Ultrafast photoexcitation dynamics of π-conjugated bodipy-anthracene-radical triad system†
Katsuichi
Kanemoto
*a,
Atsushi
Fukunaga
a,
Motoaki
Yasui
a,
Daisuke
Kosumi
a,
Hideki
Hashimoto
a,
Hirotaka
Tamekuni
b,
Yuichi
Kawahara
b,
Yohei
Takemoto
b,
Jun
Takeuchi
c,
Yozo
Miura
c and
Yoshio
Teki
*b
aCREST/JST and Department of Physics, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: kkane@sci.osaka-cu.ac.jp
bDivision of Molecular Material Science, Graduate School of Science, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: teki@sci.osaka-cu.ac.jp
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka City University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 17th May 2012
Abstract
Ultrafast photoexcitation dynamics of the bodipy-anthracene-radical triad of the photoexcited quartet state and its control compounds were investigated by fs-laser spectroscopy. The fs-spectroscopy successfully unveiled complicated dynamical processes consisting of electron-transfer, energy-transfer, and charge separated ion-pair generation. Based on the results, a pathway leading to the high-spin state generation is discussed.
Spin manipulation in organic molecular materials offers a new and expansive interest in molecular magnetism1–3 and nascent molecular spintronics,4–6 from both the fundamental and technological points of view. In spin manipulation, the photo-control of magnetism7 and/or conducting property is a challenging topic and has attracted much interest. For realizing photo-controlled molecular spin devices, fundamental processes such as intramolecular spin alignment, electron transfer (ET), energy transfer (EnT), and spin dynamics need to be thoroughly examined.7 To study such processes, a triad system with a radical moiety is ideal and has indeed been employed for the research.8–10 Particularly, the system is now attracting much attention as it can be used for studying quantum teleportation.11
As a model compound to study the fundamental processes, we designed and synthesized 1 (A-D-R) (inset of Fig. 1), in which a bodipy (bodipy: 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene) acceptor moiety (A) is covalently linked via a phenyl-anthracene moiety as donor (D) to the verdazyl radical (R). We previously reported evidence of the photo-excited quartet state of 1 with a unique dynamic electron spin polarization (DEP) (Fig. S1†).12 Although similar phenomena have been reported for the triplet state of the photo-system I and II reaction centers13 and of their model systems,14 our report presented the first observation of the photoinduced quartet high-spin state suggested to be generated by a pathway through the charge-separated ion-pair (CSIP) state (A−-D+-R).12 However, there are some issues that should be addressed in order to reveal the mechanism of generation of such photoinduced high-spin states with unique DEP. First, although the existence of the CSIP state was suggested from the unique DEP shown by time-resolved electron spin resonance (TRESR), direct detection of the CSIP state was not obtained. Moreover, although bodipy is well known as an efficient through-bond energy acceptor for anthracene,15 the participation of the CSIP state suggests the possibility of bodipy acting also as an electron acceptor for the anthracene moiety in the photo-excited state. This suggests the coexistence of ET and EnT during the generation process of the high-spin state, and the dynamical processes including them should be clarified.
 | ||
| Fig. 1 (a) Transient absorption spectra (ΔOD) of 1 in butyronitrile upon excitation at 390 nm. The inset indicates the molecular structure of 1. (b) UV-Vis absorption spectrum of 1 at room temperature in butyronitrile. The absorption band in the region of 360–460 nm and the band with a peak at 500 nm are assigned to those of the anthracene and bodipy moieties, respectively. | ||
In this work, ultrafast photoexcitation dynamics of the model compound 1 and its control compounds were investigated by femtosecond (fs) laser spectroscopy in order to address the above issues. The fs-spectroscopy successfully revealed the presence of the CSIP state and unveiled the dynamics and correlation of the ET and EnT processes. Based on the results, a pathway leading to the high-spin state generation is discussed.
The laser system used for the fs pump–probe measurement was composed of a Ti![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :sapphire laser (Spectra Physics, Hurricane-X) with a repetition rate of 1 kHz at 780 nm (∼100 fs pulse duration). The pump pulses at 390 nm were produced from the second harmonic generation (SHG) output of the Ti:sapphire amplifier. A white-light continuum was produced in the sapphire plate using a portion of the amplified 780 nm beam and was then used as probe pulses of variable wavelength. Details of the experimental setup for the pump–probe measurements were described in previous references.16,17 The pump–probe experiments were performed at room temperature for the butyronitrile or toluene solutions of 1 (2.73 × 10−5 mol L−1) that were used after bubbling with N2 gas. Although both solutions exhibited similar trends in photoexcitation dynamics, spectral variations were more striking in the butyronitrile solution probably because of its higher dielectric constant. We thus present the results of the butyronitrile solution to discuss the photoexcitation of 1 in this article. The typical pump–probe spectra in the toluene solution are presented in the supplementary information (see Fig. S2†).
:sapphire laser (Spectra Physics, Hurricane-X) with a repetition rate of 1 kHz at 780 nm (∼100 fs pulse duration). The pump pulses at 390 nm were produced from the second harmonic generation (SHG) output of the Ti:sapphire amplifier. A white-light continuum was produced in the sapphire plate using a portion of the amplified 780 nm beam and was then used as probe pulses of variable wavelength. Details of the experimental setup for the pump–probe measurements were described in previous references.16,17 The pump–probe experiments were performed at room temperature for the butyronitrile or toluene solutions of 1 (2.73 × 10−5 mol L−1) that were used after bubbling with N2 gas. Although both solutions exhibited similar trends in photoexcitation dynamics, spectral variations were more striking in the butyronitrile solution probably because of its higher dielectric constant. We thus present the results of the butyronitrile solution to discuss the photoexcitation of 1 in this article. The typical pump–probe spectra in the toluene solution are presented in the supplementary information (see Fig. S2†).
The typical pump–probe spectra obtained are shown in Fig. 1a, in which the change of the optical density (ΔOD) is plotted for the wavelength of the probe light. The laser excitation at λ = 390 nm corresponds to the selective excitation of the anthracene moiety (Fig. 1b). Positive and negative ΔOD peaks appear at 575 and 505 nm, respectively, immediately after the excitation, as shown in the 0.02 ps spectrum. The positive peak band is typical of a singlet excitation of anthracene,18 and a similar peak was observed from the fs-pump–probe experiment using the same experimental setup for an anthracene derivative prepared as a model donor molecule (see Fig. S3†). The positive band is thus assigned to the transient absorption (TA) band of the excited singlet state of the phenylanthracene moiety (SD*). The peak wavelength of the negative peak is close to the absorption and fluorescence peaks of the bodipy moiety (see, Fig. 1b and S4,† respectively) and is thus assigned as the overlap of stimulated emission (SE) and bleaching bands (hereafter, abbreviated as SE). The TA band decreases after 0.1 ps, whereas the SE band goes on increasing until about 0.7 ps, details of which are shown in Fig. 2. Analyses of the time profiles for the data between 0.1 ps and 0.7 ps revealed that the time constants of the TA band-decrease (575 nm) and the SE band-increase (505 nm) are close to each other (∼0.3 ps). This indicates that rapid intramolecular EnT occurs from the anthracene to the bodipy moieties with this time scale. In particular, the disappearance of the singlet peak in the 2 ps spectrum in Fig. 1 demonstrates that the rapid EnT is very efficient.
 | ||
| Fig. 2 The time profiles of the pump–probe signals in 1 measured at 505, 575 and 600 nm. The inset is an enlarged image of the 600 nm-profile in the long time range. | ||
We now focus on the time profiles in Fig. 2 that turn to a rise after several picoseconds. In addition to the turning behaviors of the TA (575 nm) and SE (505 nm) bands, the positive ΔOD signal at 600 nm, where absorption in 1 is negligible (Fig. 1b), grows after 7 ps, as shown in the inset of Fig. 2. This demonstrates that the tail from the TA band centering at 575 nm disappears and a new electronic state is formed after 7 ps. To clarify the origin, we calculated a difference spectrum between the 100 ps and 7 ps pump–probe spectra, the result of which is shown in Fig. 3. The difference spectrum gives three peaks at 500 nm, 525 nm and 575 nm. Related to this, we present the absorption spectrum of electrochemically oxidized phenyl-anthracene linked to the verdazyl radical (D-R).19 The spectrum has two peaks around 525 nm and 575 nm and its intensity rapidly decreases above 600 nm. These features coincide well with those of the pump–probe difference spectrum, which indicates that the spectral features of the difference spectrum stem from oxidized species except for the 500 nm-peak. It has been shown via the observation of triplet species that, when the phenylanthracene-verdazyl radical (D-R) system is oxidized, the anthracene moiety becomes the cationic form with a triplet state T(D+-R) by the intramolecular ET from D to R after the oxidation of the R moiety.19 This is the result of the more stabilized electronic structure of the triplet state, T(D+-R), than the singlet one, S(D-R+). It is thus concluded that the cationic moiety at anthracene (D+-R) is generated after 7 ps in 1.
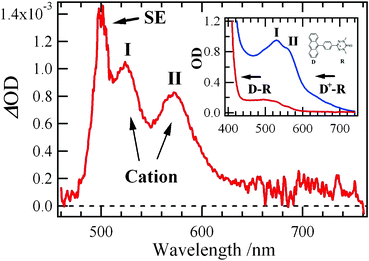 | ||
| Fig. 3 A difference spectrum between the 100 ps and 7 ps pump–probe spectra in 1. The inset indicates the UV-vis absorption spectra of phenyl-anthracene linked to the verdazyl radical (D-R) and its cationic species (D+-R) generated by electrochemical oxidation. The electrochemical oxidation occurs only partly, and hence neutral and cationic states coexist in the spectrum of D+-R. | ||
The 500 nm-peak of the difference spectrum is close to the absorption and fluorescence peaks of bodipy. The appearance of the 500 nm-peak is thus attributed to the reduction of the SE signal. We note that the fluorescence of bodipy decays with a time constant of 2.5 ns, as shown by a time-resolved fluorescence measurement for a model acceptor molecule (Fig. S4†). This indicates that the reduction of the SE signal in the difference spectrum of 1 is much more rapid than the intrinsic SE decay. We thus conclude that the reduction of the SE signal is attributed to the generation of the cationic moiety at anthracene. Namely, the cation-generation and the SE-reduction indicate the occurrence of the ET from the bodipy moiety to the anthracene moiety.
The above finding means that the SE-reduction is caused by the generation of the anionic moiety at the bodipy site. This and the above result indicate the generation of the cationic moiety and hence indicate that the CSIP state (A−-D+-R) is generated after 7 ps of photoexcitation. To confirm these assignments, we examined the fs-dynamics of the D-R system, which are shown in Fig. 4. It shows that the system exhibits no spectral features suggesting the generation of cationic species. This indicates that the cation generation at the anthracene donor moiety in 1 occurs by the presence of the acceptor moiety. These results support our assignments concerning the generation of the CSIP state in 1 by the mechanism proposed above. In contrast, the signal of the anion of the bodipy moiety (A−) was not observed in Fig. 3. It is probably because the anion only gives a very weak absorption around 590 nm,20 which is masked by the stronger cation band II of the donor in Fig. 3.
 | ||
| Fig. 4 Typical fs-pump–probe spectra of phenyl-anthracene linked to the verdazyl radical (D-R) in butyronitrile measured at room temperature. The center peak at 575 nm corresponds to the transient absorption signal from the singlet excitons of the anthracene moiety. This result indicates that the singlet absorption signal simply decays with time and the spectral features of the cationic species are not observed from this system. | ||
Based on the above results, we propose the following mechanisms concerning the photoexcitation dynamics in 1, which are depicted in Fig. 5 and are consistent with the mechanism that we proposed previously.12
 | ||
| Fig. 5 The mechanisms and ultrafast dynamics clarified by this work. The parallel spin configuration decreases the total energy. This is schematically depicted with an unrestricted Hartree–Fock (UHF) picture by lowering the energy level of TD*in the quartet state. | ||
(a) The starting process is the EnT from the photoexcited D to the A with a time constant of 0.3 ps, which is dominated by a Dexter-type electron exchange mechanism.
(b) The electron in the HOMO of D transfers to the HOMO of A and the CSIP state is generated. The time constant of this ET was determined to be 120 ps from the rise time of the 600 nm-time profile over 7 ps. The fluorescence of A is quenched by its ionization. The CSIP state consists of three unpaired spins.
In the observed photoexcitation dynamics, a signature of the quartet (Q) state was not identified from the CSIP state, although the presence of the Q state was confirmed by the TRESR technique measured at 30 K (Fig. S1†).7 Whereas the difference of the measuring temperature between the fs-transient absorption and TRESR measurements might result in different dynamical processes, the presence of the CSIP state demonstrated here by the fs-transient absorption was also suggested by the TRESR measurements.12 This suggests that the Q state observed in the TRESR measurements would be generated after the formation of the CSIP state. In this case, the primary mechanism of the Q state generation is the unique intersystem crossing (ISC) through the quantum mixed states of the LUMO electron in A−.17 It was shown that the Q state is comprised of the triplet state of the anthracene moiety (TD*) and the doublet state of R.21 Therefore, the ISC occurs via back electron transfer (BET) from A− to D (charge recombination (CR)). This mechanism of the Q state generation suggests that the direct photoexcitation of A, instead of that of D, can also generate the Q state. It was indeed identified,22,23 which confirms the rationality of the mechanism.
The model D-R system exhibited a rapid transient absorption-decay of the excited singlet state of the phenylanthracene (SD*) moiety (Fig. 4). We focus here on a peculiar spectral feature around 470 nm which grows following the decay of the SD* band. This feature is due to the formation of the high spin quartet state or a low-lying doublet state energetically located closely above the quartet state, which is generated by the exchange coupling between TD* and R. This suggests that the 470 nm-feature would stem from TD* although it is red-shifted from the triplet band of anthracene observed around 420–430 nm24 probably due to the slight delocalization of the π-conjugation into the ethynyl-phenyl group. Related to this assignment, we previously reported that the model D-R system generates a high-spin state by photoexcitation.25 We then explained that the high-spin state is generated via an enhanced ISC (EISC) owing to the presence of the R moiety, and a similar EISC was also reported from the viewpoint of the ISC rate by Giacobbe et al. for a series of perylene derivatives covalently linked to a nitroxide radical.8c One might then consider that the EISC competes with the generation of the CSIP state. However, no clear spectral features were obtained around 470 nm from compound 1, as shown in Fig. 1a. This indicates that the generation of the CSIP state occurs prior to ISC due to the presence of the bodipy-acceptor. This is a unique point of our high-spin triad system consisting of donor–acceptor and radical moieties.
In conclusion, the fs-spectroscopic technique demonstrated that the model system of A-D-R gives rise to the ultrafast photoinduced EnT (0.3 ps), which is then followed by the electron transfer (120 ps) leading to the generation of the CSIP state in the BuCN liquid phase. Based on the present results, the Q state with the unique DEP observed in the frozen glass matrix by TRESR was strongly suggested to arise from the CSIP state by the ISC with an electron transfer from A− to D, although the fs-pump–probe experiment in the frozen glass matrix is difficult at the moment. This model advances the understanding of photoexcitation dynamics in molecular high spin systems and is useful to optimize molecular design toward realizations of photo-controlled molecular spin devices.
We acknowledge Mr. Yusuke Kawanaka for providing the purified 4,4-difluoro-1,3,5,7-tetramethyl-8-(p-ethynylphenyl)-4-bora-3a,4a-diaza-s-indacene. Y. T. is grateful to the Grant-in-Aid for Scientific Research on General Research (B) (No. 21350081) from JSPS, Japan.
References
- Proceedings of the 9th International Conference on Molecule Based Magnets, Polyhedron, 2005, 24, 2063–2912 Search PubMed.
- (a) C. Kollmar and O. Kahn, Acc. Chem. Res., 1993, 26, 259 CrossRef CAS; (b) O. Kahn, Molecular Magnetism, VCH, New York, 1993 Search PubMed.
- (a) J. S. Miller, Adv. Mater., 2002, 14, 1105 CrossRef CAS; (b) J. S. Miller and A. J. Epstein, Angew. Chem., 1994, 106, 399 CrossRef CAS; (c) J. S. Miller and A. J. Epstein, Angew. Chem. Int. Ed. Engl., 1994, 33, 385 CrossRef.
- (a) M. Ruben, E. Breuning, J.-M. Lehn, V. Ksenofontov, F. Renz, P. Gütlich and G. B. M. Vaughan, Chem.–Eur. J., 2003, 9, 4422 CrossRef CAS; (b) A. Dei, D. Gatteschi, C. Sangregorio and L. Sorace, Acc. Chem. Res., 2004, 37, 827 CrossRef CAS.
- (a) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS; (b) E. Coronado and A. J. Epstein, Molecular spintronics and quantum computing, J. Mater. Chem., 2009, 19, 1661 RSC.
- M. M. Matsushita, H. Kawakami and T. Sugawara, Phys. Rev. B, 2008, 77, 195208 CrossRef.
- (a) K. Matsuda and M. Irie, J. Am. Chem. Soc., 2001, 123, 9896 CrossRef CAS; (b) H. Matsuzaki, W. Fujita, K. Awaga and H. Okamoto, Phys. Rev. Lett., 2003, 91, 017403 CrossRef CAS.
- (a) Q. Mi, E. T. Chernick, D. W. McCamant, E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2006, 110, 7323 CrossRef CAS; (b) E. T. Chernick, Q. Mi, A. M. Vega, J. V. Lockard, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. B, 2007, 111, 6728–6737 CrossRef CAS; (c) E. M. Giacobbe, Q. Mi, M. T. Colvin, B. Cohen, C. Ramanan, A. M. Scott, S. Yeganeh, T. J. Marks, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 3700 CrossRef CAS; (d) M. T. Colvin, E. M. Giacobbe, B. Cohen, T. Miura, A. M. Scott and M. R. Wasielewski, J. Phys. Chem. A, 2010, 114, 1741 CrossRef CAS.
- (a) K. Ishii, J. Fujiwara, Y. Ohba and S. Yamauchi, J. Am. Chem. Soc., 1996, 118, 13079 CrossRef CAS; (b) J. Fujiwara, Y. Iwasaki, Y. Ohba, S. Yamauchi, N. Koga, S. Karasawa, M. Fuhs, K. Möbius and S. Weber, Appl. Magn. Reson., 2001, 21, 483 CrossRef , and references cited therein; (c) K. Ishii, Y. Hirose and N. Kobayashi, J. Phys. Chem. A, 1999, 103, 1986 CrossRef CAS , and references cited therein.
- (a) M. Asano-Someda, A. van der Est, U. Krüger, D. Stehlik, Y. Kaizu and H. Levanon, J. Phys. Chem. A, 1999, 103, 6704 CrossRef CAS; (b) A. van der Est, M. Asano-Someda, P. Ragogna and Y. Kaizu, J. Phys. Chem. A, 2002, 106, 8531 CrossRef CAS; (c) P. K. Poddutoori, M. Pilkington, A. Alberola, V. Polo, J. E. Warren and A. van der Est, Inorg. Chem., 2010, 49, 3516 CrossRef CAS , and references cited therein.
- (a) K. M. Salikhov, J. H. Golbeck and D. Stehlik, Appl. Magn. Reson., 2007, 31, 237 CrossRef CAS; (b) Y. E. Kandrashkin and K. M. Salikhov, Appl. Magn. Reson., 2010, 37, 549 CrossRef.
- (a) Y. Teki, H. Tamekuni, J. Takeuchi and Y. Miura, Angew. Chem., 2006, 118, 4782 CrossRef; (b) Angew. Chem. Int. Ed., 2006, 45, 4666 Search PubMed; (c) Y. Teki, H. Tamekuni, K. Haruta, J. Takeuchi and Y. Miura, J. Mater. Chem., 2008, 18, 381 RSC.
- H. Levanon and J. R. Norris, Chem. Rev., 1978, 78, 185 CrossRef CAS.
- (a) K. Hasharoni, H. Levanon, S. R. Greenfield, D. J. Gosztola, W. A. Svec and M. R. Wasielewski, J. Am. Chem. Soc., 1995, 117, 8055 CrossRef CAS; (b) K. Hasharoni, H. Levanon, S. R. Greenfield, D. J. Gosztola, W. A. Svec and M. R. Wasielewski, J. Am. Chem. Soc., 1996, 118, 10228 CrossRef CAS.
- C. W. Wan, A. Burghart, J. Chen, F. Bergestroem, L. Johansson, M. F. Wolford, T. G. Kim, M. R. Topp, R. M. Hochstrasser and K. Burgess, Chem.–Eur. J., 2003, 9, 4430 CrossRef CAS , and references cited therein.
- D. Kosumi, T. Kusumoto, R. Fujii, M. Sugisaki, Y. Iinuma, N. Oka, Y. Takaesu, T. Taira, M. Iha, H. A. Frank and H. Hashimoto, Chem. Phys. Lett., 2009, 483, 95 CrossRef CAS.
- D. Kosumi, K. Abe, H. Karasawa, M. Fujiwara, R. J. Cogdell, H. Hashimoto and M. Yoshizawa, Chem. Phys., 2010, 373, 33 CrossRef CAS.
- (a) A. Hartschuh, A. Port and H. C. Wolf, J. Lumin., 2001, 94–95, 441 CrossRef; (b) A. Port and A. Hartschuh, J. Lumin., 2004, 110, 315 CrossRef; (c) I. B. Ramsteiner, A. Hartschuh and H. Port, Chem. Phys. Lett., 2001, 343, 83 CrossRef CAS.
- I. Matsumoto, I. Ciofini, P. P. Lainé and Y. Teki, Chem.–Eur. J., 2009, 15, 11210 CrossRef CAS.
- T. J. Wijesinghe, M. E. El-Khouly, J. D. Blakemore, M. E. Zandler, S. Fukuzumi and F. D'Souza, Chem. Commun., 2010, 46, 3301 RSC.
- Y. Teki and T. Matsumoto, Phys. Chem. Chem. Phys., 2011, 13, 5728 RSC.
- Y. Teki, M. Nakatsuji and Y. Miura, Mol. Phys., 2002, 100, 1385 CrossRef CAS.
- Y. Takemoto and Y. Teki, ChemPhysChem, 2011, 12, 104 CrossRef CAS.
- R. H. Compton, K. T. V. Grattan and T. Morrow, J. Photochem., 1980, 14, 61 CrossRef CAS.
- Y. Teki, S. Miyamoto, M. Nakatsuji and Y. Miura, J. Am. Chem. Soc., 2001, 123, 294 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: ESR spectra, pump–probe spectra of model compounds, and time-resolved photoluminescence spectrum. See DOI: 10.1039/c2ra20473h/ |
| This journal is © The Royal Society of Chemistry 2012 |