DOI:
10.1039/C2RA20515G
(Paper)
RSC Adv., 2012,
2, 5997-6004
Self-assembly of nanoparticles from cyclotriphosphazenes grafted with hexa-[p-(carbonyl tryptophan ethyl ester) phenoxy)] group
Received
20th March 2012
, Accepted 19th April 2012
First published on 29th May 2012
Abstract
A new cyclotriphosphazene derivative substituted by tryptophan ethyl ester groups, namely, hexa-[p-(carbonyl tryptophan ethyl ester) phenoxy)] cyclotriphosphazene (HEPCP), was synthesized. The nanoparticles self-assembled from the hydrophobic HEPCP were prepared by a facile desolvation method. The nanoparticles were characterized by SEM, TEM, FTIR, XRD, TGA, CLSM and fluorescence spectrum. Based upon the molecular conformation of HEPCP and the spatial arrangements of side substituent groups, a growth mechanism was proposed for the formation of the nanoparticles. The obtained nanoparticles were highly thermally stable and exhibited strong fluorescent emission, and could be potential candidates for drug-loading carriers and tracer drug delivery.
1. Introduction
Polymeric micelles or nanoparticles have been extensively explored for the past decades because of their high potential utility in the biomedical field as drug carriers, drug delivery systems, and surface modifiers.1–6 It is well known that amphiphilic di- or triblock copolymers consisting of both hydrophilic and hydrophobic segments in an appropriate ratio could be self-assembled to form micelles or nanoparticles in aqueous solution.7–10 The hydrophobic blocks of the copolymer form the core of the micelle, while the hydrophilic blocks form the outer shell. A variety of drugs, including genes, proteins, and hydrophobic drugs, such as paclitaxel,11 can be incorporated into the hydrophobic core of the micelles.
Among various polymers, cyclotriphosphazenes as inorganic–organic hybrid polymers are excellent resources to use to develop new polymeric micelles and have attracted an increasing interest in recent years. With a cyclic backbone of alternating phosphorus and nitrogen atoms, cyclotriphosphazene derivatives are biocompatible materials and possess a wide variety of physicochemical as well as tumor targeting properties.12–14 Also, the phosphazene trimer backbone of cyclotriphosphazenes is monodisperse and it is very easy to control the purity and molecular weight of the side groups, which makes the cyclotriphosphazene micelles ideal carriers for drug delivery systems. Numerous cyclotriphosphazene derivatives and polymers have been developed for fabricating micelles or nanoparticles.15–18 As a matter of fact, Lee et al. have succeeded in the synthesis of pure amphiphilic cyclotriphosphazenes by stepwise substitutions of hexachlorocyclotriphosphazene with equimolar hydrophilic PEG and hydrophobic amino acid.17 Most cyclotriphosphazenes micelles were prepared from amphiphilic cyclotriphosphazenes. Polyethylene glycol (PEG) is most widely used as the hydrophilic block, and poly (propylene oxide) and poly(lactic acid) are employed as the hydrophobic block.3,19 However, it was surprisingly found that amphiphilic cyclotriphosphazene grafted on to the oligopeptide ethyl esters instead of amino acids as a hydrophobic side group, made great differences in the physicochemical properties of the micelles.20 Recently, it was discovered by Jun et al.6 and Toti et al.21 that cyclotriphosphazenes grafted with equimolar amounts of oligopeptide as a hydrophobic group and poly(ethylene glycol) as a hydrophilic group were able to form thermodynamically very stable micelles by self-assembly in aqueous solution. The only difference was the use of the strong hydrophobic tri- or hexa-peptide instead of a simple amino acid as a hydrophobic group. The oligopeptide-grafted cyclotriphosphazene micelles were found to offer additional advantages as drug carriers, such as strong solubilizing power for highly hydrophobic drugs based on micellar encapsulation, and easy control of their molecular weight. It was found that micelles formed initially from very hydrophobic (dicyclohexylamine)Pt-cyclotriphosphazene conjugates were subject to further aggregation to large nanoparticles, which exhibited strong stability in phosphate-buffered saline solution and outstanding tumor selectivity.22 These results demonstrated that the substitutions, especially hydrophobic groups grafted on the cyclotriphosphazenes, have a profound effect on the physicochemical properties of micelles. Therefore, it is necessary to develop a new type of cyclophosphazenes grafted with hydrophobic substitutions and explore the effect of substitutions on the micelle properties.
In this paper, a novel cyclotriphosphazene derivative armed with six tryptophan ethyl ester groups, namely, hexa-[p-(carbonyl tryptophan ethyl ester) phenoxy)] cyclotriphosphazene (HEPCP), was synthesized. The self-assembly of nanoparticle from the hydrophobic HEPCP was carried out by a desolvation technique.23 The morphology and structure of the as-obtained nanoparticles were characterized by SEM, TEM and XRD. A possible self-assembly mechanism for the nanoparticle was proposed based on the molecular conformation and the spatial arrangements of substituent groups of HEPCP. The obtained nanoparticles were highly thermal stable and exhibited strong fluorescent emission, which could be potential candidates for drug-loading carriers and tracer drug delivery.
2. Experimental
2.1. Materials
HCPCP and tryptophan ethyl ester hydrochloride were dried at 70 °C under vacuum for 48 h prior to use. Tetrahydrofuran (THF) was distilled from Na under dry N2 purge. Triethylamine was distilled from sodium hydroxide. Thionyl chloride and petroleum ether were used directly.
2.2. Preparation of hexa[p-(carbonyl tryptophan ethyl ester) phenoxy] cyclotriphosphazene (HEPCP)
The target compound of HEPCP was synthesized according to the following steps. Firstly, hexa-[p-(carboxyl) phenoxy] cyclotriphosphazene (N3P3(p-O–C6H4–COOH)6, HCPCP) was prepared according to the literature24 and activated in thionyl chloride, then the acid chloride was reacted with tryptophan ethyl ester (EtTrp) for 48 h in THF under a circumfluence condition, triethylamine was used to neutralize the byproduct hydrochloride. After the reaction, most of the solvent was evaporated under vacuum and the solution was dropped into petroleum ether, the product was separated out and washed by excess deionized water. The straw yellow powder was dried at 70 °C under vacuum for 48 h, the yield was 90%.
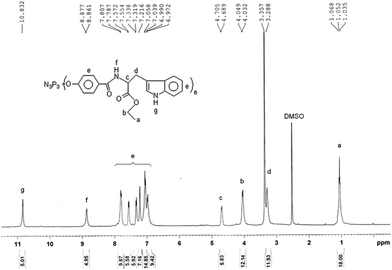
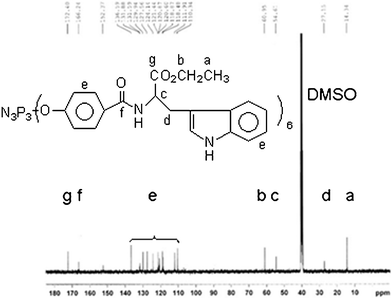
Anal. calc. (%) for C120H114N15O24P3(m/z = 2266.004, Mw + Na): C, 64.25; H, 5.12; N, 9.37. Found C, 64.79; H, 4.99; N, 9.08. IR (KBr): 3412 cm−1 (vs, –N–H), 3052 cm−1 (vs, Ph–H), 2974 cm−1 (vs, –CH3), 1731 cm−1 (vs, CH3CH2O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–), 1645 cm−1 (vs, NH–CO), 1601, 1494 cm−1 (vs, benzene), 1531 cm−1 (δ, trans-, N–H), 1204 cm−1 (–PN–),1097 cm−1 (vs, O–C–O). 31P NMR (DMSO-d6, 161.9 MHz, ppm): δ = 7.47(single). 1H NMR (DMSO-d6, 400 MHz, ppm): δ = 1.05 (18H, –CH3), 4.03 (12H, –OCH2–), 3.29 (12H, –CH2–), 4.69 (6H, –CH–),8.86 (6H, –CONH), 6.97–7.80(36 H, Ar–H), 10.83 (4H, –NH). 13C NMR (DMSO-d6, 100 MHz, ppm): δ = 14.3(–CH3), 27.1(–CH2–), 54.6(–CH–), 60.9(–OCH2–), 172.4(–COO–), 166.2(–CONH–), 110.3–136.5 (–C6H4).
O–), 1645 cm−1 (vs, NH–CO), 1601, 1494 cm−1 (vs, benzene), 1531 cm−1 (δ, trans-, N–H), 1204 cm−1 (–PN–),1097 cm−1 (vs, O–C–O). 31P NMR (DMSO-d6, 161.9 MHz, ppm): δ = 7.47(single). 1H NMR (DMSO-d6, 400 MHz, ppm): δ = 1.05 (18H, –CH3), 4.03 (12H, –OCH2–), 3.29 (12H, –CH2–), 4.69 (6H, –CH–),8.86 (6H, –CONH), 6.97–7.80(36 H, Ar–H), 10.83 (4H, –NH). 13C NMR (DMSO-d6, 100 MHz, ppm): δ = 14.3(–CH3), 27.1(–CH2–), 54.6(–CH–), 60.9(–OCH2–), 172.4(–COO–), 166.2(–CONH–), 110.3–136.5 (–C6H4).
2.3. Preparation of self-assembled nanoparticles
The typical process of self-assembly nanoparticles was as follows. 10 milligrams of HEPCP were firstly dissolved in N,N-dimethylformamide (DMF, 4 mL), which is a good solvent for HEPCP. Subsequently, Milli-Q water as a precipitant for HEPCP was dropped with a peristaltic pump into the HEPCP/DMF solutions at a rate of 1 mL min−1 with vigorous stirring. Micellization occurred, as indicated by the appearance of turbidity in the solution, and then the sample morphology was quenched by the addition of a large excess of water. The resulting colloid solution was putted into a dialysis bag (molecular weight cutoff: 8000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) and dialyzed against Milli-Q water for removal of the organic solvent, and the water was removed by freeze-drying under vacuum for 24 h. In the present studies, the initial sample concentration in DMF was 2.5 mg mL−1.
000 g mol−1) and dialyzed against Milli-Q water for removal of the organic solvent, and the water was removed by freeze-drying under vacuum for 24 h. In the present studies, the initial sample concentration in DMF was 2.5 mg mL−1.
2.4. Characterization
Fourier transform Infrared (FTIR) spectrum was recorded on an Avator360 instrument (Nicolet, USA) using KBr pellets. 1H (400 MHz), 13C (100 MHz) 31P (161.9 MHz)NMR spectra were recorded by using Bruker Avance spectrometer (Switzerland) in the Fourier transform mode with DMSO-d6 as solvent, respectively. Chemical shifts were relative to tetramethylsilane at δ = 0 ppm for protons. The configuration of HCPCP and EtTrp was optimized from the corresponding minimum energy process using the DFT (B3LYP/6-31g*) method in Gaussian 03. Fluorescence spectra were recorded on a LS55 luminescence spectrometer (Perkin-Elmer), and the fluorescent images of the samples were analyzed using confocal laser scanning microscopy 510 Meta (CLSM). The morphology of the nanoparticles was observed with a FEI QuanTa 200 scanning electron microscopy (SEM). The samples for the SEM observations were the powder products obtained by freeze-drying. To minimize sample charging, the dried samples were coated with gold prior to the SEM imaging. The transmission electron microscopy (TEM) was performed with a Mic-H7650 microscope operating at an accelerating voltage of 100 kV. For the preparation of the TEM samples, a 200-mesh copper grid coated with carbon was dipped into the sample solution for 10 min, and then was taken out and put on the filter paper for removing the excess water before observation. Thermogravimetric analysis (TGA) was performed using a Perkin–Elmer TGA Pyris 1 analyzer. The measurement was carried at a heating rate of 10 K min−1 under nitrogen atmosphere. The X-ray diffraction (XRD) pattern was recorded with a D/MAX 2200 diffractometer equipped with a rotating anode and a Cu-Kα source (λ = 0.15406 nm). The particle size measurement was performed on dynamic light scattering (DLS) with angle detection at 90°. The turbidity measurement was carried out on UV-vis spectrophotometer at a wavelength of 740 nm where the absorption was lowest for the polymer and solutions. The solutions at different concentrations were prepared by dissolving HEPCP in DMF, and the turbidity measurement was started at CW = 10% (v.). In defined time intervals, a certain content of Milli-Q water was added dropwise to the investigated solutions, and the sample solution was stirred for 10 min prior to each measurement.
3. Results and discussion
3.1. Synthesis and molecular structure of HEPCP
The synthesis route for the HEPCP compounds was shown in Fig. 1. The cyclotriphosphazene compound HCPCP, as a starting material, was prepared according to the literature24 and firstly activated in thionyl chloride. The object compound appended with six p-(carbonyl tryptophan ethyl ester) phenoxy-containing groups, HEPCP, was obtained by reaction of acid chloride with primary amine of EtTrp. The as-prepared HEPCP was characterized by MS, 31P NMR, 1H NMR, 13C NMR, and FT-IR. All the results were consistent with the predicated structure, as shown in the experimental section.
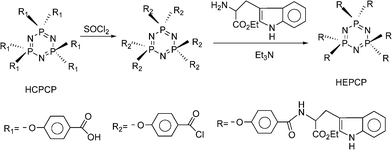 |
| | Fig. 1 The synthesis route of HEPCP. | |
Fig. 2 shows the 31P NMR spectra of HCPCP and HEPCP. The spectra were similar and a singlet with shift positions at 7.73 ppm (HCPCP) and 7.47 ppm (HEPCP), respectively, corresponding to the phosphorous atom with six identical side groups. Compared with HCPCP, the electronic conjugative effect of the amide group in HEPCP is stronger than the carboxyl, so its chemical shift decreased.
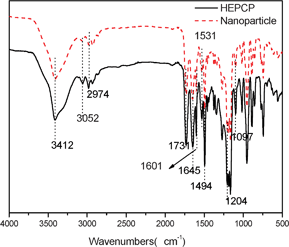
Fig. 3 shows the 1H NMR spectrum of HEPCP. According to the 1H NMR spectrum, the protons were observed at 8.83 ppm (CONH) and 10.83 ppm (NH), and the aromatic protons appeared between 7.13 and 7.75 ppm. It can be concluded from 1H NMR results that the hydroxyl groups were substituted by EtTrp groups. The 13C NMR spectrum (Fig. 4) was also consistent with the structure of HEPCP. In FTIR spectrum (Fig. 11), it was found that infrared spectrum showed evidence for retention of the cyclotriphosphazene ring in HEPCP, with a maximum absorption peak of 1204 cm−1 (v, –PN–). The characteristic peaks of a secondary acylamide at 1645 cm−1 (v, NH–CO), 1531 cm−1 (δ, trans-, N–H), 3412 cm−1 (v, –N–H), and 1731 cm−1 (v, CH3CH2O–CO–) were also detected, which confirmed the substitution of EtTrp groups in HEPCP. Further investigation by mass spectrometry (Fig. S1) shows that the peak of the molecular weight was 2266.004, Mw + Na, corresponding to the side groups grafted with six EtTrp groups in HEPCP. All these above results support the complete substitution of hexa-tryptophanethylester-phenoxy-group on the cyclotriphosphazene ring.
Self-assembly of the nanoparticles from HEPCP was performed through a simple desolvation method and the typical process is schematically illustrated in Fig. 5. Briefly, a buff transparent solution was firstly obtained by dissolving HEPCP in DMF (a good solvent for HEPCP). Then the Milli-Q water as a desolvation was dropped into the above solution, and any change could not be observed for the yellow transparent solution as the water content was low. Until the water was added up to 26 v%, a slight increase in turbidity indicated that molecular aggregations occurred. It can be deduced from the phenomenon that self-assembly of HEPCP was started at a water content of 26 v% because the solubility of HEPCP molecule was decreased, resulting in the aggregation of HEPCP molecules via molecular stacking. Increasing the water content by dropping, a relatively cloudier solution was observed, and cloudy solution was obtained with the water content up to 40 v%. On further increasing water content, the change of the solution was slight, which indicated that the self-assembly process of HEPCP was achieved.
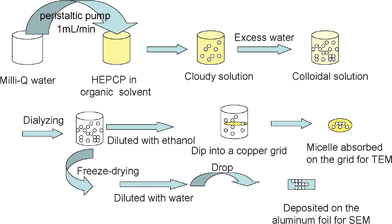 |
| | Fig. 5 A schematic illustration of the self-assembly procedure of HEPCP. | |
Turbidity measurement is a simple and efficient method to monitor the process of self-assembly micellization. Fig. 6 shows the turbidity curves of absorbance as a function of water content at different concentrations. It can be observed that it was difficult to form nanoparticles in dilute solution below the concentration of 1.3 mg mL−1, and the variation of turbidity at different concentrations can be divided into two stages at a certain content. In the first stage, the absorbance of the solution was not changed when the water content was below 26 v%, which indicated that the nanoparticles or aggregations were not formed. Increasing the water content up to 26–40 v%, the absorbance was increased rapidly, which was indicative of the formation of nanoparticles by a self-assembly process. It should be noted that the variation of turbidity was relatively slight as the water content was added up to 50 v%, implying that the self-assembly process for nanoparticles was basically completed. The critical water content (cwc), defined as the water content at which micellization starts, could be obtained by observation of the onset of turbidity of the transparent solutions. According to the results shown in Fig. 5, it can be found that the cwc for the self-assembly of HEPCP to nanoparticles in different concentrations was the same as 26 v%.
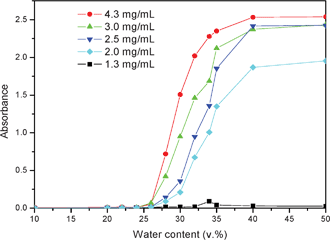 |
| | Fig. 6 Turbidity curves of absorbance as a function of water content at different concentrations. | |
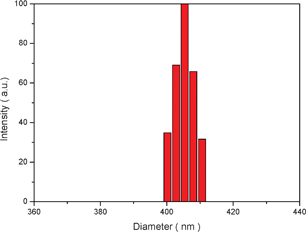
The cloudy solution with the water content of 50 v% was allowed to age for 0.5 h and then quenched by addition of a large excess of water to freeze the morphologies. The resulting colloidal solution was dialyzed in a dialysis bag for 24 h for removal of the DMF, as illustrated in Fig. 5. Then the white aqueous suspensions obtained were diluted for morphology observation. Fig. 7 shows the typical SEM image of the as-prepared nanoparticles. It can be seen that the product was uniform spherical particles with smooth surfaces. Also, it can be observed from the TEM image (Fig. 8) that the products are well-defined solid nanoparticles. The particle size was measured by DLS and Fig. 9 shows the histogram for the particle size distribution of HEPCP nanoparticles. The average particle size is 405 ± 7 nm, indicative of mono-dispersed nanoparticles. These results demonstrate that the uniform stable nanoparticles could be prepared through self-assembly of hydrophobic hexa-tryptophanethylesterphenoxy-grafted cyclotriphosphazene derivatives (HEPCP) by a simple desolvation method. It should be noted that the self-assembly process of HEPCP to nanostructure was different to the conventional solvent exchange process, which was transferring the molecule from a “good” solvent into a “poor” solvent.25 In the present study, when the HEPCP molecule was transferring from DMF (“good” solvent) into water (“poor” solvent), the product was powder rather than nanoparticles.
3.3. Characterization and properties of the nanoparticles
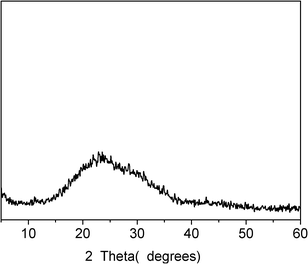
The structure, thermal stability and fluorescent property of the as-prepared nanoparticles were investigated by XRD, DSC, FTIR, TG, CLSM and LS. Fig. 10 shows the XRD result of the nanoparticles. From the XRD pattern, a wide peak at 15–35° could be seen, while no diagnostic diffraction peak can be observed, indicating that the obtained nanoparticles were amorphous structures. The DSC data further shows that there was no obvious melting-temperature for the nanoparticles, which was indicative of the amorphous structure. Fig. 11 shows the FTIR spectra of the HEPCP molecules and the nanoparticles. It can be seen that the characteristic peaks of the nanoparticles are consistent with that of HEPCP molecules, especially, there was no difference at the peak of 3412 cm−1, which indicated that no extra hydrogen bonds were formed during the self-assembly process. The identity of HEPCP molecules and the nanoparticles in FTIR spectrum suggested that the nanoparticles were self-assembled from HEPCP molecules through weak intermolecular interactions.
The thermal stability of HEPCP molecules and the as-prepared nanoparticles was investigated by thermogravimetric analysis (TGA). Fig. 12 gives the TGA and DTG curves of HEPCP molecules and the nanoparticles. It can be seen that HEPCP had two weight loss sections in the whole thermal degradation progress (Fig. 12a). The initial decomposition temperature of HEPCP was at 255 °C, as shown in Fig. 12b, which could be ascribed to the decomposition of the ester bond and followed by the breaking of the acylamide bond.24 The main decomposition appeared at 360 °C due to the decomposition of cyclotriphosphazene and phenyl structures. For the as-prepared nanoparticles, there are also two weight loss sections during the thermal degradation process, but the initial decomposition occurred at 305 °C, an increase of 50 °C, which was indicative of a new interaction force between the HEPCP molecules during the self-assembly process. Furthermore, the maximum decomposition temperature of nanoparticles was at 371 °C, increased by 11 °C. The nanoparticles presented higher thermal stability, which indicates that the self-assembly of HEPCP molecules was driven by the weak noncovalent interactions between side chain substitutions.
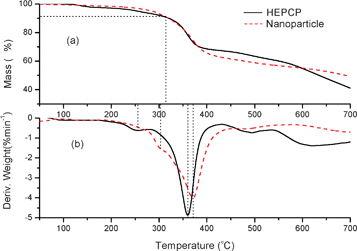 |
| | Fig. 12 TGA (a) and DTG (b) curves of HEPCP powder and the nanoparticles formed from HEPCP. | |
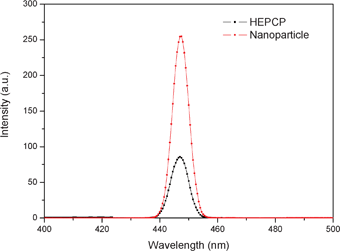
In addition, the fluorescent properties of the nanoparticles was also investigated. Fig. 13 shows the fluorescence spectra of HEPCP and as-prepared nanoparticles. It can be observed that both HEPCP and the nanoparticles exhibited a maximum luminescence peak centered at 447 nm, while the luminescence emission intensity of the nanoparticles was stronger than that of HEPCP. Thus, the self-assembled nanoparticles could be easily imaged with a fluorescence microscope due to the enhanced luminescence emission, as demonstrated in Fig. S2. The strong fluorescent emission of nanoparticles could be contributed to the self-assembly of HEPCP molecules. It has been reported that better arrangement of cyclic phosphazenes through self-assembly could result in stronger fluorescent emission for the assembled nanoparticles.26 During the course of self-assembly of HEPCP, the substitutional groups in the same plane of cyclotriphosphazene could be arranged by molecular stacking, which perhaps could induce higher fluorescence emission of the suprananostructures compared with the molecular phosphazene compounds.
On the basis of the above experimental results, the cyclotriphosphazene compound grafted with six hydrophobic tryptophan ethyl ester groups could be self-assembled to form uniform nanoparticles by simple adding “poor” solvent into its “good” solvent. The driving forces for the self-assembly were investigated based upon the molecular conformation and the spatial arrangements of side tryptophanethylesterphenoxy groups on HEPCP, and a possible mechanism was proposed through the analysis. Firstly, the molecular configurations of HCPCP and EtTrp were optimized from the energy-optimized conformation of the molecule (DFT calculation, B3LYP/6-31g*), respectively. Fig. 14 (a) shows the molecular structure of HCPCP optimized by DFT calculation (B3LYP/6-31g*) using Gaussian 03. It can be seen that the HCPCP features a planar cyclotriphosphazene (PN)3 ring structure that provides a rigid support for the six P-bonded phenoxyl substituents, which is equipped with additional carbonyl groups in axial positions, three anchored at either side of the ring plane. Arrays of three p-carbonylphenoxyl groups that are bonded directly to P centers at one face of the cyclotriphosphazene ring furnish a basket-type arrangement. Thus, the HCPCP which contains three carbonyl groups in a planar six-membered ring offers a unique array of three binding sites for further substituents at either ring face. Fig. 14 (b) shows the molecular conformation of EtTrp optimized by DFT method. It can be observed that the EtTrp molecule contains a rigid indole ring (an aromatic ring connected with a pyrrole ring27) in addition to a flexible alkyl chain, which is exhibited more stability.28 The primary amine group at the alkyl chain could be easily reacted with other active groups, such as the acid chloride obtained by the carboxyl groups of HCPCP.
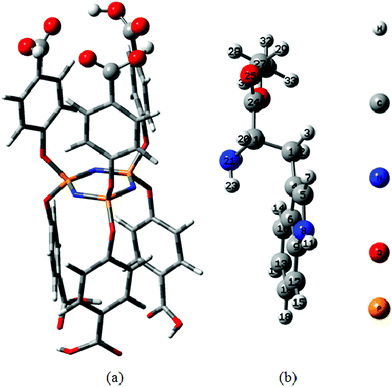 |
| | Fig. 14 Energy-minimized configuration of HCPCP (a) and EtTrp (b) obtained by DFT calculation (B3LYP/6-31g*) using Gaussian 03. | |
According to the results of structure characterisation and the relative cyclotriphosphazene derivative conformations defined by X-ray diffraction methods in the crystalline state,29,30 the molecular model of HEPCP was simulated as shown in Fig. 15. It can be seen that the HEPCP molecule retains the basic configuration features of HCPCP, which is a planar cyclotriphosphazene ring support for six EtTrp substituents. The arrays of three EtTrp groups at one face of the (PN)3 ring also form a basket-type cofacial arrangement. It should be noted that the EtTrp side group is folded structure determined by the characteristic of trans-acylamide bond, which makes the indole ring approach furthest from the aromatic ring of the cyclotriphosphazene arm. It has been recognized that the close contact of the indole ring and phenyl group induced weak nonconventional interactions like π–π interactions, and the importance of aromatic π–π interactions as stabilizing forces has been demonstrated in benzene dimers.27,28 Thus, the folded structure of the EtTrp side group in HEPCP has close contact between the indole ring and aromatic ring, inducing a stable HEPCP molecular conformation. According to the calculated results of benzene dimers,31 the intermolecular potential energy is lowest as the vertical and horizontal separation between indole ring and benzene was fixed at 3.4 Å and 1.6 Å, respectively, causing the conformational stability of the compound. For the HEPCP molecule simulated as shown in Fig. 15, the vertical displacement between the benzene plane in the indole ring and the benzene in the cyclotriphosphazene arm is about 3.6 Å, and the parallel displacement between the two aromatic ring planes is about 1.73 Å, which is close to the calculated value. The contact structure between the indole ring and aromatic ring of HEPCP molecules could induce weak noncovalent interactions, such as π–π and CH⋯π interactions. The characteristic properties of typical π–π interactions, as reported in previous experimental and theoretical studies has been revealed as follows:32–34 (1) the distance between the C–H hydrogen and the center of the π-system is in the range of 2.6–3.0 Å, (2) the distance between the carbon atom of the C–H donor and the center of the π-system is ≤4.5 Å. Thus, the distance between benzene, pyrrole rings in the indole ring and benzene in the cyclotriphosphazene arm is close enough to result in the π–π and CH⋯π interactions.
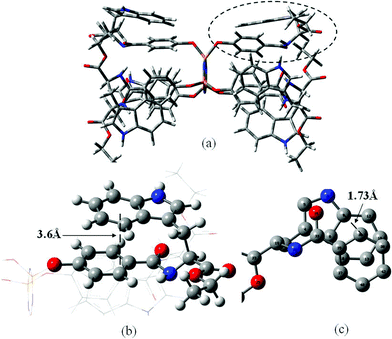 |
| | Fig. 15 The simulated molecular model of HEPCP: (a) configuration of HEPCP molecule, (b) enlarged top view of the molecular structure at circled part, showing the vertical displacement between the benzene plane in indole ring and the benzene in cyclotriphosphazene arm, (c) enlarged side view of the molecular structure at circled part, showing the parallel displacement between the two aromatic ring planes. | |
Based upon the above analysis, we proposed a possible self-assembly mechanism of HEPCP molecules to form nanoparticles. First, the movement of HEPCP existing in pure DMF in the molecular state is free, and then, it was limited when the “poor” solvent was added and the distance between HEPCP molecules was shortened. When the water content was increased up to cwc, a cloudy change demonstrated the distance was shortened enough to lead to the molecular aggregation induced by noncovalent interactions, such as π–π and CH⋯π interactions between intermolecular indole rings. Despite the weakness of each individual interaction, they provide a significant contribution to the formation and stability of supramolecular aggregates due to their extensive nature and multiplicity.27,35 It has been demonstrated in Fig. 5 that the cwc was the same at different concentrations for the formation of aggregates. On further adding Milli-Q water, the other HEPCP molecules continue to be arranged around the seeds via noncovalent interactions, leading to the formation of larger aggregations.
The self-assembly of HEPCP is controlled by the molecular shape and the spatial arrangements of side groups that are able to interact intermolecularly via noncovalent interactions, such as van der Waals forces and π–π interactions.36,37 The intermolecular noncovalent interactions (π–π and CH⋯π interactions) between HEPCP molecules leads to the self-assembly of nanoparticles. The change of chemical bonds did not occur during the self-assembly process, resulting in the consistency of molecular structure of HEPCP molecules and nanoparticles, as revealed by the FTIR results (Fig. 11). But, the thermal stability of the self-assembled nanoparticles was increased, especially for the first decomposition peak, as demonstrated by Fig. 12. This was because the noncovalent interactions mainly occurred in the indole rings of side EtTrp groups. It should be noted that the “poor” solvent of water did not directly participate in the formation of noncovalent interactions, due to the fact that the hydrophilic N–H group was protected by EtTrp groups. It can be reasonably concluded that the self-assembly of HEPCP could proceed in other solvent systems, such as acetone, THF, DMSO and DMAc, et al. Our subsequent experiments have proven this supposition. Such methods provides a facile way for fabricating polymeric self-assemblies through noncovalent interactions.
4. Conclusions
In conclusion, a new cyclotriphosphazene derivative armed with six tryptophan ethyl ester groups, namely, hexa-[p-(carbonyl tryptophan ethyl ester) phenoxy)] cyclotriphosphazene (HEPCP), was synthesized. The HEPCP with six rigid aromatic side groups could be successfully self-assembled to form nanoparticles through a desolvation method. The as-prepared nanoparticles were highly thermal stable materials with uniform and spherical morphology, demonstrating strong fluorescent emission. The self-assembly of HEPCP to nanoparticles is controlled by the intermolecular noncovalent interactions originating from the molecular conformation and the spatial arrangements of substituent tryptophan ethyl ester groups. The hydrophobic, thermally stable nanoparticles with strong fluorescent emission could act as optical drug-loaded biomaterials and tracer drug delivery systems for nanomedical platforms.
Acknowledgements
Financial support was given by the Foundation of State Key Laboratory of Theoretical and Computational Chemistry, the Fundamental Research Funds for the Central Universities (DL10BB26, DL11CB04), the China Postdoctoral Science Foundation (20100480959, 201104406, LBH-10275), the Specialized Research Fund for the Doctoral Program of Higher Education of China (20100062120007).
References
- K. Yu and A. Eisenberg, Macromolecules, 1996, 29, 6359 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169 CrossRef CAS.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2003, 55, 403 CrossRef CAS.
- H. R. Allcock, E. S. Powell and Y. Chang, Macromolecules, 2004, 37, 7163 CrossRef CAS.
- Y. J. Jun, U. S. Toti, H. Y. Kim, J. Y. Yu, B. Jeong, M. J. Jun and Y. S. Sohn, Angew. Chem., Int. Ed., 2006, 45, 6173 CrossRef CAS.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3 CrossRef CAS.
- A. RIsler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95 CrossRef.
- J. X. Zhang, L. Y. Qiu, K. J. Zhu and Y. Jin, Macromol. Rapid Commun., 2004, 25, 1563 CrossRef CAS.
- J. X. Zhang, L. Y. Qiu, X. Li, Y. Jin and K. J. Zhu, Small, 2007, 3, 2081 CrossRef CAS.
- Y. J. Jun, J. H. Min, D. E. Ji, J. H. Yoo, J. H. Kim, H. J. Lee, B. Jeong and Y. S. Sohn, Bioorg. Med. Chem. Lett., 2008, 18, 6410 CrossRef CAS.
- G. Giavaresi, M. Tschon, V. Borsari, J. H. Daly, J. J. Liggat, M. Fini, V. Bonazzi, A. Nicolini, A. Carpi, M. Morra, C. Cassinelli and R. Giardino, Biomed. Pharmacother., 2004, 58, 411 CAS.
- Y. J. Jun, J. I. Kim, M. J. Jun and Y. S.Sohn, J. Inorg. Biochem., 2005, 99, 1593 CrossRef CAS.
- J. Y. Yu, Y. J. Jun, S. H. Jang, H. J. Lee and Y. S. Sohn, J. Inorg. Biochem., 2007, 101, 1931 CrossRef CAS.
- A. Uslu, A. Kılıc and S. Guvenaltın, Polyhedron, 2010, 29, 2516 CrossRef CAS.
- Y. K. Chang, E. S. Powell and H. R. Allcock, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2912 CrossRef CAS.
- S. B. Lee, S. C. Song, J. I. Jin and Y. S. Sohn, J. Am. Chem. Soc., 2000, 122, 8315 CrossRef CAS.
- J. Z. Xu, S. H. Moon, B. Jeong and Y. S. Sohn, Polymer, 2007, 48, 3673 CrossRef CAS.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137 CrossRef CAS.
- S. C. Song, S. B. Lee, B. H. Lee, H. W. Ha, K. T. Lee and Y. S. Sohn, J. Controlled Release, 2003, 90, 303 CrossRef CAS.
- U. S. Toti, S. H. Moon, H. Y. Kim, Y. J. Jun, B. M. Kim, Y. M. Park, B. Jeong and Y. S. Sohn, J. Controlled Release, 2007, 119, 34 CrossRef CAS.
- J. Y. Yu, Y. J. Jun, S. H. Jang, H. J. Lee and Y. S. Sohn, J. Inorg. Biochem., 2007, 101, 1931 CrossRef CAS.
- C. Weber, J. Kreuter and K. Langer, Int. J. Pharm., 2000, 196, 197 CrossRef CAS.
- B. C. Bing and B. Li, Sci. China, Ser. B: Chem., 2009, 52, 2186 CrossRef CAS.
- S. Z. Liu, X. Wu, A. Q. Zhang, J. J. Qiu and C. M. Liu, Langmuir, 2011, 27, 3982 CrossRef CAS.
- F. Wurthner, Chem. Commun., 2004, 1564 RSC.
- S. Tsuzuki, T. Uchimaru and M. Mikami, J. Phys. Chem. A, 2011, 115, 11256 CrossRef CAS.
- L. F. Holroyd and T. V. Mourik, Chem. Phys. Lett., 2007, 442, 42 CrossRef CAS.
- W. C. Marsh and J. Trotter, J. Chem. Soc. A, 1971, 169 RSC.
- V. L. Furer, A. E. Vandyukov, J. P. Majoral, A. M. Caminade and V. I. Kovalenko, Chem. Phys., 2006, 326, 417 CrossRef CAS.
- S. Tsuzuki, T. Uchimaru, K. Matsumura, M. Mikami and K. Tanabe, Chem. Phys. Lett., 2000, 319, 547 CrossRef CAS.
- M. Brandl, M. S. Weiss, A. Jabs, J. Suhnel and R. Hilgenfeld, J. Mol. Biol., 2001, 307, 357 CrossRef CAS.
- J. G. Hill, J. A. Platts and H. Werner, Phys. Chem. Chem. Phys., 2006, 8, 4072 RSC.
- N. Mohan, K. P. Vijaylakshmi, N. Koga and C. H. Suresh, J. Comput. Chem., 2010, 31, 2874 CAS.
- A. Gavezzotti, Acta Crystallogr., Sect. B: Struct. Sci., 2010, B66, 396 CAS.
-
J. M. Lehn, Supramolecular Chemistry. Wiley-VCH, Germany, 1995 Search PubMed.
-
J. W. Steed and J. L. Atwood, Supramolecular Chemistry. Wiley, Chichester, UK, 2000 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.