Composite fluorescent vesicles based on ionic and cationic amphiphilic calix[4]arenes
Paul K.
Eggers
a,
Thomas
Becker
b,
Marissa K.
Melvin
a,
Ramiz A.
Boulos
a,
Eliza
James
a,
Natalie
Morellini
c,
Alan R.
Harvey
d,
Sarah A.
Dunlop
c,
Melinda
Fitzgerald
c,
Keith A.
Stubbs
e and
Colin L.
Raston
*a
aCentre for Strategic Nano-Fabrication, The University of Western Australia, 35 Stirling Hwy, Crawley 6009, Australia. E-mail: colin.raston@uwa.edu.au; Fax: +618 6488 3045; Tel: +618 3045 8683
bNanochemistry Research Institute, Curtin University, Kent Street, Bentley, 6102, Australia. E-mail: T.Becker@curtin.edu.au; Fax: +618 9266 4699; Tel: +618 9266 7806
cSchool of Animal Biology, The University of Western Australia, 35 Stirling Hwy, Crawley 6009, Australia. E-mail: sarah.dunlop@uwa.edu.au; Fax: +618 6488 7527; Tel: +618 6488 1403
dSchool of Anatomy and Human Biology, The University of Western Australia, 35 Stirling Hwy, Crawley 6009, Australia. E-mail: alan.harvey@uwa.edu.au
eSchool of Chemistry and Biochemistry, The University of Western Australia, 35 Stirling Hwy, Crawley 6009, Australia. E-mail: keith.stubbs@uwa.edu.au; Fax: +618 6488 7330; Tel: +618 6488 2725
First published on 11th June 2012
Abstract
Amphiphilic calixarenes bearing ionisable phosphonic acid groups or cationic trimethylamine groups attached to the upper rim of calix[4]arenes in the cone conformation, with dodecyl moieties attached to the lower rim, have a high uptake in PC-12 cells, with the calixarenes being localised cytoplasmically. The calixarenes form tightly packed layers, as established using scanning tunnelling microscopy (STM) on atomically flat surfaces, and can form vesicles or micelles. The vesicles have been wrapped in a peptide–glycol coat for enhanced stability.
Introduction
The ability to deliver a multi-component package consisting of drugs, enzymes and possibly genetic material to a specific location is the “Holy Grail” in medicine. Such a system would allow known ratios of enzymes and drugs to be delivered, for regeneration of injury sites as well as for preventing toxic drugs from affecting healthy tissue in the treatment of diseases such as cancer.One possible approach for packaging functional materials is to use amphiphilic phospholipids. The human body uses the self-assembly of amphiphiles to create membranes that separate various components and provide a barrier against external environments.1 In principle, these same compounds in the form of vesicles offer scope to deliver packages2 to specific locations using biotags.
Vesicles readily form through self-assembly, enclosing the contents of the surroundings in which they formed, and can be small enough to be delivered into the body in a variety of ways.2 However, in reality the lipids which the body uses are not stable enough in their vesicle form for long duration drug delivery. The cells within our body reinforce the lipids with various complex protein structures to ensure their structural integrity.3 A range of strategies have been enacted with greater or lesser success in order to increase the stability of lipid vesicles within the body. Such strategies include: (i) high-melting-temperature lipids which result in wax-like, instead of fluid-like, bilayers at body temperature, (ii) the addition of cholesterol which stabilises the standard lipids, (iii) polymerisation of head groups and/or tails to stabilise the vesicles, and (iv) the PEGylation of the head groups to inhibit fusion and multilamellar vesicles which use multiple bilayers in order to reduce the probability of lysis.4 Another strategy which could be used involves a virus capsid which also uses self-assembly associated with repeating protein units to form a rigid shell.5 These shells are highly ordered helical or icosahedral structures.6 The capsid of viruses is extremely stable under a variety of conditions and has proven time and again to be an effective delivery system.7,8 Furthermore, viruses often use a coat or envelope on top of their capsid to assist in delivery. However, the capsid proteins are complex, often immunogenic and are extremely difficult to synthesise and manipulate, and thus are not necessarily suitable for routine enzyme/drug delivery.
Our packaging approach herein is to design a hybrid system that combines the simplicity of the amphiphilic lipids with the stability of the virus capsid. We use positively and negatively charged amphiphilic calix[4]arenes to form a robust bilayer. The symmetry and the opposing charges of the amphiphilic calixarenes reduce the fluidity of the membrane, hence creating structural rigidity, while derivatisation of the calixarene head groups allows the incorporation of a biotag, a fluorescent tag and/or an extra-vesicular matrix framework to be attached without disrupting the integrity of the vesicles. The charged amphiphiles are based on p-phosphonated calix[4]arenes which readily self assemble via interactions between the polar head groups and C18 alkyl chains.9,10 We report the formation of vesicles using these amphiphilic calixarenes and the surface structure resulting from their self-assembly. In addition, we report the toxicology of these compounds, the location of the calixarenes after incubation with PC-12 cells via a fluorescent tag attached to the calixarene, and demonstrate a method to create a tertiary structure on the surface of the vesicle for enhancing their stability.
The amphiphilic calix[4]arenes possess four lower rim C12 alkyl chains, with the incorporation of more than one such surfactant moiety in a single molecule resulting in a greater viscosity and stability of the self assembled vesicles.11,12 Lower rim O-alkyl calix[4]arenes in general can form well defined bilayer structures, including the case where the alkyl moieties are interdigitated.13 The choice of calix[4]arenes rather than the larger ring systems which are also readily accessible, especially for calix[6 and 8]arenes, relates to the ability to lock the calix[4]arenes in a cone conformation with all the alkyl chains on the same side of the plane of the lower rim phenolic O-atoms. This occurs for O-propyl groups and longer alkyl groups with the smaller groups resulting in a conformation change via threading through the annulus of the molecules.14
Results and discussion
Amphiphile design and synthesis
The amphiphilic calix[4]arenes used in this study, 1–4, are shown in Scheme 1.![The amphiphilic calix[4]arenes used in the present study, with R = C12H25.](/image/article/2012/RA/c2ra20491f/c2ra20491f-s1.gif) | ||
| Scheme 1 The amphiphilic calix[4]arenes used in the present study, with R = C12H25. | ||
With calix[4]arenes as the structural unit of the amphiphiles, one out of the four upper rim head groups may be used for attaching a biotag, a fluorescent molecule as in compound 3, or the support point for an extra vesicular matrix. The other head groups can maintain the ability of the calixarene to assemble into bilayers without producing significant defects, i.e. an assembly mechanism similar to the calixarene where all head groups are the same. Incorporation of the azide group as the functional group for the attachment of a fluorescent molecule, as in compound 4, was undertaken because of the bioorthogonal, biocompatibility15 and high yields associated with click chemistry (see below), and the polarity of the azide group maintaining the amphiphilic nature of the calixarene. The choice of benzylic head groups in all the compounds in the present study relates to both the ease of their synthesis and the extra degree of freedom the methylene unit attached to hetero-atoms allows in packing the bilayers of calixarenes.
The two different head groups chosen were trimethylamine, compound 1, and a phosphonate, compound 2, due to their opposite charge and biocompatibility, and thus the lower likelihood of imparting significant toxicity. Calix[4]arenes 1 and 2 and the associated analogues with a fluorescent molecule or an azide group attached, compounds 3 and 4, have O-dodecyl alkyl chains on the lower rim. Targeting such an intermediate length C12 alkyl chain relates to our recent studies on the toxicology of p-phosphonated calix[4]arenes, where the head groups are attached directly to the phenol rings. Longer alkyl chains result in higher toxicity16 and shorter alkyl chains can result in a higher degree of calixarene directed self-assembly; that is the formation of micelles and vesicles is controlled more by the calixarene itself rather than through the interplay of the alkyl chains.
Synthesis of calix[4]arenes 1, 2, 3 and 4
The synthesis of compounds 1–4 is shown in Scheme 1. The synthetic steps up to and including the alkylation have been described previously.16 The formylation reaction, which was the lowest yielding reaction (70% yield), was adapted from the method of Dondoni et al.17 All subsequent reactions were completed in close to quantitative yields. Click chemistry was chosen for in situ modifications as it has been shown to be biocompatible, is highly selective and consistently achieves high yields.15,18–20 The click reaction to attach the fluorescent molecule was monitored by fluorescence spectroscopy showing a 40 nm shift in the fluorescence peak and an increase in the fluorescence intensity after attachment.21Effects of calix[4]arenes 1 and 2 on cell viabilities
In order to establish whether compounds 1 and 2 had any toxicity, cell viability studies using a LIVE/DEAD cell-based assay were performed on rat PC-12 cells. The concentrations assessed were 0.001, 0.01, 0.1, 0.3, 1 and 3 mg mL−1. As can be seen from Fig. 1, 1 has a significant effect on PC-12 cell viability from 0.01 mg mL−1, with 74% of the cells still viable at this concentration; 2 has a noticeable effect from 0.001 mg mL−1, with 78% of the cells still viable at this concentration. Both compounds had an IC50 (50% of cells still viable) of approximately 1 mg mL−1. This level of toxicity could be beneficial for a delivery system targeting cancer cells as long as the delivery system maintains its integrity until it reaches the targeted cells. This then would give dual-action toxicity with both the packaging system as well as the contents of the package being able to kill the cancer cells. Nevertheless, the present study is an analysis of the structure of the packaging system and its plausibility, not an attempt to demonstrate efficacy in drug delivery. Furthermore, we have recently shown that the toxicity of these compounds is related to chain length and that p-phosphonated calix[4]arenes with four lower rim O-octyl groups are less toxic.16 Thus, future work will also be undertaken using a slightly shorter chain which is less toxic. See below for toxicity studies on compound 1 and 2.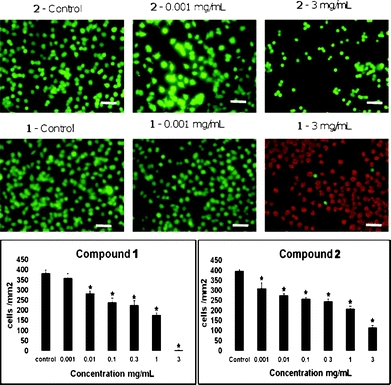 | ||
| Fig. 1 Toxicology data for compounds 1 and 2. The effects of the preparations of 1 and 2 on PC-12 cell viabilities (mean ± standard error of the mean (SEM)) after 24 h in culture and representative images of untreated (control) and treated PC-12 cells (objective 40×). Statistically significant reductions in the viability of the cultures treated with the calixarene preparations compared to the untreated cells are indicated by * (p ≤ 0.05). Scale bar = 100 μm. | ||
Localisation studies
As a follow on to the toxicology, an imaging study on the localisation of the fluorophore calixarene 3 within cells was undertaken. Compound 3 has an excitation wavelength of 350 nm and an emission wavelength of 440 nm (Fig. 2, red squares). The precursor fluorophore to compound 3 has a fluorescence peak at 404 nm, with the shift to 440 nm indicative of a successful ‘click’ reaction.21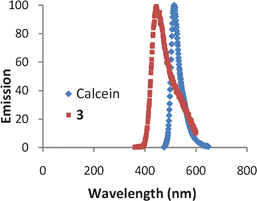 | ||
| Fig. 2 Fluorescence spectra for calcein (excitation 475 nm) and compound 3 (excitation 350 nm). | ||
Compound 3 was added to the media of PC-12 cultures 24 h prior to imaging. PC-12 cells are originally derived from rat pheochromocytoma and are used as a model system for primary neuronal cells.22 These cells are relatively small, have a small amount of cytoplasm, a large nucleus and their doubling time exceeds five days.22 As a control to clarify the intracellular localisation of the calixarene, calcein acetoxymethyl (Calcein-AM) was added 30 min prior to imaging. Calcein-AM is a P-glycoprotein substrate, cell permanent and a non-fluorescent compound.23 Once inside the cell Calcein-AM is rapidly hydrolysed by intracellular esterases to form the strongly green fluorescent calcein anion. The calcein ion is retained in live cells and has an excitation wavelength of 475 nm and an emission wavelength of 525 nm (Fig. 2, blue diamonds). The distinctly different excitation wavelengths of 3 and calcein allow both fluorophores to be located in the cell and excited separately.
Fig. 3a and d show differential interference contrast images of the PC-12 cells 24 h after compound 3 was added. In these images, the cells have extended multiple processes indicating that they are in a healthy state. In an effort to gain insight into the cellular localisation of 3, we undertook photon microscopy experiments. Fig. 3b and e show two photon microscopy images of the same area as Fig. 3a and d but depict the calcein emission wavelength. These images show that calcein is retained predominantly in the cytoplasm. Fig. 3c and f are two photon microscopy images of the same area as Fig. 3b and e but depicting the emission wavelength of 3. It can be seen from Fig. 3c and f that 3 has the same localisation as calcein in that it penetrates the membrane into the cytoplasm and does not appear to enter the nucleus. Furthermore there is also very little evidence of the fluorescence of 3 outside the PC-12 cells indicating close to 100% uptake of the added calixarene.
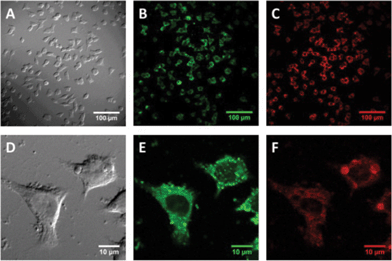 | ||
| Fig. 3 The same field of view of PC-12 cultures was imaged by differential interference contrast (a and d), and two photon fluorescence for calcein, (b and e) or compound 3 (c and f). | ||
Surface organisation
To determine the surface organizational structure and the capability of these bilayers to produce a physical barrier, the vesicles (as identified by dynamic light scattering (DLS) and described later in this paper) were allowed to fuse with a decanethiol self-assembled monolayer (SAM) on an atomically smooth Au(111) surface, forming a biomimetic bilayer.24 This technique is well established and produces consistent reproducible results. The biomimetic bilayer is produced through a thermodynamic driving force which results from the increase in entropy achieved when water is removed from the non-polar distal head groups of the alkanethiols by the non-polar tails of the amphiphilic calixarenes.24 Studies on the assembly of a mixture of compounds 1, 2 and 4 are detailed below.Impedance
Impedance studies were undertaken as an integrated approach to understanding the self assembly of compound 2. Impedance studies give information on the capability of the layer to separate charge and hence will show if a biomimetic layer is formed and if that layer is uniform.If an insulating compound covers a metal surface forming a uniform layer without defects, then that layer can be represented solely as a Helmholtz capacitor in the equivalent circuit. Thus, the equivalent circuit for the electrochemical cell would be a capacitor, representing the uniform coverage of the surface of a metal, and a resistor, representing the resistance of the solution. At low frequencies the phase angle is dominated by a Helmholtz capacitor with values of less than 90° representing defects in the capacitor, at high frequencies the phase angle is dominated by the solution resistance and thus should be close to zero.25–27 If the layer is completely uniform and is thick enough not to have any resistive character, then at 1 Hz the phase angle of the system will be 90°.25,26 Thus, if there is a resistive component such as from defects in the uniform coverage or electron tunnelling through the layer, at 1 Hz the impedance measurement will be less than 90°. As can be seen in Fig. 4, the decanethiol SAM has the lowest phase angle at 85° at 1 Hz followed by the bilayer formed from 2 which has a phase angle of 87°.
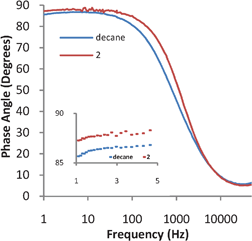 | ||
| Fig. 4 Impedance spectra for a SAM of decanethiol and a biomimetic bilayer of 2 formed on top of the decanethiol SAM. | ||
It is expected that a decanethiol SAM even with uniform coverage would have a lower phase angle than a bilayer due to an increase in the thickness created by the coverage of calixarene lipids. Although an increase in thickness decreases the capacitance, it also decreases the resistance of the SAM (due to electron tunnelling). This increases the capacitive character of the layer resulting in a phase angle closer to 90°. As indicated in Fig. 4, the bilayer made from 2 has an increase in capacitive character relative to the decanethiol SAM. Since the phase angle is greater than the decanethiol SAM, the impedance measurements imply that biomimetic bilayers have formed.24
STM results
The Au(111) on mica substrates were studied using scanning tunnelling microscopy (STM). Hydrogen flamed gold on mica substrates show the triangular terracing expected for atomically flat Au(111), Fig. 5a.28 As shown in both Fig. 5a and b, the images of the decanethiol SAMs depict the expected pitting caused by the formation of alkanethiol SAMs. The STM image in Fig. 5b, as a close up of the decanethiol monolayer shows the typical hexagonal packing of an alkanethiol SAM on gold.28,29 | ||
| Fig. 5 STM images of decanethiol SAM on atomically flat Au(111) for decreasing magnification from (a) to (b). | ||
Fig. 6a and b show STM images of 2 on top of a layer of decanethiol. The closest packing of the SAM has a separation between 0.8 to 0.9 nm with the next closest packing between 1 to 1.1 nm. These distances agree with the model in Fig. 6d and the expected size of a calixarene. The model in Fig. 6d is more chaotic than the STMs of compound 2, however the packing distances are approximately the same. This indicates that each spot in the STMs of compound 2 contain four phosphate groups. The packing is very close to the hexagonal packing of an alkanethiol monolayer which suggests that the underlying layer is influencing the packing.
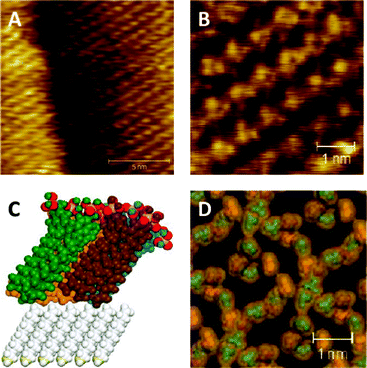 | ||
| Fig. 6 (a and b) STM images of a biomimetic bilayer of 2 formed on top of a SAM of decanethiol on Au(111), and (c and d) a simulation of the same system, different colours represent different calixarenes. | ||
Molecular simulation studies were undertaken to investigate the effect of the length and width of the unit cell on the packing arrangement and energy of the p-phosphonated calix[4]arenes sitting on decanethiols. The optimized structure corresponding to the lowest energy had a unit cell containing four calixarenes with a width and length of 22 Å. As shown in Fig. 6c, this corresponds to the calixarenes being tilted, which maximises packing, optimises the van der Waals interactions and the electrostatic forces between the calixarenes, and interactions between the calixarenes and the decanethiols. The tilt angle is approximately 30°, which is the same angle of the underlying alkane layer. However, the hexagonal packing may not strictly be due to the underlying decanethiol monolayer. In this context we note that previous studies of crystallised calixarenes with O-alkyl chains have also shown a hexagonal lattice with a similar tilt with respect to the plane of the bilayer.30,31
In relation to the structure of the bilayers, the STM images and impedance results show that 2 can self-assemble into a highly ordered tight packed layer, completely covering the substrate.
Vesicles
Compounds 1 and 2 were designed to form vesicles when mixed and this has been confirmed experimentally using dynamic light scattering (DLS). Compound 2 and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and 2 formed vesicles with a mean hydrodynamic diameter of 107 nm and standard deviations of 5 nm and 4 nm, respectively. This was the expected vesicle size after extruding using a 100 nm pore filter with an Avanti Polar Lipid Mini-Extruder. Thus, these two calixarene systems are predisposed to self-assembling into vesicles. However, compound 1 by itself resulted in 5.4 nm particles with a standard deviation of 0.5 nm. The length of 1 from the tip of the alkane chain to the methyl of the trimethylamine equates to approximately 2.4 nm. This is approximately half the diameter of the micelles formed from 1 at 5.2 nm. Thus, compound 1 by itself spontaneously self-assembles into micelles in preference to vesicles. Interestingly, shorter chain analogues of compound 2 have also been shown to form micelles by first solubilising the compounds at high pH, and then on reducing the pH, the micelles maintained their integrity to pH < 3.16
:1 mixture of 1 and 2 formed vesicles with a mean hydrodynamic diameter of 107 nm and standard deviations of 5 nm and 4 nm, respectively. This was the expected vesicle size after extruding using a 100 nm pore filter with an Avanti Polar Lipid Mini-Extruder. Thus, these two calixarene systems are predisposed to self-assembling into vesicles. However, compound 1 by itself resulted in 5.4 nm particles with a standard deviation of 0.5 nm. The length of 1 from the tip of the alkane chain to the methyl of the trimethylamine equates to approximately 2.4 nm. This is approximately half the diameter of the micelles formed from 1 at 5.2 nm. Thus, compound 1 by itself spontaneously self-assembles into micelles in preference to vesicles. Interestingly, shorter chain analogues of compound 2 have also been shown to form micelles by first solubilising the compounds at high pH, and then on reducing the pH, the micelles maintained their integrity to pH < 3.16
Extravesicular matrix
As mentioned in the introduction, vesicles containing lipids alone do not have long-term stability within the body. In attempting to increase the stability of the vesicles, we incorporated a pillar compound where one of the four trimethylamine groups of 1 is replaced by an azide, compound 4. In this situation it is envisaged that the three trimethylamine units hold the compound within the bilayer with the azide group available to attach the extravesicular framework. In demonstrating that it is possible to form an extravesicular matrix for coating the vesicles, click chemistry was used to first attach a synthetic peptide consisting of (glycine-glycine-glyine-propargylglycine)n to the outside of the vesicle, and then a 1000n ethyleneglycol was attached onto the peptide coat of the vesicle. The ratio of amphiphilic calixarenes used to make the vesicles was 8:8:1 for 1:2:4, respectively. This ratio was decided upon to ensure that each molecule of 4 was surrounded by compounds 1 and 2. As can be seen from Fig. 7a the initial vesicle had a mean diameter of approximately 70 nm. This is smaller than the diameter of the vesicles reported in the previous section, which is a consequence of using two filters during the extrusion instead of one. As shown in Fig. 7b, after the peptide and the glycol was attached to the vesicle, the peak at 70 nm disappeared and a peak at 150 nm appeared. This is strongly suggestive that the peptide–glycol coat wraps around the vesicle. It should be noted that the peak also tailed toward larger sizes which may indicate that the peptide binds some vesicles together.
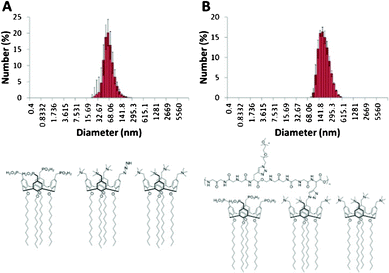 | ||
| Fig. 7 DLS data for (a) vesicles prepared from an 8:8:1 ratio of 1:2:4, and (b) the same system wrapped in a peptide–ethylene glycol coat. | ||
In a phosphate buffered solution coating the vesicles significantly enhanced their stability. Without the coating, vesicles made up of compounds 1 and 2 would start to agglomerate and precipitate out of solution after 48 h. The solution of coated vesicles was stable with no observed precipitation over the course of a month.
Conclusions
Amphiphilic calixarenes bearing ionisable phosphonic acid groups or cationic trimethylamine groups attached to the upper rim of calix[4]arenes locked in the cone conformation, and with dodecyl moieties attached to the lower rim, have an extremely high uptake in the cytoplasm of PC-12 cells. Also noteworthy is that the calixarenes form tightly packed layers, and can assemble into vesicles or micelles. In addition, we have shown that the vesicles can be wrapped in a peptide–glycol coat which should increase their stability in vivo.Experimental
Molecules
The amphiphilic calix[4]arenes used in the self assembly studies, 1–4, are shown in Scheme 1, with their syntheses described below.:ethyl acetate, 7:3) to give a white solid (2.73 g, 70%).
1H NMR (CDCl3, 500 MHz) δ: 0.85 (t, 3J = 7.05 Hz, 12H), 1.27 (m, 72H), 1.87 (m, 8H), 3.33, 4.48 (2d, 2J = 13.8 Hz, 2 × 4H) 3.96 (t, 3J = 7.34 Hz, 8H), 7.13 (s, 8H), 9.53 (s, 4H).
13C NMR (CDCl3, 125.8 MHz) δ: 13.9 (CH3), 22.5 (CH2), 26.1 (CH2), 29.3 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 30.2 (CH2), 30.8 (CH2), 31.8 (CH2), 75.6 (CH2), 130.1 (CH), 131.2 (C), 135.5 (C), 161.8 (C), 191.3 (CH).
1H NMR (CDCl3, 500 MHz) δ: 0.88 (t, 3J = 6.90 Hz, 12H), 1.35 (m, 72H), 1.96 (m, 8H), 3.16, 4.46 (2 d, 2J = 13.1 Hz, 2 × 4H), 3.91 (t, 3J = 6.95 Hz, 8H), 4.34 (s, 8H), 6.70 (s, 8H).
13C NMR (CDCl3, 125.8 MHz) δ: 14.1 (CH3), 22.7 (CH2), 25.6 (CH2), 26.4 (CH2), 29.5 (CH2), 29.8 (CH2), 29.9 (CH2), 30.1 (CH2), 30.4 (CH2), 31.0 (CH2), 32.0 (CH2), 64.5 (CH2), 75.3 (CH2), 127.1 (CH), 134.6 (C), 134.8 (C), 155.9 (C).
1H NMR (CDCl3, 500 MHz) δ: 0.92 (t, 3J = 7.14 Hz, 12H), 1.34 (m, 72H), 1.93 (m, 8H), 3.17, 4.44 (2 d, 2J = 13.3 Hz, 2 × 4H), 3.90 (t, 3J = 7.43 Hz, 8H), 4.32 (s, 8H), 6.68 (s, 8H).
13C NMR (CDCl3, 125.8 MHz) δ: 14.2 (CH3), 22.8 (CH2), 26.3 (CH2), 29.4 (CH2), 29.5 (CH2), 29.7 (CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (CH2), 30.3 (CH2), 30.9 (CH2), 32.0 (CH2), 46.6 (CH2), 75.4 (CH2), 128.6 (CH), 130.9 (C), 135.2 (C), 156.8 (C).
1H NMR (CDCl3, 500 MHz) δ: 0.72 (t, 3J = 7.04 Hz, 12H), 1.20 (m, 72H), 1.84 (m, 8H), 2.79 (m, 36 H), 3.17, 4.27 (2 d, 2J = 12.9 Hz, 2 × 4H), 4.01 (8H), 4.39 (s, 8H), 7.00 (s, 8H).
13C NMR (CDCl3, 125.8 MHz) δ: 13.8 (CH3), 22.4 (CH2), 26.1 (CH2), 29.2 (CH2), 29.6 (CH2), 29.7 (CH2), 29.9 (CH2), 30.3 (CH2), 31.7 (CH2), 51.9 (CH3), 68.1 (CH2), 75.8 (CH2), 121.7 (C), 133.4 (CH), 135.2 (C), 157.8 (C).
TOF MS ES+ (m/z): Expected for C92H160Cl3N4O4+, 1490.1505. Found: 1490.1520.
:methanol, 10:1) to give a white solid (2.67 g, 96%).
1H NMR (CDCl3, 500 MHz) δ: 0.73 (t, 3J = 6.95 Hz, 12H), 1.15 (m, 120H), 1.70 (m, 8H), 2.62 (d, 2J = 21.2 Hz, 8H), 2.91, 4.20 (2 d, 2J = 13.5 Hz, 2 × 4H), 3.66 (t, 3J = 7.25 Hz, 8H), 3.80 (m, 16H), 6.36 (s, 8H).
13C NMR (CDCl3, 125.8 MHz) δ: 13.8 (CH3), 16.0 (d, CH3, 7.5 Hz), 22.4 (CH2), 26.1 (CH2), 29.2 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 29.5 (CH2), 29.7 (CH2), 30.0 (CH2), 31.6 (CH2), 33.0 (CH2, d, 138 Hz), 63.3 (CH2), 74.9 (CH2), 123.8 (C), 129.2 (CH), 134.7 (C), 155.5 (C).
1H NMR (MeOD/CDCl3, 600.1 MHz) δ: 0.79 (t, 3J = 6.90 Hz, 12H), 1.25 (m, 72H), 1.87 (m, 8H), 2.82 (m, 8H), 3.03, 4.30 (2 d, 2J = 12.7 Hz, 2 × 4H), 3.72 (t, 3J = 7.55 Hz, 8H), 6.67 (s, 8H).
13C NMR (MeOD/CDCl3, 150.9 MHz) δ: 13.9 (CH3), 22.6 (CH2), 26.2 (CH2), 29.3 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 29.9 (CH2), 30.3 (CH2), 31.8 (CH2), 32.6 (CH2), 33.5 (CH2), 75.4 (CH2), 125.5 (C), 129.6 (CH), 134.7 (C), 154.9 (C).
TOF MS ES− (m/z): Expected for C80H130O16P42−, 735.4155. Found: 735.4182.
13C NMR (CDCl3, 125.8 MHz) δ: 13.8 (CH3), 22.4 (CH2), 29.2 (CH2), 29.5 (CH2), 29.6 (CH2), 29.7 (CH2), 29.8 (CH2), 29.9 (CH2), 30.0 (CH2), 30.1 (CH2), 30.2 (CH2), 30.3 (CH2), 30.7 (CH2), 31.6 (CH2), 52.0 (m, CH3), 54.2 (m, CH2), 68.3 (m, CH2), 75.8 (m, CH2), 121.7 (m, C), 129.3 (m, CH), 133.4 (m, CH), 135.2 (m, C), 157.8 (m, C).
Fluorescence
Excitation: 350 nm. Emission: 450 nm (fluorescence at 400 nm prior to click reaction)21Vesicle preparation
Three different vesicle compositions were prepared. They were 1 alone, 2 alone and a 1:1 mixture of 1 and 2. The vesicles were formed by dissolving 4 mg, of the amphiphilic calixarenes in 10 mL of a methanol–chloroform mixture. This solution was transferred to a 50 mL round bottom flask (RBF) and dried under vacuum in a rotary evaporator such that the compound was spread evenly on the sides of the RBF. The RBF was then placed under high vacuum for 8 h. 5 mL of 10 mM pH 7.2 phosphate buffer made up to an ionic strength of 154 mM with NaCl was added to the RBF. The RBF was then connected to the rotary evaporator, the water bath was set to 37 °C and the flask was rotated at ambient pressure for 3 h. The resulting dispersion was then extruded 11 times using an Avanti Polar Lipids Mini-Extruder with 100 nm filters.
Cell culture
Rat pheochromocytoma cells (PC-12) were obtained from the Mississippi Medical Centre (Jackson, MS) and grown in a RPMI medium supplemented with 10% horse serum (HS), 5% foetal bovine serum (FBS), 2 mM L-glutamine, 2 mM penicillin-streptomycin, 1 mM MEM sodium pyruvate and 0.1 mM MEM non-essential amino acids (GIBCO®, Invitrogen, Carlsbad, California, USA). Cells were grown on flasks coated with poly-L-lysine (PLL; 10 μg mL−1; Sigma, St Louis, Missouri, USA) and housed in an incubator at 37 °C, 5% CO2. PC-12 cells were seeded at 2 × 105 cells mL−1 in 96 well plates or chamber slides coated with PLL as above. Compounds were assessed by dissolving each calixarene preparation (3 mg mL−1) in DMSO (final maximum concentration of DMSO in solution 1%) by heating in a thermomixer (Eppendorf, Crown Scientific Pty Ltd. Minto, NSW, Australia) at 70 °C, 750 rpm and serially diluted to give final concentrations of 1, 0.3, 0.1, 0.01 and 0.001 mg mL−1. This procedure typically results in micelles forming in solution2 prior to the addition to cells, for example a micelle diameter of 6.4 nm with a standard deviation of 0.9 nm was measured by DLS for compound 1. The effects of each concentration of calixarene on cell viabilities were assessed in triplicate as described.16 Control cultures were incubated in complete media + 1% DMSO.Electrode preparation
The gold ball electrodes were prepared by melting 1 mm diameter 99.99+% gold wire to a 0.4 mm drop using a hydrogen flame. Melting of the wire was done according to the procedure published by Darwish et al.32 Both the gold on mica substrates and the gold ball electrodes were hydrogen flamed immediately prior to use.SAM formation
Immediately after hydrogen flaming the gold ball electrodes and the gold on mica substrates were placed in a 1 mM decanethiol ethanolic solution. After 18 h the substrates were rinsed with ethanol and MilliQ water then immersed in the vesicle containing solution for a further 18 h. The gold ball electrodes and the gold on mica substrates with the same biomimetric bilayers were prepared at the same time and in the same flasks/solutions.Dynamic light scattering (DLS)
The DLS measurements were carried out on a Malvern Instruments Zetasizer Nano S (ZEN1600). The measurements were taken for 30 s and 7 serial 30 s measurements were combined to form a subset, and 100 subsets were used to calculate the vesicle size and standard deviation.Electrochemistry
The impedance studies were done on a Gamry Reference 3000 Potentiostat/Galvanostat/ZRA, using a gold ball working electrode, a platinum counter electrode and a Thermo Orion 900200 Ag/AgCl double junction reference half-cell with epoxy body (sure-flow junction) with filling solutions 900002 (inner) and 900003 (outer) for the reference electrode. The electrolyte for the impedance was 10 mM KCl and the potential was held at 0 V vs. the reference electrode. Impedance measurements were performed with an amplitude of 10 mV.STM Measurements
The STM measurements were performed at room temperature with a PicoPlus scanning probe system (Agilent, California, USA) equipped with a 2 μm STM scanner (model N9501A). STM tips were cut from a 0.25 mm Pt/Ir (80/20) wire immediately prior to the experiment. Typically, a bias voltage of −0.7 V and tunnelling current of 0.9 nA was used. The feedback parameters of the system were adjusted for operation in constant current mode. After installation of the probe and sample the instrument was left for 30 min to equilibrate before images were recorded.Molecular modelling
The starting geometry of decanethiols sitting on gold nanoparticles were obtained from published literature data.33 A hexagonal geometry base with sulphur atoms 4.97 Å apart was built. Decanethiols were attached to the sulphur atoms on one side and gold particles attached on the other side. Following construction, the structure was cleaned to correct for unorthodox bond lengths and angles. The angle between the decanethiols chains and the normal was then reduced to 30° to match the experimental value.The crystal structure of p-phosphonated calix[4]arene was imported from CCDC and used as the basis for the calix[4]arene structure used in the modelling. Dodecyl chains were attached to the lower rim of the calix[4]arene structure. The unit cell was set to a Å by a Å by 59.7 Å where a took on values from 20–28 to ascertain the effect of the calix[4]arene density on the total energy and morphology of the system post minimisation.
Acknowledgements
Support of this study by the Australian Research Council is acknowledged, along with the facilities, scientific, and technical assistance, in particular from Dr Lindsay Byrne, of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, which is funded by The University of Western Australia, State and Commonwealth Governments. M.F. is supported by the Neurotrauma Research Program of Western Australia and S.A.D. by the NHMRC (APP1002347). We would like to thank Anja Werner and The Corrosion Centre for Education, Research & Technology at Curtin University for their help with the impedance measurements and Cameron Evans who assisted in obtaining the confocal microscope images.References
- M. S. Bretscher, Science, 1973, 181, 622–629 CAS.
- P. Walde and S. Ichikawa, Biomol. Eng., 2001, 18, 143–177 CrossRef CAS.
- V. Bennett and A. J. Baines, Physiological Reviews, 2001, 81, 1353–1392 CAS.
- J. J. Moon, H. Suh, A. Bershteyn, M. T. Stephan, H. Liu, B. Huang, M. Sohail, S. Luo, S. Ho Um, H. Khant, J. T. Goodwin, J. Ramos, W. Chiu and D. J. Irvine, Nat. Mater., 2011, 10, 243–251 CrossRef CAS.
- F. L. Homa and J. C. Brown, Rev. Med. Virol., 1997, 7, 107–122 CrossRef CAS.
- G. Vernizzi and M. Olvera de la Cruz, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 18382–18386 CrossRef CAS.
- Y. Ren, S. M. Wong and L.-Y. Lim, Bioconjugate Chem., 2007, 18, 836–843 CrossRef CAS.
- C. R. Parrish, Curr. Top. Microbiol. Immunol., 2010, 343, 149–176 CrossRef CAS.
- A. D. Martin, R. A. Boulos, L. J. Hubble, K. J. Hartlieb and C. L. Raston, Chem. Commun., 2011, 47, 7353–7355 RSC.
- A. D. Martin, R. A. Boulos, K. A. Stubbs and C. L. Raston, Chem. Commun., 2011, 47, 7329–7331 RSC.
- F. M. Menger and J. S. Keiper, Angew. Chem., Int. Ed., 2000, 39, 1906–1920 CrossRef.
- Z. Raoul, Adv. Colloid Interface Sci., 2002, 97, 205–253 CrossRef.
- A. D. Martin, A. N. Sobolev and C. L. Raston, CrystEngComm, 2010, 12, 2666–2668 RSC.
- A. Ikeda and S. Shinkai, Chem. Rev., 1997, 97, 1713–1734 CrossRef CAS.
- C. Besanceney-Webler, H. Jiang, T. Zheng, L. Feng, D. Soriano del Amo, W. Wang, L. M. Klivansky, F. L. Marlow, Y. Liu and P. Wu, Angew. Chem., Int. Ed., 2011, 50, 8051–8056 CrossRef CAS.
- A. D. Martin, E. Houlihan, N. Morellini, P. K. Eggers, E. James, K. A. Stubbs, A. R. Harvey, M. Fitzgerald, C. L. Raston and S. A. Dunlop, ChemPlusChem, 2012, 77, 308–313 CrossRef CAS.
- A. Dondoni, A. Marra, M.-C. Scherrmann, A. Casnati, F. Sansone and R. Ungaro, Chem.–Eur. J., 1997, 3, 1774–1782 CrossRef CAS.
- L. V. Lee, M. L. Mitchell, S.-J. Huang, V. V. Fokin, K. B. Sharpless and C.-H. Wong, J. Am. Chem. Soc., 2003, 125, 9588–9589 CrossRef CAS.
- X.-L. Sun, C. L. Stabler, C. S. Cazalis and E. L. Chaikof, Bioconjugate Chem., 2005, 17, 52–57 CrossRef.
- J. L. Brennan, N. S. Hatzakis, T. R. Tshikhudo, V. Razumas, S. Patkar, J. Vind, A. Svendsen, R. J. M. Nolte, A. E. Rowan and M. Brust, Bioconjugate Chem., 2006, 17, 1373–1375 CrossRef CAS.
- M. Sawa, T.-L. Hsu, T. Itoh, M. Sugiyama, S. R. Hanson, P. K. Vogt and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12371–12376 CrossRef CAS.
- K. Kelly-Spratt, QIAGENNews, 1998, 3–4 Search PubMed.
- E. H. Kerns and L. Di, Drug like properties: concepts, structure design and methods: from ADME to toxicity optimization, Academic Press, 2008 Search PubMed.
- A. L. Plant, Langmuir, 1993, 9, 2764–2767 CrossRef CAS.
- E. Boubour and R. B. Lennox, Langmuir, 2000, 16, 7464–7470 CrossRef CAS.
- E. Boubour and R. B. Lennox, J. Phys. Chem. B, 2000, 104, 9004–9010 CrossRef CAS.
- W. Wang, S. Zhang, P. Chinwangso, R. C. Advincula and T. R. Lee, J. Phys. Chem. C, 2009, 113, 3717–3725 CAS.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- P. K. Eggers, H. M. Zareie, M. N. Paddon-Row and J. J. Gooding, Langmuir, 2009, 25, 11090–11096 CrossRef CAS.
- T. E. Clark, M. Makha, J. J. McKinnon, A. N. Sobolev, M. A. Spackman and C. L. Raston, CrystEngComm, 2007, 9, 566–569 RSC.
- T. E. Clark, M. Makha, C. L. Raston and A. N. Sobolev, CrystEngComm, 2006, 8, 707–711 RSC.
- N. A. Darwish, P. K. Eggers, W. Yang, M. N. Paddon-Row and J. J. Gooding, Nanoscience and Nanotechnology (ICONN), 2010 International Conference on, 2010, pp. 302–305 Search PubMed.
- H. Schönherr and G. J. Vancso, Langmuir, 1999, 15, 5541–5546 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |