Photoinduced charge separation in donor–acceptor spiro compounds at metal and metal oxide surfaces: application in dye-sensitized solar cell†
Lorena
Macor
a,
Miguel
Gervaldo
a,
Fernando
Fungo
*a,
Luis
Otero
*a,
Thomas
Dittrich
b,
Chih-Yen
Lin
c,
Liang-Chen
Chi
c,
Fu-Chuan
Fang
c,
Shu-Woei
Lii
c,
Ken-Tsung
Wong
*c,
Chih-Hung
Tsai
d and
Chung-Chih
Wu
d
aDepartamento de Química, Universidad Nacional de Río Cuarto, Agencia Postal N° 3, X5804BYA, Río Cuarto, Argentina. E-mail: lotero@exa.unrc.edu.ar; ffungo@exa.unrc.edu.ar
bHelmholtz Center Berlin for Materials and Energy, Institute of Heterogeneous Materials, Hahn-Meitner-Platz 1, D-14109 Berlin, Germany
cDepartment of Chemistry, National Taiwan University, Taipei 106, Taiwan. E-mail: kenwong@ntu.edu.tw
dDepartment of Electrical Engineering, Graduate Institute of Photonics and Optoelectronics, and Graduate Institute of Electronics Engineering, National Taiwan University, Taipei 10617, Taiwan
First published on 24th April 2012
Abstract
Modulated charge separation has been investigated by surface photovoltage spectroscopy in the fixed capacitor arrangement for four different fluorene compounds adsorbed from highly diluted solutions at ultra-thin nanoporous TiO2 (np-TiO2), Au and ITO surfaces. Donor (–N(C6H5)2) and acceptor moieties (–CN, –COOH) were linked by fluorene or spirobisfluorene cores and the chain length has been changed by introducing thiophene. Modulated charge separation by electron injection and intramolecular transport has been separated and directed adsorption of spiro compounds at Au surfaces has been demonstrated. Striking differences between the interaction of linking –CN and –COOH groups and the different substrates were observed. The capability as TiO2 spectral sensitizer of spirobisfluorene donor–acceptor molecules with extended conjugation has been demonstrated. Solar cells exhibit 5.6% energy conversion efficiency, confirming that spiro-configured geometry is a promising route to the design of metal free dyes.
Introduction
Separation of photo-generated charge carriers at organic/inorganic interfaces depends sensitively on the nature of chemical bonds and interactions at interfaces.1 Especially local charge transfer and stable molecular alignment in space are crucial for numerous applications in organic and hybrid electronic devices, such as dye-sensitized solar cells (DSSCs), organic light emitting diodes (OLEDs) and organic transistors. In the past, various approaches for oriented molecule adsorption at inorganic surfaces have been developed, for example the formation of self-assembled mono-layers from organic molecules with –SH end-groups at Au surfaces2 or the electro-chemical grafting of organic molecules at Si surfaces from diazonium salt solutions.3 In particular, development of DSSCs has attracted great research interest, due to their potential use as an environmentally friendly energy source,4–8 and one of the most important issues in DSSCs is the nature of the photosensitizers and their interaction with metal oxide surfaces. Generally, metal-free organic dyes possess molecular structures constituted of a donor part (D) and an acceptor counterpart (A) bridged by a π-conjugated linkage. As a class of the donor part, triphenylamine (TPA) and its derivatives have shown promising applications in the development of photovoltaic devices,9,10 whereas, cyanoacetic acid moiety is one of the most used units as electron acceptor/anchoring group in the design of organic dyes for highly efficient DSSCs. The D and A moieties combined with π-conjugated central cores in the DSSC dyes determine the absorption region of the solar spectrum. In order to efficiently capture the solar energy it is necessary to have the extended π-conjugation core to bridge D and A units to red shift the absorption spectra. As a consequence of this molecular design strategy, the rod-like molecular structures are prolonged, which can have a strong tendency to generate dye aggregation and consequently lead to self-quenching between dye molecules in the excited state. Therefore, new molecular design approaches towards the DSSC dyes that are capable of suppressing dye aggregation will be highly desired. In this regard, the spiro linkage emerges as a potential skeleton for new DSSC dyes.11–13 When the constituted branches composed of D–A terminus with extended π–system core are perpendicularly bonded via a common sp3-hybridized atom, the 3-dimensional structure inhibits intermolecular interactions, decreasing the tendency to the aggregate formation.12 Recently we reported a spiro-configured D–A sensitizer used in DSSCs, where the two anchoring groups give efficient binding to TiO2 surface and the spiro-configuration efficiently suppressed dye aggregation, making the dye a promising candidate to use in efficient DSSCs.13In this work spirobisfluorene D–A systems were compared with a non-spiro D–A compound. Also the nature of A groups, as well as the length of the conjugated molecular linkages, were varied. For this purpose four different molecules (named SSD1, SSD2, CN2 and NSD2, see Fig. 1) with a spirobisfluorene (SSD1, SSD2, CN2) and non-spiro fluorene (NSD2) cores have been synthesized. Diphenylamine was attached as D directly at one side of the fluorene cores (SSD1, CN2, NSD2) or at an intermediate thiophene group in order to increase the linkage length (SSD2). As acceptors, dicyano (CN2) or cyanoacrylic acid (SSD2, SSD2, NSD2) moieties were attached to one site of the fluorene (SSD2, SSD2, CN2) or to the central part of the fluorene (NSD2) cores via a carbon atom with a conjugated link. The molecules were adsorbed from highly diluted solutions at nanoporous TiO2 (np-TiO2), Au and ITO surfaces for getting information about interactions of the molecules at the different surfaces.
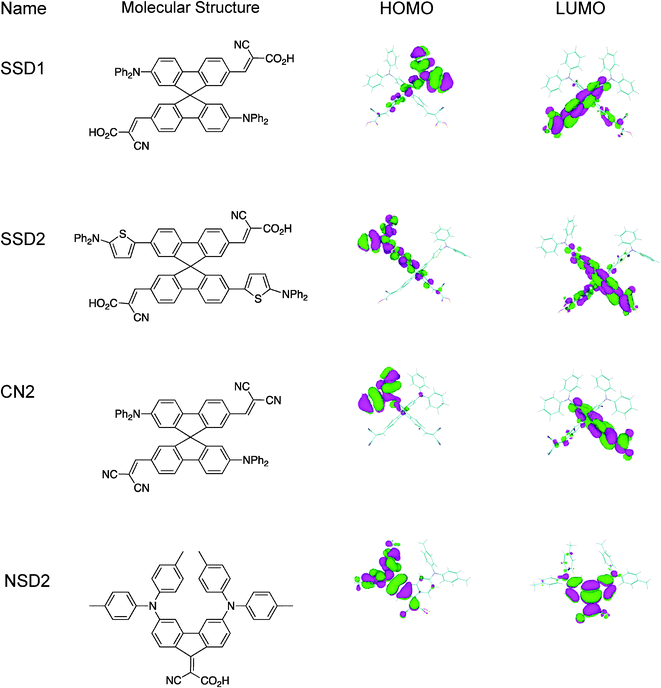 | ||
| Fig. 1 Structure of the studied molecules. Geometric optimization (AM1 semi-empirical calculations) of the dye structures. HOMO–LUMO frontier molecular orbitals. | ||
Modulated charge separation was investigated by surface photovoltage (SPV) spectroscopy in the fixed capacitor arrangement.14 Spectral information about the direction and amplitude of modulated charge separation and about changes in the trends of time response can be obtained with high sensitivity from modulated SPV. The latter can be detected by investigating the change between the in-phase and 90° phase-shifted SPV signals.
The SPV can be described by the amount of separated charge (Q) and by the distance (d) between the centers of separated positive and negative charge carriers. Q depends on the photo-generation of charge carriers. Furthermore, both Q and d depend on time (t) due to the time dependence of charge separation, transport and recombination processes, whereas different electronic states or electronic transitions can be involved. The situation becomes even more complicated considering a time-dependent relative dielectric constant (εrel), which may be caused, for example, by charge separation in layer systems with different dielectric properties SPV can be described as:
 | (1) |
DSSCs with spirobisfluorene D–A dye were constructed and evaluated. High incident photon-to-current conversion and energy conversion efficiencies confirm that spiro-configured geometry is a promising route to the design of new high efficient dyes.
Material and methods
General
All the chemicals used were provided by Sigma-Aldrich in spectroscopic or electrochemical grade without additional purification. The synthesis and characterization of SSD1 and CN2 molecules have been described previously.13,15Optical absorption spectra were recorded for the molecules in acetonitrile solution. The dye molecules were adsorbed from highly diluted solution (0.05 μM in methylene chloride) on Au (evaporated on glass substrates), ITO (PGO) and np-TiO2 surfaces under identical conditions. The FTIR spectra (Bruker Tensor 27) were measured for dye molecules in KBr pellets and for molecules absorbed on TiO2.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:7) to afford compound 1 (0.63 g, 72%) as an orange solid. mp 166–167 °C; IR (KBr) ν 3060, 2922, 1695, 1599, 1491, 1471, 1450, 1274, 1241. 1203, 695; 1H NMR (Fig. 1a, ESI†) (d6-DMSO, 400 MHz) δ 9.78 (s, 2H), 8.25 (d, J = 8.0 Hz 2H), 8.17 (d, J = 8.0 Hz, 2H), 7.97 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz, 2H), 7.28 ∼ 7.24 (m, 10H), 7.11 (s, 2H), 7.05 ∼ 7.01 (m, 12H), 6,83(s, 2H), 6.55(d, J = 4.0 Hz, 2H); 13C NMR (Fig. 1b, ESI†) (CDCl3, 100 MHz) δ 191.7, 152.0, 149.8, 148.8, 147.8, 139.3, 137.0, 136.4, 136.2, 131.5, 129.4, 125.8, 124.8, 123.5, 123.0, 122.9, 122.2, 121.5, 120.7, 93.3, 65.6; MS (m/z, FAB+) 871.1 (9); HRMS (m/z, FAB+) Calcd. C59H38N2O2S2 870.2375, found 870.2377.
:1:7) to afford compound 1 (0.63 g, 72%) as an orange solid. mp 166–167 °C; IR (KBr) ν 3060, 2922, 1695, 1599, 1491, 1471, 1450, 1274, 1241. 1203, 695; 1H NMR (Fig. 1a, ESI†) (d6-DMSO, 400 MHz) δ 9.78 (s, 2H), 8.25 (d, J = 8.0 Hz 2H), 8.17 (d, J = 8.0 Hz, 2H), 7.97 (d, J = 8.0 Hz, 2H), 7.71 (d, J = 8.0 Hz, 2H), 7.28 ∼ 7.24 (m, 10H), 7.11 (s, 2H), 7.05 ∼ 7.01 (m, 12H), 6,83(s, 2H), 6.55(d, J = 4.0 Hz, 2H); 13C NMR (Fig. 1b, ESI†) (CDCl3, 100 MHz) δ 191.7, 152.0, 149.8, 148.8, 147.8, 139.3, 137.0, 136.4, 136.2, 131.5, 129.4, 125.8, 124.8, 123.5, 123.0, 122.9, 122.2, 121.5, 120.7, 93.3, 65.6; MS (m/z, FAB+) 871.1 (9); HRMS (m/z, FAB+) Calcd. C59H38N2O2S2 870.2375, found 870.2377.
Surface photovoltage analysis
The measurements of modulated SPV were performed in the fixed capacitor arrangement with chopped light (modulation frequency 6 Hz) from a quartz prism monochromator (SPM2) and a halogen lamp (100 W). The SPV signals were detected with a high impedance buffer (measurement resistance 10 GΩ) and a two-phase lock-in amplifier (EG&G, model 5210). The measurements were carried out in vacuum. The SPV spectra were not normalized to the photon flux.The in-phase (equivalent to the sine) and 90° phase-shifted (equivalent to the cosine) signals are measured with the two-phase lock-in amplifier. The phase shift has been calibrated with a Si photodiode. The sign of the in-phase SPV signal is positive (negative) if the photo-generated electrons are preferentially separated towards the internal (external) surface. The tangent of the phase angle is defined by the quotient between the in-phase and 90° phase-shifted signals. The amplitude of the modulated SPV signal is defined as the square root of the sum of the squared in-phase and 90° phase-shifted SPV signals.
The absolute value of the in-phase signal is in maximum and the 90° phase-shifted signal is zero, i.e. the phase angle is 0° or ±180°, if the time constants of the increasing and decreasing signals are much shorter than the modulation period. In contrast, the in-phase signal is zero and the absolute value of the 90° phase-shifted signal is in maximum, i.e. the phase angle is ±90° or 270°, if the time constants of the increasing and decreasing signals are much longer than the modulation period. In this case, charge separation, transport processes and recombination are very slow, for example, limited by trapping with long trapping lifetimes.
Fabrication of dye-sensitized solar cells
To prepare the DSSC working electrodes, the FTO glass plates were first cleaned in a detergent solution using an ultrasonic bath for 15 min, and then rinsed with water and ethanol. A layer of 20-nm-sized anatase TiO2 nanoparticles for the transparent nanocrystalline layer was first coated onto the FTO glass plates by the doctor-blade method. After drying the film at 120 °C, another layer of 400-nm-sized anatase TiO2 nanoparticles was then deposited as the light scattering layer of the DSSC. The resulting working electrode was composed of a 12-μm-thick transparent TiO2 nanoparticle layer (particle size: 20 nm) and a 4-μm-thick TiO2 scattering layer (particle size: 400 nm). The nanoporous TiO2 electrodes were then sequentially heated at 150 °C for 10 min, at 300 °C for 10 min, at 400 °C for 10 min, and finally, at 500 °C for 30 min. After cooling, the nanoporous TiO2 electrodes were immersed into a acetonitrile–tert-butanol mixture (1:1) solution containing SSD2 organic dye (0.5 mM) with deoxycholic acid (0.5 mM, DCA) as a co-adsorbent at room temperature for 24 h. Counter-electrodes of the DSSC were prepared by depositing 40-nm-thick Pt films on the FTO glass plates by e-beam evaporation. The dye-adsorbed TiO2 working electrode and a counter electrode were then assembled into a sealed DSSC cell with a sealant spacer between the two electrode plates. A drop of electrolyte solution [0.6 M 1-butyl-3-methylimidazolium iodide (BMII), 0.05 M LiI, 0.03 M I2, 0.5 M 4-tert-butylpyridine, and 0.1 M guanidinium thiocyanate in a mixture of acetonitrile-valeronitrile (85:15, v/v)] was injected into the cell through a drilled hole. Finally, the hole was sealed using the sealant and a cover glass. An anti-reflection coating film was adhered to the DSSC. A mask with an aperture area of 0.125 cm2 was applied to a testing cell during photocurrent-voltage and incident photon-to-current conversion efficiency measurements.
Photovoltaic characterization of DSSCs
The photocurrent-voltage characteristics of the DSSCs were measured under illumination of AM1.5G solar light from a 300 W xenon lamp solar simulator. The incident light intensity was calibrated at 100 mW cm−2. Photocurrent-voltage curves were obtained by applying an external bias voltage to the cell and measuring the generated photocurrent.Incident monochromatic photon-to-current conversion efficiency (IPCE) spectra were measured by using a 75 W xenon arc lamp as the light source coupled to a monochromator. The IPCE measurements were taken by illuminating monochromatic light on the solar cells (with a wavelength sampling interval of 10 nm from 300 nm to 800 nm) and measuring the short-circuit current of the solar cells. The IPCE measurements were performed with a lock-in amplifier, a low speed chopper, and a bias light source under full computer control.
Results
Synthesis of fluorene and spirobisfluorene core D–A dyes
Scheme 1 depicts the synthesis of two new dye molecules SSD2 and NSD2. The Stille coupling reaction of 2,2′-dibromo-7,7′-bisdiformyl-9,9′-spirobisfluorene with 2-diphenylamino-5-tributylstannanylthiophene, which was prepared in situ by the deprotonation of 2-diphenylaminothiophene with n-BuLi and followed by quenching the lithiated intermediate with Bu3SnCl, gave intermediate compound 1 with an isolated yield of 72%. Finally, the dialdehyde 1 was condensed with cyanoacetic acid to yield the target compound SSD2 in 86% yield via Knöevenagel reaction in the presence of ammonium acetate. The synthesis of dye NSD2 started from the N-arylation of 3,6-dibromo-9-fluorenone using typical Buchwald–Hartwig C–N bond coupling reaction conditions, giving the intermediate fluorenone 2 in 82%. The Knöevenagel reaction of 2 with cyanoacetic acid was accomplished in the presence of strong Lewis acid TiCl4 and followed by the deprotonation with pyridine gave NSD2 in 71%. | ||
| Scheme 1 Synthesis of SSD2 and NSD2 dye molecules. | ||
Optical absorption
The molar extinction coefficient spectra of the investigated molecules are shown in Fig. 2. The optical absorption characteristics are depicted in Table 1. All spectra exhibit a prominent peak between 390 and 500 nm with molar extinction coefficients around 3 and 5 × 104 M−1 cm−1 in the peak regions (Table 1). These bands are attributed to charge-transfer transitions from the amino donor to the dicyano (CN2) or cyanoacrylic acid acceptor moieties. This assumption is supported by the fact that the dyes show negative solvatochromism.16,17 Moreover frontier molecular orbitals obtained through AM1 calculations18 (Fig. 1) show that the highest occupied molecular orbitals (HOMO) of the dyes are localized mostly on the diphenylamino groups, whereas the lowest unoccupied molecular orbitals (LUMO) are localized on the cyanoacrylic acid (SSD1, SSD2 and NSD2) or dicyano (CN2) units. Thus, photoinduced transitions can produce directional charge transfer states, with electron population around the acceptor groups that interacts with solid surface (vide infra). As expected, the absorption of SSD2 is red-shifted by ca. 22 nm compared to that of SSD1 because of the presence of thiophene groups. Thus, SSD2 should anticipate having more efficient light harvesting capability. However, replacing the carboxyl group with a more electron-withdrawing cyano group in CN2 dye produces a red-shift of 68 nm in charge transfer band maximum with respect to SSD1. The dye NSD2, which lacks the spiro structure, is more panchromatic due to the molecular rigidity and efficient electronic interactions between the D and A subunits.19,20 | ||
| Fig. 2 Molar extinction coefficient spectra of the synthesized molecules SSD1, SSD2, CN2, and NSD2, (a–d, respectively). | ||
| Molecule | SSD1 | SSD2 | CN2 | NSD2 |
|---|---|---|---|---|
| λ max (nm)/log ε | 392/4.66 | 414/4.71 | 460/4.63 | 494/4.51 |
FT-IR spectroscopy
Carboxylic acid is mainly used for anchoring of dye molecules at TiO2 surfaces in dye-sensitized solar cells due to its high stability and simple synthesis. Carboxylic or carboxylate groups provide efficient electronic coupling with the Ti(3d) conduction band orbital manifold at TiO2 surfaces.21,22 In the present case, SSD1, SSD2 and NSD2 possess –COOH groups that provide anchoring capability over the TiO2 surface. More importantly, SSD1 and SSD2 hold two carboxylic groups, which enable the formation of bidentate binding states with the oxide semiconductor.13 The anchoring behavior of carboxylate groups on TiO2 surface can be verified by comparing the FT-IR spectra before and after the adsorbing processes.13,23 FT-IR spectra of NSD2 in both KBr and TiO2 (Fig. 3) reveal that the bands at 1684 and 1726 cm−1 assigned to the carboxylic acid group completely disappear as the dyes are adsorbed on the TiO2 surface, providing clear evidence for the anchoring behavior. However, it has been proposed that carboxyl groups in D–π–A sensitizers are not essential as electron acceptors, but serve as anchoring groups of the dyes onto the semiconductor surface.24,25 Nevertheless, D–π–A dyes with electron-withdrawing groups such as –CN were also shown to be efficient in the generation of photoelectric effects.26 In our case, CN2 molecule possesses four cyano groups, whose strong electron accepting effect leads to a red shift in the absorption band (Fig. 2). The interaction of –CN groups with TIO2 surface was analyzed by FT-IR spectroscopy. Fig. 3 shows that the CN stretching band at 2225 cm−1 disappears when the dye is adsorbed over the semiconductor oxide. It is reasonable to speculate that this effect is due to the interaction between the semiconductor and the electron-acceptor moieties on the CN2 dye.27 On the other hand, in dyes containing cyanoacrylic acid CN stretching remains nearly unaltered after dye adsorption on TiO2. This would indicate that the presence of carboxylic groups interferes with CN–TiO2 interaction, probably due to a geometric impediment.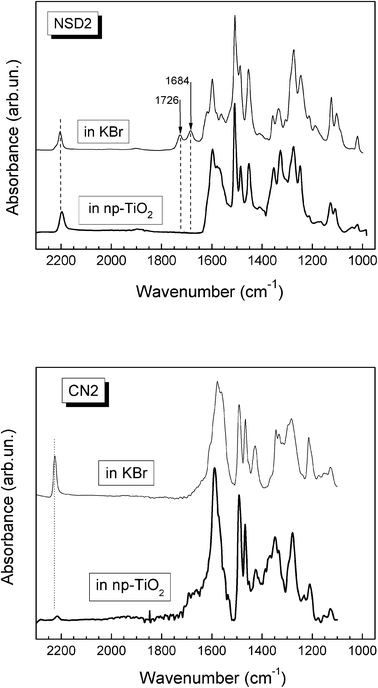 | ||
| Fig. 3 FTIR spectra of NSD2 and CN2 molecules in KBr and np-TiO2. | ||
Surface photovoltage – in-phase and 90° phase-shifted signals of SSD1
Fig. 4 (a) shows the spectra of in-phase and 90° phase-shifted SPV signals for SSD1 molecules adsorbed at np-TiO2. The in-phase SPV signal is positive. Therefore photo-generated electrons are separated preferentially towards the internal interface. Differently to optical absorption in solution, the onset of the SPV signals was at a longer wavelength of about 620 nm (2 eV). The wavelength at the maximum SPV signal around 470 nm (2.65 eV) cannot be related to a peak in optical absorption since the SPV signal is given by the absorption and the photon flux, as well as by the dependence of charge transfer and recombination on light intensity and electronic states involved. The amplitudes of the in-phase and 90° phase-shifted SPV signals are about 1.35 and 1.13 mV, respectively, for SSD1 molecules adsorbed at np-TiO2. The amplitudes of the in-phase and 90° phase-shifted signals are of the same order in the region of the peak, i.e. the phase angle is about −40°. In contrast, the in-phase SPV signal is much smaller than the 90° phase-shifted SPV signal at photon energies close to the onset of the SPV signals, i.e. the phase angle is close to −90°.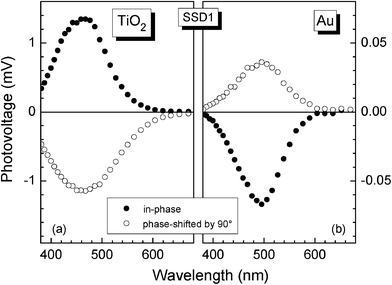 | ||
| Fig. 4 Spectra of the in-phase and 90° phase-shifted surface photovoltage signals for SSD1 molecules adsorbed at np-TiO2 (a) and Au (b) surfaces. | ||
The sign of the in-phase and phase-shifted SPV signals changed for SSD1 molecules adsorbed at Au (Fig. 4b) in comparison to SSD1 molecules adsorbed at np-TiO2, i.e. photo-generated electrons are separated preferentially towards the external surface. The amplitudes of the in-phase and 90° phase-shifted SPV signals are about 67 and 35 μV, respectively, for SSD1 molecules adsorbed at Au. Furthermore, the amplitude of the in-phase SPV signal is larger than the amplitude of the 90° phase-shifted SPV signal by about 2 times. Therefore the phase angle is close to 160°. The ratio between the in-phase and 90° phase-shifted SPV signals was nearly constant over the spectrum, i.e. the phase angle is nearly constant.
Surface photovoltage – spectra of the modulated SPV amplitudes
Fig. 5 (a–d) depicts the spectra of the modulated SPV amplitude for SSD1, SSD2, CN2 and NSD2, respectively, adsorbed at np-TiO2 (stars), ITO (double crosses) and Au (filled circles). The noise level was several μV for the given measurements. The spectra are strongly red-shifted in comparison to the absorption spectra of the molecules in solution. The maximum SPV amplitudes were reached for the SSD2, NSD2 and SSD1 molecules adsorbed at np-TiO2 and amounted to very similar values of 1.4, 1.4 and 1.5 mV, respectively, at around 500 nm (2.5 eV). The SPV amplitude increases exponentially at lower photon energies for SSD2, NSD2 and SSD1 molecules adsorbed at np-TiO2 and the characteristic energy parameters (Et) are 0.11, 0.06 and 0.11 eV, respectively. The SPV amplitude was only 0.18 mV for the CN2 molecules adsorbed at np-TiO2 and the spectrum contains at least two contributions at photon energies below 560 nm (2.2 eV) while the lower Et was 0.19 eV. | ||
| Fig. 5 Spectra of the modulated SPV amplitude for SSD1, SSD2, CN2, and NSD2 (a–d respectively) adsorbed at np-TiO2 (stars), ITO (double crosses) and Au (filled circles). | ||
The maximum SPV amplitudes were 34, 75 and 13 μV for SSD2, SSD1 and CN2 molecules, respectively, adsorbed at Au. No SPV signal could be measured for NSD2 molecules adsorbed at Au.
SPV signals could be obtained for all molecules adsorbed at ITO. The maximum SPV amplitudes were 38, 19, 25 and 71 μV for SSD2, NSD2, SSD1 and CN2 molecules, respectively.
There was one striking difference in the shapes of the spectra between molecules adsorbed at np-TiO2 and molecules adsorbed at Au or ITO. At higher photon energies the SPV amplitude decreased much stronger for the spectra of molecules adsorbed at Au or ITO than of molecules adsorbed at np-TiO2. For example, the SPV amplitude was 19 μV or 1.3 mV at a wavelength of 560 nm (2.2 eV) and 7 μV or 1.3 mV at a wavelength of 460 nm (2.7 eV) for NSD2 adsorbed at ITO or np-TiO2, respectively. For CN2 the situation is even more striking with SPV amplitudes of 71 μV or 0.14 mV at wavelength of 515 nm (2.4 eV) and of 10 μV or 0.15 mV at 415 nm (3.0 eV) for adsorption at ITO or np-TiO2, respectively. Furthermore the spectra of the SPV amplitude can be a bit more broadened to higher photon energies for SSD2 or SSD1 adsorption at ITO in comparison to adsorption of the molecules at Au.
Surface photovoltage – spectra of the phase angle
The phase angle spectra of SSD1, SSD2, CN2, and NSD2 molecules are given in Fig. 6 (a–d) respectively, for adsorption at np-TiO2 (stars), ITO (double crosses) and Au (filled circles). All phase angles ranged between −96° and −38° for the molecules adsorbed at np-TiO2. A strong change of the phase angle has been observed for the SSD2, NSD2 and SSD1 molecules adsorbed at np-TiO2. For example, for SSD2 molecules adsorbed at np-TiO2 the phase angle increased from −84° to −96° with wavelength decreasing from 688 to 620 nm (1.8 to 2.0 eV) and decreased up to −42° at 485 nm (2.55 eV). The phase angle remained nearly constant for wavelengths between 490 and 430 nm (2.55 and 2.9 eV) and increased further with increasing photon energy −57° at 370 nm (3.35 eV) for SSD2 molecules adsorbed at np-TiO2. For CN2 molecules adsorbed at np-TiO2 the qualitative behavior was similar but the maximum change of the phase angle was only from −58° at 565 nm (2.2 eV) to −50 at 485 nm (2.55 eV).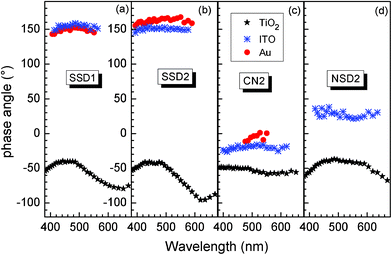 | ||
| Fig. 6 Spectra of phase angle for SSD1, SSD2, CN2, and NSD2 (a–d respectively) adsorbed at np-TiO2 (stars), ITO (double crosses) and Au (filled circles). | ||
The phase angles remained practically constant within one spectrum for the molecules adsorbed at Au or ITO. Scattering in the phase angles is caused by the noise at low SPV signals. For SSD2, SSD1 and CN2 adsorbed at Au the phase angles were about 158…165°, 145…153° and 1…−10°, respectively. For SSD2, NSD2, SSD1 and CN2 adsorbed at ITO the phase angles were about 152…145°, 22–38°, 151…159° and −24…−17°, respectively.
Photovoltaic characterization of SSD2 DSSC
The photovoltaic characteristics of SSD2 as a sensitizer for DSSCs were evaluated using a sandwich DSSC (details of the device preparation and characterization are described in the Materials and methods section). The incident monochromatic photon-to-current conversion efficiency (IPCE) spectrum of the DSSC is shown in Fig. 7. The IPCE spectrum of the device using SSD2 sensitizer exceeds 70% in the spectral region ranging from 342 to 512 nm, (3.62 to 2.42 eV) and reaches a maximum of 87% at 460 nm (2.7 eV). | ||
| Fig. 7 IPCE spectrum of a DSSC based on SSD2 organic dye. | ||
Fig. 8 shows the current–voltage (J–V) curve of the DSSC under standard global AM 1.5G solar irradiation. For the solar cell based on SSD2, short-circuit photocurrent density (JSC) was 10.94 mA cm−2, open-circuit voltage (VOC) was 0.71 V, and the fill factor (ff) was 0.72, yielding an overall conversion efficiency (η) of 5.6%.
 | ||
| Fig. 8 Photocurrent density vs. voltage for a DSSC based on SSD2 organic dye under AM 1.5 G simulated solar light (100 mW cm−2). | ||
Discussion
The main parameters obtained from the SPV spectra (SPVmax – the maximum SPV amplitude, Et – the characteristic energy parameter describing the exponential increase of the SPV amplitude at lower photon energies, and phase angle) are summarized in Table 2 for the SSD1, SSD2, CN2 and NSD2 molecules adsorbed at np-TiO2, Au and ITO surfaces. As it was remarked, the parameter Et is given by the exponential absorption tails which are induced by disorder. Therefore larger values of Et are related to larger disorder. In the following the correlation of SPVmax, Et and phase with specific variations of the molecules, i.e. (i) existence of spiro linkage, (ii) length of the molecular chain, and (iii) nature of the electron acceptor, will be discussed separately for the different surfaces.| molecule | np-TiO2 | Au | ITO | ||||
|---|---|---|---|---|---|---|---|
| SPVmax (mV) | E t (eV) | phase (°) | SPVmax (mV) | phase (°) | SPVmax (mV) | phase (°) | |
| SSD1 | 1.75 | 0.11 | −79…−40 | 0.075 | 152 | 0.025 | 155 |
| SSD2 | 1.64 | 0.11 | −95…−42 | 0.032 | 165 | 0.038 | 150 |
| CN2 | 0.19 | 0.19 | −57…−48 | 0.013 | −4 | 0.071 | −20 |
| NSD2 | 1.4 | 0.06 | −83…−37 | — | − | 0.019 | 30 |
Directed adsorption of spiro compounds at Au surface
SPV signals could be observed only for adsorbed at Au molecules containing spirobisfluorene (SSD1, SSD2, CN2) but not for the molecule without the spiro group (NSD2). Intramolecular charge separation between the donor and acceptor moieties causes modulated SPV signals in layers of molecules adsorbed at Au. The nature of the donor and acceptor moieties is similar for all molecules. Therefore, the absence of SPV signals for NSD2 adsorbed at Au is caused by the geometry of the flat adsorbed molecules avoiding charge separation perpendicular to the surface. The presence of SPV signals for SSD2, SSD1 and CN2 molecules adsorbed at Au gives evidence for directed preferential adsorption of the molecules.Photo-generated electrons are separated preferentially towards the external surface in the modulated SPV signals for SSD1 and SSD2 molecules adsorbed at Au. This means that partial negative charge of the molecule is directed to the Au surface. We assume that charged acceptor moieties of SSD1 and SSD2 molecules are directed to the Au surface. In this case electrons will be separated preferentially towards the donor moieties. The modulated SPV signals are larger for the SSD1 than for SSD2 molecules adsorbed at Au by more than a factor of 2. This may be caused by a lower density of adsorbed SSD2 molecules and/or by a shorter effective charge separation length.
The modulated SPV amplitude was very small for the CN2 molecule adsorbed at Au and photo-generated holes were preferentially separated towards the external surface. This is opposite to the direction of non-modulated charge separation of CN2 molecules adsorbed at Au.6 A certain interpretation is problematic since the direction of modulated charge separation can be driven by occupied or unoccupied, or by unoccupied or occupied, donor and acceptor states, respectively.
Directed adsorption and molecular interactions at ITO surface
A positive in-phase SPV signal appeared for NSD2 adsorbed at ITO and the SPV amplitude increased by more than 5 times for CN2 adsorbed at ITO in comparison to adsorption at Au. ITO is a highly doped metal oxide with an extension of the space charge region less than 1 nm. Injection of photo-generated electrons from dye molecules adsorbed at a FTO (SnO2:F) surface into the space charge region of highly doped FTO has been shown by transient SPV measurements.28 In this work, it has been shown that injection of electrons from adsorbed NSD2 and CN2 molecules into the space charge region of ITO can dominantly contribute to modulated charge separation.The phase angles of the SPV spectra measured for SSD1 and SSD2 adsorbed at ITO did not change significantly in comparison to adsorption at Au. This means that injection of photo-generated electrons does not dominate the modulated charge separation of SSD1 and SSD2 adsorbed at ITO. However, the modulated amplitude of SSD1 adsorbed at ITO decreased by 3 times in comparison to adsorption at Au, i.e. there is an influence of the substrate on the intramolecular charge separation.
Molecular interactions at the TiO2 surface
All spectra showed quite large modulated SPV amplitudes with same signs of the in-phase and 90° phase-shifted signals. This behavior points to one common dominating mechanism of charge separation. Modulated charge separation is dominated by electron injection from adsorbed dye molecules into TiO2 nanoparticles. The SPV spectra of the SSD1, SSD2 and NSD2 molecules showed similarly largest values of SPVmax and largest variations of the phase angles. In contrast, SPVmax and the variation of the phase angle were much smaller for CN2 molecules than for SSD2, NSD2 and SSD1 molecules adsorbed at np-TiO2. At the same time the values of Et can differ strongly for the different molecules. Related differences are caused by specific differences in the interaction between adsorbed molecules and TiO2.In difference to the CN2 molecule the SSD2, NSD2 and SSD1 molecules bind via the carboxylic groups to the TiO2 surface. There is no correlation between Et or SPVmax and the variation of the phase angles. Therefore, the variation of the phase angle can not be simply assigned to a transport process limited by the occupation of traps in the exponential tail states. The strong change of the phase angle is probably related to the influence of electrons accumulated in TiO2 nanoparticles on long-range charge-transfer complexes by changing the state of oxidation of Ti4+ to Ti3+ at the TiO2/molecule interface containing O–Ti–O–C bonds as suspected recently.29 The nature of chemical surface bonds changes in the case of adsorbed CN2 molecules and the state of oxidation of titanium has practically no influence on the spectrum of the phase angle.
SSD2 and SSD1 molecules contain two carboxylic groups whereas NSD2 binds only via one carboxylic group to TiO2. It is very interesting that Et is only 0.06 eV if the molecule binds with only one carboxylic group but Et is 0.11 eV if two carboxylic groups bind to the TiO2 surface. It seems that the density of bidentate surface bonds has a strong influence on disorder in the near surface region of TiO2 nanoparticles.
Modulated SPV amplitude is limited by the injection and back electron transfer processes; if there is no regeneration of the neutral state of the dye within the modulation period, there would be no modulated SPV signal.30 If we assume similar injection kinetics for all dyes, which usually is very fast (ps),31–33 the observed modulated SPV behavior can be ascribed to the difference of distances between charge carriers centers and/or slow back electron transfer kinetics. For CN2, the anchoring interaction with TiO2 surface is weaker than the carboxylic acid containing dyes; thus it is possible that the molecule is laying on the semiconductor surface, which leads to a shorter distance between the photo-generated positive (in dye) and negative (in np-TiO2) charge carriers. In contrast, we attributed the high photoelectric efficiency of SSD# dyes to their particular orthogonal geometry, giving a suitable structural configuration for efficient surface adherence to a semiconductor oxide and subsequent electron transfer. These results indicate the possibility that SSD2 dye is potentially able to be used in solar energy conversion devices with efficiencies similar to the obtained with SSD1,13 but with an extended response in the solar spectrum. On the other hand, NSD2 dye produces the largest photovoltage responding to low photon energy due to the more efficient intramolecular charge transfer structural features. This effect could be due to the dye molecular structural characteristics. In this case, the dye holds two amino groups rigidly bridged through coplanar fluorene to the acceptor, which drives to a more efficient intramolecular photoinduced charge transfer state, due to a better stabilization of positive charge. However, we observed that the adsorbed NSD2 was slowly released from TiO2 surface in contact with common organic solvents used for the electrolyte in the DSSC. This observation will strongly limit the future application of NSD2 as an efficient DSSC dye. On the contrary, SSD1 and SSD2 remained on the electrode surface after several solvent rinses, without dye aggregation at high dye concentration on the electrode surface.
Photovoltaic perfomance of SSD2-DSSC
The SSD2-DSSC IPCE spectrum starts at ∼620 nm (2 eV), in agreement with dye absorption and photovoltaic characteristics, and it is red shifted around 70 nm with respect to SSD1-DSSC IPCE spectrum.13 The 75–90% efficiency reached in the 350–500 nm (3.54–2.48 eV) range indicates that the photoinduced electron injection and electron collection efficiencies of the DSSCs assembled with SSD2 are high and similar to those devices based on organometallic complex dyes,34 which exhibit very good performance in part because their ability to keep the positive charge away from the TiO2 surface, avoiding deleterious back electron transfer.35 In the cases of SSD113 and SSD2 dyes the photogenerated positive charges are located on the diphenylamine units, which are positioned distant from the TiO2 surface (see Fig. 1). In addition, the two carboxylate anchoring groups of SSD# dyes do not only align with the TiO2 coordination sites to provide strong bivalent binding (Fig. 3), but also separate the electrode surface from the photo-oxidized electron donor moieties, diminishing electron/hole recombination. All these characteristics are reflected in the satisfactory short-circuit photocurrent density, open circuit photovoltage and energy conversion efficiency obtained under AM 1.5 solar illumination condition, regardless that SSD2 is not panchromatic as Ru organo-metallic complexes, and only part of the solar spectrum is absorbed, but this fact could be an advantage in the design and construction of semitransparent power-producing windows. Moreover, due to the high molar extinction coefficient of SSD2, it is possible to construct DSSCs with thin nanostructured oxide semiconductor films, and with adequate light-harvesting efficiencies. These are very important factors in the development of solid state DSSCs.Conclusions
A series of spirobisfluorene and non-spiro fluorene-centered D–A systems were synthesized and adsorbed at np-TiO2, Au and ITO surfaces. Short and rigid spirobisfluorene centered D–A systems are well suitable for directed molecule adsorption at Au and ITO surfaces by simple dipping procedure in highly diluted solution. This may become helpful for the development of simplified engineering procedures of functionalization of organic/inorganic interfaces. The interaction between the np-TiO2 substrate and binding carboxylic group has a strong influence on the phase angle of modulated SPV spectra in such a way that charge back transfer can be strongly retarded. The mechanism behind this behavior is not clear yet. Furthermore, the capability of spirobisfluerene D–A dye with extended conjugation for their utilization in DSSCs has been demonstrated. The cells exhibit 5.6% energy conversion efficiency, confirming that the TiO2–dye anchoring characteristic using dyes with carboxylic groups in spiro-configured molecules is a promising route to the design of new DSSC devices. The results we obtained demonstrate the high potential of spiro-configured D–A organic dyes for application in photovoltaic devices.Acknowledgements
We thank Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Universidad Nacional de Río IV (UNRC), National Science Council of Taiwan and DAAD (Germany) for financial support.References
- F. Fungo, M. E. Milanesio, E. N. Durantini, L. Otero and Th. Dittrich, J. Mater. Chem., 2007, 17, 2107 RSC.
- A. Ulman, J. F. Kang, Y. Shnidman, S. Liao, R. Jordan, G. -Y. Choi, J. Zaccaro, A. S. Myerson, M. Rafailovich, J. Sokolov and C. Fleischer, Rev. Mol. Biotechnol., 2000, 74, 175 CrossRef CAS.
- J. Rappich, A. Merson, K. Roodenko, Th. Dittrich, M. Gensch, K. Hinrichs and Y. Shapira, J. Phys. Chem. B, 2006, 110, 1332 CrossRef CAS.
- L. Moreira Gonçalves, V. Bermudez, H. Aguilar Ribeiro and A. Magalhães Mendes, Energy Environ. Sci., 2008, 1, 655–667 Search PubMed.
- D. Wei, Int. J. Mol. Sci., 2010, 11, 1103 CrossRef CAS.
- J. Preat, D. Jacquemin and E. A. Perpète, Energy Environ. Sci., 2010, 3, 891 CAS.
- J. N. Clifford, E. Martínez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS.
- D. H. Lee, M. J. Lee, H. M. Song, B. J. Song, K. D. Seo, M. Pastore, C. Anselmi, S. Fantacci, F. De Angelis, M. K. Nazeeruddin, M. Gräetzel and H. K. Kim, Dyes Pigm., 2011, 91, 192 CrossRef CAS.
- C. -P. Hsieh, H. -P. Lu, C. -L. Chiu, C. -W. Lee, S. -H. Chuang, C. -L. Mai, W. -N. Yen, S. -J. Hsu, E. W. -G. Diau and C. -Y. Yeh, J. Mater. Chem., 2010, 20, 1127 RSC.
- P. Zabel, Th. Dittrich, Y. -L. Liao, C. -Y. Lin, K. -T. Wong, F. Fungo, L. Fernandez and L. Otero, Org. Electron., 2009, 10, 1307 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS.
- D. Heredia, J. Natera, L. Otero, F. Fungo, C. -Y. Lin and K. -T. Wong, Org. Lett., 2010, 12, 12 CrossRef CAS.
- V. Duzhko, V. Yu. Timoshenko, F. Koch and Th. Dittrich, Phys. Rev. B: Condens. Matter, 2001, 64, 075204 CrossRef.
- C. -L. Chiang, C. -F. Shu and C. -T. Chen, Org. Lett., 2005, 7, 3717–3720 CrossRef CAS.
- Y-. L. Liao, C. -Y. Lin, K. -T. Wong, T. -H. Hou and W. -Y. Hung, Org. Lett., 2007, 9, 4511–4514 CrossRef CAS.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS.
- M. P. Balanay, C. V. P. Dipaling, S. H. Lee, D. H. Kim and K. H. Lee, Sol. Energy Mater. Sol. Cells, 2007, 91, 1775–1781 CrossRef CAS.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495 RSC.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS.
- V. Shklover, Y. E. Ovchinnikov, L. S. Braginsky, S. M. Zakeeruddin and M. Grätzel, Chem. Mater., 1998, 10, 2533–2541 CrossRef CAS.
- A. Hagfeldt and M. Grätzel, Molecular photovoltaics, Acc. Chem. Res., 2000, 33, 269–277 CrossRef CAS.
- A. Abbotto, N. Manfredi, C. Marinzi, F. De Angelis, E. Mosconi, J. -H. Yum, Z. Xianxi, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2009, 2, 1094–1101 CAS.
- Y. Ooyama, Y. Shimada, Y. Kagawa, Y. Yamada, I. Imae, K. Komaguchi and Y. Harima, Tetrahedron Lett., 2007, 48, 9167–9170 CrossRef CAS.
- Y. Ooyama, Y. Shimada, A. Ishii, G. Ito, Y. Kagawa, I. Imae, K. Komaguchi and Y. Harima, J. Photochem. Photobiol., A, 2009, 203, 177–185 CrossRef CAS.
- Y. Hao, X. Yang, J. Cong, H. Tian, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 4031–4033 RSC.
- J. Fujisawa, T. Nagatani, Y. Sanehira, J. Nakazaki, S. Uchida, T. Kubo and H. Segawa, Chem. Soc. Jpn. 88th Annual Meeting, 2008, 3L6–05 Search PubMed.
- B. Mahrov, G. Boschloo, A. Hagfeldt, L. Dloczik and Th. Dittrich, Appl. Phys. Lett., 2004, 84, 5455–5457 CrossRef CAS.
- Th. Dittrich, B. Neumann and H. Tributsch, J. Phys. Chem. C, 2007, 111, 2265–2269 CAS.
- C. Sahin, Th. Dittrich, C. Varlikli, S. Icli and M. Ch. Lux-Steiner, Sol. Energy Mater. Sol. Cells, 2010, 94, 686–690 CrossRef CAS.
- G. Benko, J. Kallioinen, J. E. I. Korppi-Tommola, A. P. Yartsev and V. Sundstrom, J. Am. Chem. Soc., 2002, 124, 489–493 CrossRef.
- G. Ramakrishna, D. A. Jose, D. K. Kumar, A. Das, D. K. Palit and H. N. Ghosh, J. Phys. Chem. B, 2005, 109, 15445–15453 CrossRef CAS.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J. -E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Gratzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS.
- S. -J. Wua, C. -Y. Chena, J. -G. Chenb, J. -Y. Lia, Y. -L. Tungc, K. -C. Hob and C. -G. Wu, Dyes Pigm., 2010, 84, 95–101 CrossRef.
- X. Li, J. Gui, H. Yang, W. Wu, F. Li, H. Tian and C. Huang, Inorg. Chim. Acta, 2008, 361, 2835–2840 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00995a/ |
| This journal is © The Royal Society of Chemistry 2012 |