. The resulting pore size of PA gel was ∼300 nm, as determined from the SEM image of the freeze-dried gel (Fig. s1†), which was larger than the reported pore size of ∼150 nm in bulk PA gel.24 Compared to the gel of higher solid content and with smaller pore size of tens of nanometers,24,25 the 300 nm pore size in the current gel facilitated a faster protein loading and releasing rate. Before usage, the PA gels were rinsed with DI water thoroughly to wash away soluble substances inside the gel, including residue monomers, initiators, and short-chain oligomers. Then the chip carrying the gel was incubated at 50 °C to dehydrate the gel.
 | ||
| Fig. 2 a) A micrograph of the microwells carrying dry gel; b) a micrograph of the dispensing yellow dye solution into the microwells carrying dry gel via microchannels; c) a micrograph of microwells containing the gel rehydrated in yellow dye solution after peeling off the microchannels; d) a micrograph of the microchambers generated by binding the microwells containing the gel loaded yellow dye solution to the microwells containing blue dye solution (scale bar: 1 mm). | ||
We tested the capability of the rehydratable gel to load liquid during the liquid dispensing process using food dye solutions. Yellow dye solution was aspirated into the microwells via degassed PDMS microchannels, in the meanwhile rehydrating the gel (Fig. 2b). The gel was completely rehydrated when the yellow dye solution filled the whole microwell. Afterwards the PDMS slab was carefully peeled off against the glass chip on which the yellow dye solution stored in the gel was left in the microwells (Fig. 2c). Extra solution in the microchannels was carried away with the PDMS slab. The microwells carrying the gel containing yellow dye solution was aligned to the microwells carrying blue dye solution. Every two aligned microwells bound into a close microchamber in which the solutions were mixed, generating a green color (Fig. 2d).
The dehydration and rehydration kinetics of the PA gel in microwells were studied by monitoring the gel volume change over time (Fig. 3). We assumed that the swelling and shrinking of the gels were isotropic and therefore calculated the volume change from the change of the lateral dimension. Incubating the gel in an oven of 50 °C quickly dehydrated the gel. The particles shrank down to 3% of the original volume in 5 min. Rehydration of the PA gel is a reversal process of dehydration. The particles began to rehydrate as soon as aqueous liquids were dispensed into the microwells and contacted the gel. A complete rehydration process took 5 min. After rehydration, the gel swelled to 95% of the original volume. It was reported that a 60–120 μm slab of PA gel took 2–3 min to be fully rehydrated when immersed in buffer.20 In our experiment, rehydration of the gel was slower, because an extra time was required to dispense the liquid into the microwells. Furthermore, the gel dehydration/rehydration-in-water was repeated 5 times with its loading capacity unaffected (Fig. s2†). The reversible gel rehydration could be helpful when different reagents are loaded into the gel in succession.
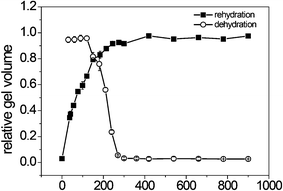 | ||
| Fig. 3 Kinetics of gel dehydration at 50 °C and rehydration in water at room temperature. | ||
3.2. Protein loading rate and capacity of the gel
To demonstrate the fast loading of proteins in PA gel during gel rehydration, the rehydration process of eGFP in the gel labeled with rhodamine B was monitored by confocal microscopy. After the 5-min rehydration, the red fluorescence from rhodamine B was observed across the entire microwells (Fig. 4b), indicating the stretching of the PA chains as the gel regained their original shape. Green fluorescence from eGFP penetrated into the gel networks at the same time. In particular, the intensity of green fluorescence was homogeneous in all the three dimensions of the gel (Fig. 4c), indicating that the eGFP molecules were evenly distributed in the rehydrated gel. | ||
| Fig. 4 Uptake of eGFP into the rehydratable gel. a) Confocal micrograph of the dehydrated PA gel stained by rhodamine B. The boundary of the microwells is indicated by dashed lines. b) and c) Confocal micrographs after the gel in a) was rehydrated in eGFP with Red and Green filters respectively. Images a–c were obtained using the z-scan mode of a laser scanning confocal microscope to provide a comprehensive vision of the gel rehydration laterally as well as vertically (scale bar: 800 μm). | ||
We further studied the protein loading rate and capacity by determining the relative protein concentration in gel based on the following equation:
 | (1) |
c gel,r is the relative protein concentration in the gel, which denotes the percentage ratio of the protein concentration in gel (cgel) to the averaged protein concentration in the total volume of the gel and the protein solution (ct); ci and cr are the protein concentrations in the initial protein solution and in the leftover protein solution, which were derived from the absorbance at 280 nm; Vi, Vr, and Vgel are the volumes of the initial protein solution, the leftover protein solution and the gel after rehydration, respectively. Since the initial volume of the dehydrated gel was negligible compared with the volume of the initial protein solution, estimations were made that ct ≈ ci and Vgel ≈ Vi − Vr.
Fig. 5a illustrates the loading kinetics of lysozyme into the PA gel during gel rehydration. After only 1-min rehydration, the relative lysozyme concentration in gel had already reached 80%, though the gel had not swelled to the maximum volume. Maximum lysozyme retention in gel at a relative concentration of 91% was achieved in 5 min, in accordance with the confocal images. Therefore in the following experiments we considered 5 min as the cut-off time for both the gel rehydration and protein loading.
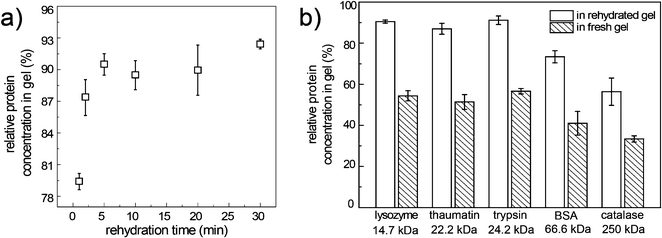 | ||
| Fig. 5 a) Relative protein concentrations of lysozyme in the rehydrated gel as a function of rehydration time; b) relative protein concentrations of the five proteins in the rehydrated gel and in the fresh gel after 5-min incubation in the stock protein solutions. | ||
In addition to lysozyme, four other proteins with the molecular weight in the range of 20 kDa to 250 kDa were also tested (Fig. 5b). For the three proteins with molecular weights smaller than 25 kDa, the relative concentrations in the rehydrated gel all reached ∼90%. Larger proteins revealed a smaller relative protein concentration in the gel (Fig. 5b). For the 250 kDa catalase, a relative protein concentration of 56% was achieved in the rehydrated gel, suggesting that the protein loading was affected by the molecular weight. It is known that proteins with molecular weights larger than 500 kDa had low mobility in the mechanically stable PA gel.26 Therefore we deduced that the rehydratable gel method would be limited to proteins with molecular weights smaller than 500 kDa.
To further prove the potency of the rehydratable gel in loading protein solutions efficiently, a control experiment was carried out that measured the relative protein concentrations in fresh gel (see ESI† for details). In fresh gel, the initial water content was significantly higher than that in the dry gel, which would dilute the uptaken protein solution. To exclude the dilution effect of the fresh gel, experiments were designed so that the total amount of protein, water and solid content of the gel were the same in both experiments using the rehydratable gel and the fresh gel. The relative protein concentration decreased as the molecular weight of the protein increased in both experiments using the rehydratable gel and the fresh gel (Fig. 5b), implying that the protein loading in both experiments relied on the protein diffusivity in gel. Nevertheless, for all the testing proteins, the relative protein concentrations in the rehydrated gel were much higher than the relative protein concentrations in the fresh gel. Gel rehydration enhanced the protein absorption in PA gel and therefore increased the efficiency of protein loading. It is likely that other than diffusive transport of proteins into the gel, convective transport induced by expanding the gel network also contributed to the rapid loading of proteins in rehydrated gel.27
3.3. Crystallization of proteins loaded in gel
The gel array carrying proteins in microwells provides a simple platform to carry out multiplex reactions by binding the microwells containing the gels to the microwells containing other reagents to form microchambers (Fig. 1d). When the resulting glass chambers were sealed by tight clamping and were stored in oil, no loss of water was observed in the chambers during an incubation of two months, making the microchambers suitable for growing protein crystals.Protein crystallization plays a crucial role in determining the tertiary protein structures using X-ray diffraction. Recently there has been much progress in using microfluidic techniques to screen the conditions for obtaining protein crystals at low protein consumption and high throughput.6,28,29 However, these methods have not seen wide adoption in laboratories, primarily due to the complicated flow controlling systems.
Here we aim to simplify the crystallization method by using the rehydratable gel array. There is a long history of protein crystallization in the presence of gels,30 for the purpose of creating convection-suppressed environments for crystal growth or establishing concentration gradients. The PA gel is superior in its excellent chemical stability and clean chemistry of polymerization. Moreover, the PA gel has been widely used as the solid support for electrophoresis31 and NMR measurements of both globular and membrane proteins,32,33 suggesting that the PA gel is compatible with both types of proteins and is a good choice for the crystallization experiment. To confirm the viability of producing protein crystals with the PA gel array, four model proteins were crystallized in microwells with PA gel as the medium to store the protein solutions.
The results verified that PA gel was an appropriate medium for all the four proteins (Fig. 6): all the parallel trials yielded crystals for each protein. The crystals were growing on top of the gel instead of within the gel, probably due to the unfavorable pore size in the gel for crystal growth. We examined the qualities of lysozyme and thaumatin crystals by X-ray diffraction (Fig. s3). The resolution of the diffraction pattern was 1.5 Å for both proteins, reaching the detection limit of the instrument.
 | ||
| Fig. 6 Micrographs of protein crystals grown using the protein carrying gel for a) 60 mg mL−1 lysozyme, b) 40 mg mL−1 trypsin, c) 10 mg mL−1 catalase, and d) 20 mg mL−1 thaumatin (scale bar: 200 μm). | ||
Control experiments using protein solutions were carried out to compare with the proteins stored in the rehydratable gel. Though cracks were observed in some lysozyme crystals that were in contact with the gel (Fig. 6a), the crystals produced by both methods had similar morphology and diffraction resolution (data not shown), implying that the gel did not impede the ordered arrangement of protein in the crystals. The chip with 64 microwells allowed each protein to have 4 different concentrations tested at the same time, and each concentration was repeated in 16 parallel trials for statistical analysis. The average number of protein crystals produced under each condition was presented in Fig. 7. At low protein concentrations where the appearance of crystals was rare in solution conditions, the presence of the gel resulted in an increased possibility of crystal growth, as in the cases of 60 mg mL−1 lysozyme (Fig. 7a), 50 mg mL−1 and 40 mg mL−1 trypsin (Fig. 7b), 10 mg mL−1 catalase (Fig. 7c) and 15 mg mL−1 thaumatin (Fig. 7d). At higher protein concentrations, the crystal number was similar in both methods for lysozyme, trypsin and catalase. Thaumatin was an exception, which showed significantly increased number of crystals grown on the gel at 20 mg mL−1 thaumatin concentration compared with the parallel control experiments. Precipitations were observed if the thaumatin concentration was further increased in the experiments using rehydratable gel.
 | ||
| Fig. 7 Number of the crystals grown using the protein carrying gel or in solution phase for a) lysozyme, b) trypsin, c) catalase and d) thaumatin. The numbers listed below each diagram indicate the concentrations of the protein dispensed into the microwells. The bars denoted by asterisk indicate the result of zero crystal. | ||
The difference between the crystal numbers of the four proteins using the rehydratable gel and in solution conditions suggested that the gel influenced protein nucleation and crystal growth. The introduction of the gel with porous structure may increase the probability of heterogeneous nucleation of the proteins. Porous silica with 5–10 nm pores was reported to induce the nucleation of protein crystals.34,35 The studies suggested that the nucleation enhancement effect of the porous materials relied strongly on the pore size. Recent study has shown that the mesh size of a polymer microgel affects the performance of the gel in controlling the nucleation of some organic molecules.36 Though the pores in the rehydratable PA gel were as large as 300 nm, the mesh size of the gel37 was comparable to the pores of the porous silica in the aforementioned report that facilitated protein stacking. Therefore it is possible that the microstructures of PA gel promoted the nucleations of proteins at low protein concentrations. Moreover, the interaction between the gel network and proteins may have also affected the heterogeneous nucleation of proteins on the gel. In our study, the amide groups on PA gel could potentially interact with the protein molecules through hydrogen bonding, which may contribute to the significant enhancement of thaumatin nucleation by PA gel.
4. Conclusions
In this work, a method of loading and storing nanoliter liquid using a rehydratable gel was validated, which is helpful to maintain the volume of the sample solution in microwells during liquid dispensing. The use of rehydratable gel provides a simple and reliable approach to deliver minimal amounts of proteins, and this method is particularly advantageous in applications that require concentrated protein solutions. With the aid of liquid dispensing using degassed PDMS microchannels, trials of protein crystallization were carried out on the gel array and protein crystals were harvested in the microwells. In particular, the nucleation enhancement effect of the rehydratable gel derived from the microporous structure of the gel and the polymer–protein interaction on protein crystallization was observed, suggesting that the gel has the competency to control the nucleation of proteins. The simplicity of the gel production in the microwells makes it possible to scale up the method for high-throughput analysis. We envision that the method is suitable to produce large arrays of hydrated biomolecules and has potential applications in single cell analysis, biomedical therapeutics and multiplex bioassays.Acknowledgements
We thank the Chinese University of Hong Kong and the Hong Kong PhD Fellowship Scheme (PF09-08514) for the financial support. We thank Candice Chang for providing the eGFP samples, Feng Wang for SEM operation, and Dr Shannon W. N. Au for XRD analysis.References
- D. P. Little, T. J. Cornish, M. J. Odonnell, A. Braun, R. J. Cotter and H. Koster, Anal. Chem., 1997, 69, 4540–4546 CrossRef CAS.
- S. Weiss, G. T. John, I. Klimant and E. Heinzle, Biotechnol. Prog., 2002, 18, 821–830 CrossRef CAS.
- A. Schober, R. Gunther, A. Schwienhorst, M. Doring and B. F. Lindemann, Biotechniques, 1993, 15, 324–329 CAS.
- P. H. Cleveland and P. J. Koutz, Assay Drug Dev. Technol., 2005, 3, 213–225 CrossRef CAS.
- J. B. Wang, Y. Zhou, H. W. Qiu, H. Huang, C. H. Sun, J. Z. Xi and Y. Y. Huang, Lab Chip, 2009, 9, 1831–1835 RSC.
- L. Li, W. B. Du and R. F. Ismagilov, J. Am. Chem. Soc., 2010, 132, 112–119 CrossRef CAS.
- X. Zhou, L. Lau, W. W. L. Lam, S. W. N. Au and B. Zheng, Anal. Chem., 2007, 79, 4924–4930 CrossRef CAS.
- R. Moerman and G. W. K. van Dedem, Anal. Chem., 2003, 75, 4132–4138 CrossRef CAS.
- R. Moerman, J. Knoll, C. Apetrei, L. R. van den Doel and G. W. K. van Dedem, Anal. Chem., 2005, 77, 225–231 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- G. R. Hendrickson and L. A. Lyon, Soft Matter, 2009, 5, 29–35 RSC.
- N. Park, S. H. Um, H. Funabashi, J. F. Xu and D. Luo, Nat. Mater., 2009, 8, 432–437 CrossRef CAS.
- X. F. Leng, W. H. Zhang, C. M. Wang, L. A. Cui and C. J. Yang, Lab Chip, 2010, 10, 2841–2843 RSC.
- A. Y. Rubina, A. Kolchinsky, A. A. Makarov and A. S. Zasedatelev, Proteomics, 2008, 8, 817–831 CrossRef CAS.
- C. P. Tanase, R. Albulescu and M. Neagu, Expert Rev. Mol. Diagn., 2011, 11, 461–464 CrossRef.
- S. Majd and M. Mayer, J. Am. Chem. Soc., 2008, 130, 16060–16064 CrossRef CAS.
- R. C. Allen, B. Budowle, C. A. Saravis and P. M. Lack, Acta Histochem. Cytochem., 1986, 19, 637–645 CrossRef CAS.
- R. C. Allen and G. M. Graves, Bio/Technology, 1990, 8, 1288–1290 CrossRef CAS.
- M. D. Frey, A. Kinzkofer, M. B. Atta and B. J. Radola, Electrophoresis, 1986, 7, 28–40 CrossRef CAS.
- A. Guttman, J. Liq. Chromatogr. Relat. Technol., 1998, 21, 1249–1258 CrossRef CAS.
- A. E. Sherr and A. M. Swift, J. Appl. Polym. Sci., 1965, 9, 3929–3934 CrossRef CAS.
- Q. H. He, S. Chen, Y. Su, Q. Fang and H. W. Chen, Anal. Chim. Acta, 2008, 628, 1–8 CrossRef CAS.
- Y. Y. Chang, Undergraduate Thesis, The Chinese University of Hong Kong, 2011 Search PubMed.
- N. C. Stellwagen, Electrophoresis, 1998, 19, 1542–1547 CrossRef CAS.
- K. Engberg and C. W. Frank, Biomed. Mater., 2011, 6, 055006 CrossRef.
- C. M. Warren, P. R. Krzesinski and M. L. Greaser, Electrophoresis, 2003, 24, 1695–1702 CrossRef CAS.
- S. H. Gehrke, Adv. Polym. Sci., 1993, 110, 81–144 CrossRef CAS.
- C. L. Hansen, E. Skordalakes, J. M. Berger and S. R. Quake, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16531–16536 CrossRef CAS.
- B. Zheng, L. S. Roach and R. F. Ismagilov, J. Am. Chem. Soc., 2003, 125, 11170–11171 CrossRef CAS.
- A. McPherson, Crystallization of Biological Macromolecules, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 1999 Search PubMed.
- B. Wenge, H. Bonisch, J. Grabitzki, G. Lochnit, B. Schmitz and M. H. J. Ahrend, Electrophoresis, 2008, 29, 1511–1517 CrossRef CAS.
- R. Tycko, F. J. Blanco and Y. Ishii, J. Am. Chem. Soc., 2000, 122, 9340–9341 CrossRef CAS.
- D. H. Jones and S. J. Opella, J. Magn. Reson., 2004, 171, 258–269 CrossRef CAS.
- N. E. Chayen, E. Saridakis, R. El-Bahar and Y. Nemirovsky, J. Mol. Biol., 2001, 312, 591–595 CrossRef CAS.
- N. E. Chayen, E. Saridakis and R. P. Sear, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 597–601 CrossRef CAS.
- Y. D. Y. Diao, M. E. Helgeson, A. S. Myerson, T. A. Hatton, P. S. Doyle and B. L. Trout, J. Am. Chem. Soc., 2011, 133, 3756–3759 CrossRef CAS.
- L. M. Lira, K. A. Martins and S. I. C. de Torresi, Eur. Polym. J., 2009, 45, 1232–1238 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: estimation of relative protein concentration in fresh gel; SEM image of the gel porous structure; reversibility of the gel dehydration and rehydration; X-ray diffraction pattern of the protein crystals. See DOI: 10.1039/c2ra20511d/ |
| This journal is © The Royal Society of Chemistry 2012 |