Analysis of metallic nanoparticle-DNA assembly formation in bulk solution via localized surface plasmon resonance shift†
Kah Ee
Fong
and
Lin-Yue Lanry
Yung
*
Department of Chemical and Biomolecular Engineering, Faculty of Engineering, National University of Singapore, Singapore 119260. E-mail: cheyly@nus.edu.sg
First published on 16th March 2012
Abstract
Metallic nanoparticles such as gold and silver are known to exhibit localized surface plasmon resonance (LSPR). Being able to form well defined nanoassemblies of metallic nanoparticles in solution phase can produce LSPR coupling and shift, which represents a unique plasmon signature and have been used for the detection of nanoassembly formation. While most of the existing works focus on nanoassembly formation on a substrate surface, here, we investigated the formation of DNA-modified gold nanoparticle (nAu-DNA) nanoassemblies in bulk solution. Subsequently the nanoassemblies were allowed to bind on a glass substrate in order to study correlations among the LSPR wavelength shift, the plasmon color change, and the nanoassembly structure. We observed that the hybridization percentage of the complementary 50 nm nAu increased with rising nAu concentration, longer hybridization time, and longer complementary duplex DNA length. In addition, due to lower scattering yield and smaller surface area from 10 nm nAu, the 50 nm/10 nm hetero-size system displayed limited observable LSPR shift compared to 50 nm/20 nm hetero-size system, which in turn was inferior to the 50 nm/50 nm homo-size system. For the hetero-size systems, reducing the surface density of ssDNA on the 20 nm and 10 nm nAu also significantly reduced the hybridization percentages. Overall, this study allows us to understand how different experimental parameters can impact the assembly of nAu-DNA probes, particularly the limitation in using smaller size nAu (10 nm) for LSPR study.
1. Introduction
In recent years, there has been increasing research on the unique physical and chemical properties of metallic nanoparticles because of their attractive fundamental and technological applications. For example, an incident electromagnetic field on a metallic nanoparticle excites the surface conduction electrons collectively, and results in a phenomenon generally known as localized surface plasmon resonance (LSPR).1,2 Two neighboring metallic nanoparticles have been demonstrated to exhibit longitudinal LSPR delocalization that changes exponentially with the interparticle separation distance.3–6 The corresponding LSPR cross-sectional enhancement has been used routinely as plasmon ruler.7–9 The LSPR frequency shift has been used in chemical- and bio-sensing;10–14 electromagnetic field enhancement15 in Surface-Enhanced Resonant Raman Scattering (SERRS);16–18 and near-field surface plasmon coupling in low-loss nano-waveguide to transmit electromagnetic energy.19–21In the pursuit to discriminate single nucleotide polymorphism (SNP), there have been extensive studies on LSPR-aided detection using gold nanoparticles22–29 that are chemically stable and typically exhibit strong localized surface plasmon (LSPs) in the visible wavelength region. The commonly employed design mainly relies on two sets of DNA-modified nanoparticle probes, which are complementary to a portion of the non-labeled target DNA sequences (three-component assays).
On the other hand, two-component assays, which involve two sets of complementary DNA-modified nanoparticle probes, have received less attention. Most of the existing research that utilized this assay concentrated on the search for DNA-binding molecules,30–32 and the investigation of non-Watson–Crick hybridization33e.g. G-quadruplex interaction.34 Because of the faster hybridization kinetics than three-component assays,35 the two-component assays were also used to design conjugation procedures for reducing conjugation time,36 improving conjugate stability,37 controlling surface DNA density,38–40 mediating the number and space of the bound DNA,41 detecting label-free DNA by displacement,42 and engineering nanoparticle-based superlattices.43 Common readout platforms in detecting the hybridization of DNA-modified nanoparticle probes include the extinction-based colorimetric assay, electrophoresis, electron microscopy, and UV-visible spectroscopy. Optical Rayleigh scattering under dark field microscopy can be another potential readout platform,44 and a few research groups have used the scattering-based two-component assays to detect single DNA enzyme cleavage event8 and to study the LSPR coupling behavior of dimeric or higher-order metal nanoparticles.45,46
There are two commonly-used schemes constituting the two-component assays: heterogeneous (surface-bound) and homogeneous (in bulk solution) hybridization. For the former, surface bound particles enable real-time observation of the individual hybridization, but are less representative of the ensemble event.46 For the latter, ensemble hybridization provides stronger statistical significance among different populations, at the cost of sacrificing individual binding studies.33 In addition, the kinetics and thermodynamics of duplex formed with a surface-bound nanoparticle deviate from the free duplex formed in bulk solution due to additional interfacial constraints experienced by the hybridizing strand. Moreover, homogeneous hybridization enables annealing to remove any possible secondary structure in the DNA.
While most of the existing studies have focused on the heterogeneous (surface-bound) hybridization system, herein, our work opted for the homogenous (in bulk solution) system using DNA-modified gold nanoparticles (nAu). After the hybridization in solution, the hybridized particles were allowed to adsorb on a glass for easy quantification of the hybridization events. The nanoassemblies formed were visualized via Rayleigh scattering with dark field illumination. The abundance of data (i.e. individual hybridized particles on glass) allows us to provide a more statistically representative hybridization event of the ensemble. The objective of this study is to look at the ensemble hybridization behavior of different populations, which can reflect the whole population more accurately than focusing only on a limited number of individual particles. nAu are preferred over silver nanoparticles because they are chemically more stable, possess better shape controllability, and they form stronger bonds with thiol functional groups enabling easier manipulation. The effect of nAu concentration, the duplex length, the size of nAu, and the DNA surface density in hybridization were studied separately and quantified by LSPR shift. The hybridization development between these quasi-immobilized DNA reiterates the generally agreed hybridization mechanism, while highlighting the importance of surface probe densities on the hybridization efficiency and kinetics. Importantly, we demonstrated standalone small size nanoparticles, with their low light scattering yield, are less useful in probing the nanoassembly formation via dark field scattering.
2. Experimental
2.1 Materials
Gold(III) chloride trihydrate, trisodium citrate dehydrate, tannic acid, silver nitrate, 4,4′-(phenylphosphinidene)-bis-(benzenesulfonic acid) (PPBS), sodium dodecyl sulfate (SDS), sodium chloride, magnesium chloride hexahydrate were purchased from Sigma-Aldrich. Synthetic single-stranded DNA (ssDNA) strands (modified with the terminal 5′ thiol group) were purchased from Proligo. Tris buffer (pH 8.0) was purchased from 1st Base. Milli-Q water with resistance >18 MΩ/cm was used throughout the experiments.2.2 Synthesis and characterization of nAu
Prior to nAu synthesis, all the glassware and magnetic stir bars were cleaned with aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v HCl (37%): HNO3 (65%)) oxidizing solutions and then rinsed thoroughly with water before use. 10 nm nAu seeds were prepared by reducing 0.01% of gold salt solution with 0.04% of citrate and 0.001% of tannic acid in total volume of 20 mL at 70 °C. When the reacting solution turned to a clear crimson, the temperature was brought to 110 °C, boiled for 5 min, and then cooled slowly in air to room temperature. These seeds were used to prepare 50 nm nAu in a 2-stage growth process. Briefly, in the 1st stage, 50 mL of 0.1695 mM HAuCl4, refluxed at 120 °C with stirring was added with 0.5 mL of the seed solution and was left to homogenize for 30 s. 1% citrate was then injected quickly into the solution at ratio 0.9/1 of citrate to HAuCl4 and stirred for 20 min. The solution turned from blackish to purple, and finally red. Subsequently, 0.1% AgNO3 was added in a ratio 1:50 of Ag+ to Au3+ for 10 min under stirring to reshape the polycrystalline nAu into quasi-spherical shape.47,48 In the 2nd stage, another set of HAuCl4 (1.695 mL, 5 mM) and 1% citrate (ratio 0.9/1 of citrate to HAuCl4) was added into the nAu solution formed in the 1st stage for 25 min for further nAu growth. The product was then cooled slowly in air to room temperature, filtered and stored at 4 °C until further use. UV-vis extinction spectra of nAu were determined by a UV-visible spectrophotometer (Cary Varian 50 Bio). TEM characterization was acquired with a transmission electron microscope (JEOL JEM-3010) operating at 300 kV. At least 100 particles were sized from TEM micrographs via graphics software AxioVision 4.8.2 (CarlZeiss).
:1 v/v HCl (37%): HNO3 (65%)) oxidizing solutions and then rinsed thoroughly with water before use. 10 nm nAu seeds were prepared by reducing 0.01% of gold salt solution with 0.04% of citrate and 0.001% of tannic acid in total volume of 20 mL at 70 °C. When the reacting solution turned to a clear crimson, the temperature was brought to 110 °C, boiled for 5 min, and then cooled slowly in air to room temperature. These seeds were used to prepare 50 nm nAu in a 2-stage growth process. Briefly, in the 1st stage, 50 mL of 0.1695 mM HAuCl4, refluxed at 120 °C with stirring was added with 0.5 mL of the seed solution and was left to homogenize for 30 s. 1% citrate was then injected quickly into the solution at ratio 0.9/1 of citrate to HAuCl4 and stirred for 20 min. The solution turned from blackish to purple, and finally red. Subsequently, 0.1% AgNO3 was added in a ratio 1:50 of Ag+ to Au3+ for 10 min under stirring to reshape the polycrystalline nAu into quasi-spherical shape.47,48 In the 2nd stage, another set of HAuCl4 (1.695 mL, 5 mM) and 1% citrate (ratio 0.9/1 of citrate to HAuCl4) was added into the nAu solution formed in the 1st stage for 25 min for further nAu growth. The product was then cooled slowly in air to room temperature, filtered and stored at 4 °C until further use. UV-vis extinction spectra of nAu were determined by a UV-visible spectrophotometer (Cary Varian 50 Bio). TEM characterization was acquired with a transmission electron microscope (JEOL JEM-3010) operating at 300 kV. At least 100 particles were sized from TEM micrographs via graphics software AxioVision 4.8.2 (CarlZeiss).
2.3 Fabrication of nAu-DNA probes
All 33-base ssDNA sequences used in this study carry a thiol group at their 5′ terminals (Table 1). Strand P is the probe sequence to which complementary strands cP27, cP18, and cP14 hybridized to form 27 bp-, 18 bp- and 14 bp-duplexes (bp: base-pair). 50 nm nAu were used in both Scheme 1 and Scheme 2, whereas 20 nm and 10 nm nAu were only used in Scheme 2(a) and 2(b) respectively. Prior to conjugation, nAu were passivated with PPBS, and then ssDNA was incubated with 50 nm and 10 nm nAu at 1000:1 and 400:1 ratio (ssDNA:nAu) respectively. After 2 h incubation, for 50 nm nAu, NaCl concentration was increased slowly to 80 mM over 2 days in 0.01% SDS; whereas for 20 nm and 10 nm nAu, NaCl concentration was increased to 600 mM over 2 days. To prepare 20 nm and 10 nm nAu with a low surface-ssDNA density, ssDNA was incubated with 20 nm and 10 nm nAu at 5:1 ratio (ssDNA:nAu), followed by surface passivation with short five thymine bases(dT5), and then a gradual NaCl concentration increase to 600 mM over 2 days. Excess reagents were removed by repeated washing and centrifugation.
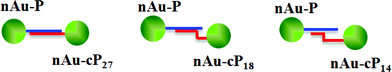 | ||
| Scheme 1 LSPR coupling between 50 nm nAu with different length in complementary sequence (not drawn to scale). The duplex regions were designed such that the separation between nAu is kept constant to eliminate distance-dependent variable. Note that in reality, each nAu is saturated with ssDNA probes on its surface. | ||
 | ||
| Scheme 2 LSPR coupling between (a) 50 nm nAu and 20 nm nAu, (b) 50 nm nAu and 10 nm nAu due to P/cP27 hybridization (not drawn to scale). Note that in reality, all 50 nm nAu are saturated with ssDNA probe; 20 nm and 10 nm nAu, either bear 5 probe ssDNA sequences each (surface-ssDNA density study) or are saturated with probe ssDNA sequences (size effect study). | ||
| Name | Oligonucleotide sequences |
|---|---|
| P | HS-5′-C6-AGC TCG ![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) -3′ -3′ |
| cP27 | HS-5′-C6-AGC TCG ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) -3′ -3′ |
| cP18 | HS-5′-C6-AGC TCG TCT TAG TCT -3′ |
| cP14 | HS-5′-C6-AGC TCG TCT TAG TCT TTC TT -3′ |
| dT5 | HS-5′-C6-TTT TT-3′ |
2.4 Formation and analysis of nAu-DNA assemblies
Two sets of nAu-DNA probes complementary to each other (denoted as nAu-P/nAu-cP27, nAu-P/nAu-cP18, and nAu-P/nAu-cP14) were diluted independently in a hybridization buffer (140 mM Tris, 140 mM NaCl, 2 mM MgCl2) according to the required concentration. Hybridization was carried out by first mixing 0.5 μL of nAu-P probe with 0.5 μL of its complements (nAu-cP27/nAu-cP18/nAu-cP14). Then, the sample was heated to 65 °C for 2 min to ensure complete melting of any duplex strands, followed by slow cooling to 24 °C at a rate of 0.5 °C min−1. All the samples were analyzed in time sequence of 1 h, 6 h, and 24 h by sandwiching 1 μL sample droplet between cover slip and microscope glass slide. Probe hybridization efficiency was examined under an up-right microscope (Nikon Eclipse 50i) with a dark-field oil condenser (N.A. 1.2–1.43, CytoViva) and a metal halide illuminator (Welch Allyn) as the white light source. Scattered light from nAu was collected by a 100× objective lens (Nikon Plan Fluor 100×, N.A. 0.5–1.3 oil iris, W.D. 0.16 mm) and directed to colour CCD camera (Nikon Digital Sight DS-Ri1) for real-colour imaging and locating the individual nanoparticles. The image acquisition (24-bit true colour TIF files, 20 ms integration time, and 5.6× gain) and analysis were performed using NIS-element AR 3.1 software (Nikon). Spectral measurements were done using an Andor SR-303i-B spectrometer (303 mm focal length, 40–100 s integration times). Statistical analysis, wherever applicable, was done using Student's t-test. Imaging and spectra experiments were conducted at ambient temperature.Three sets of studies, each aiming to investigate the influences of assembly condition on Rayleigh scattering, were carried out using complementary probes (probe = nAu-ssDNA). Scheme 1 (using 50 nm nAu only) and Scheme 2 (using 50 nm and 20 nm nAu, or 50 nm and 10 nm nAu) were repeated in triplicate and quadruplicate respectively to ensure reproducibility; Student's t-test was used for statistical analysis of study A.
3. Results and discussion
3.1 Synthesis and characterization of gold nanoparticles
TEM analysis (Fig. 1) showed that the mean particle diameters for the synthesized gold nanoparticles (nAu) were 10.03 ± 1.0 nm (denoted as 10 nm nAu) and 51.5 ± 3.7 nm (denoted as 50 nm nAu). Both displayed size distributions within 10% of their respective mean diameters. The highly homogeneous 50 nm nAu were due to (i) fairly homogeneous seeds (ii) 2-step seeding-growth, and (iii) silver addition which eliminated the formation of rod- and plate-like gold particles. Optimum silver ion concentration has been found to be 1/50 of the total gold concentration.47,48 Ag atoms, formed by reduction of Ag+ with citrate, can deposit under-potentially on the synthesized nAu (100) and (110) facets of nAu during 1st stage synthesis, and reshape the polycrystalline nAu into quasi-spherical structure.47,48 | ||
| Fig. 1 TEM images of (a) nAu with mean diameter 51.5 nm ± 3.7 nm on a wide field view, (b) its magnified view, and (c) nAu with mean diameter 10.03 nm ± 1.0 nm, which serves as a seed for the fabrication of 50 nm nAu. | ||
UV-visible spectra (Fig. 2) of 10 nm and 50 nm nAu exhibited extinction peaks at 520 nm and 527 nm respectively. The absence of a shoulder peak in the longer wavelength region indicated the absence of aggregated nAu and elongated/hexagonal particles, in accordance with TEM analysis.49 The synthesized 50 nm nAu had smaller bandwidth (II = 78 nm) than the existing protocols in the literature,48–53 signifying the high monodispersity and shape uniformity achieved.
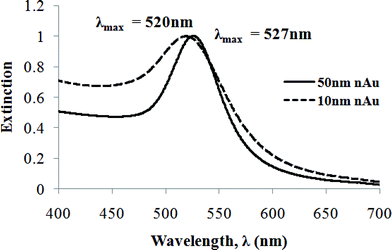 | ||
| Fig. 2 UV-visible extinction spectra of nAu solution with mean diameter 10 nm (dashed line) and 50 nm (solid line). Both are normalized to their extinction peak maxima. The bandwidth (FWHM, II) for 51.5 ± 3.7 nm nAu is 78 nm. | ||
More evidence for the shape uniformity is based on the LSPR scattering imaging. Under the dark field microscope, 99% of the 50 nm nAu exhibited green scattered light (Fig. S1†). The particles retained their stability even after several months of synthesis (data not shown). 10 nm nAu did not show up as bright as their 50 nm counterparts did because the scattering intensity of a metallic nanoparticle is proportional to the power of six of its effective radius.44
3.2 Analysis of nAu-DNA assemblies
Previous studies have demonstrated that hybridization between short-DNA-modified nAu induces surface plasmon coupling.45 This plasmon frequency shift can be detected using dark field microscopy that analyses only the scattered light. The formation of extensive and co-operative linking between nAu is expected since the nAu used in our system are saturated with oligonucleotides on the surface.54 This phenomenon brings upon enhanced plasmon shift and scattering intensities. Based on the correlation of dark field and FESEM micrographs (Table 2) as well as optical scattering spectra (Fig. 3), we used three categories to quantify conveniently the degree of hybridization, namely ‘green’, ‘yellow’, and ‘orange’ for the sake of ensemble statistics later. The ‘green’ category is composed of unhybridized nAu that scatter at a wavelength of ∼550 nm (green) strongly (Fig. 3). Small nanoassemblies classified as ‘yellow’ comprise either two hybridized 50 nm nAu or three/four 50 nm nAu hybridized in quasi-linear fashion, and with scattered wavelength of 570–580 nm; whereas nanoassemblies termed ‘orange’ consist of three 50 nm nAu in triangular configuration or more than four close-packed 50 nm nAu, and with scattered wavelength of 610–650 nm. The peak maxima of ‘orange’ nanoassemblies span wider wavelength range than ‘yellow’ nanoassemblies because of the varied number of nAu in ‘orange’ nanoassemblies. When complementary nAu-DNA are mixed in the presence of salts, individual nAu (‘green’) hybridize to their complements to form small nanoassemblies (‘yellow’), which further inter-hybridize to form nanoassemblies with significant numbers of nAu (‘orange’). In other words, as hybridization proceeds, the observed plasmon scattering turns from ‘green’ to ‘yellow’, and finally to ‘orange’. To analyze the hybridization of nAu, micrographs were captured at the bottom plane of the liquid chamber. The image processing software then discerned the scattering points into one of the aforementioned categories. All the hybridization percentages tabulated are the average of either three or four experimental results, as noted in each figure. Unless stated otherwise, P/cP27, P/cP18, and P/cP14 represent hybridization of different complementary nAu-DNA pairs. | ||
| Fig. 3 Typical light scattering spectra of the particles/nanoassemblies categorized as ‘green’, ‘yellow’, and ‘orange’ showed in Table 2, on glass-slides and in buffer (140 mM Tris, 140 mM NaCl, 2 mM MgCl2). The spectra were obtained with a dark field microscope, and exposure time 40–100 s on the EMCCD-monochromator. The ranges of peak maxima for each category are around 550 nm, 570–580 nm, and 610–650 nm. | ||

We favor larger size nAu (50 nm) which have inherently sufficient scattering intensity and yet not too bulky to slow down the particle diffusion (and hence hybridization). In addition, they are relatively stable in the salt buffer required for the hybridization. Table 1 shows that a dimeric 50 nm nAu pair exhibit sufficient peak shift to cause a distinct plasmonic color change. We also studied the LSPR coupling ability of small nAu in the subsequent sections (section 3.4: size effect and section 3.5: surface-ssDNA density studies), where 10 nm nAu was chosen for its better size contrast against 50 nm nAu, and 20 nm nAu for its lower plasmon scattering contrast against 50 nm nAu.
3.3 Probe concentration and hybridized duplex length study (Scheme 1)
In this part of the work, complementary probes P/cP27, P/cP18, and P/cP14 were at stoichiometric ratio of 1/1, and the amount of nanoassemblies was determined at various probe concentrations. Fig. 4a shows a typical dark field micrograph obtained by hybridization of 400 fM complementary probes P/cP27 at 1 h, whereas Fig. 4b represents the control that comprises only single type of probe. LSPR shift analysis showed a general trend of increased hybridization percentage with time (Fig. 5a–c) relative to the control (Fig. 5d). For all three duplex structures studied, the percentage of ‘yellow’ category remained low over time (Fig. 5a–c), suggesting that small nanoassemblies (‘yellow’) are a transition state from unhybridized nAu (‘green’) to nanoassemblies (‘orange’). This also implies that nAu form very good co-operative linking among each other, due to the high density of ssDNA functionalized on the nAu surface. At all time points, higher nAu concentration induced more hybridization, which can also be clearly seen in the dark field micrographs (Fig. S2†). With more particles per unit volume, the number of effective collisions among nAu increases, leading to a higher chance of DNA hybridization and nanoassembly formation. | ||
| Fig. 4 Probe concentration and hybridized duplex length study (Scheme 1). Dark field micrographs on (a) complementary probes nAu-P/nAu-cP27 hybridized at constant ratio of 1/1 and concentration 400 fM, (b) control comprises only single type of probe, both recorded after 1 h hybridization. The scattering color of control remains more than 95% green even after 24 h. Images were taken from the bottom plane of the liquid chamber where the particles had settled. Buffer: 140 mM Tris, 140 mM NaCl, 2 mM MgCl2. | ||
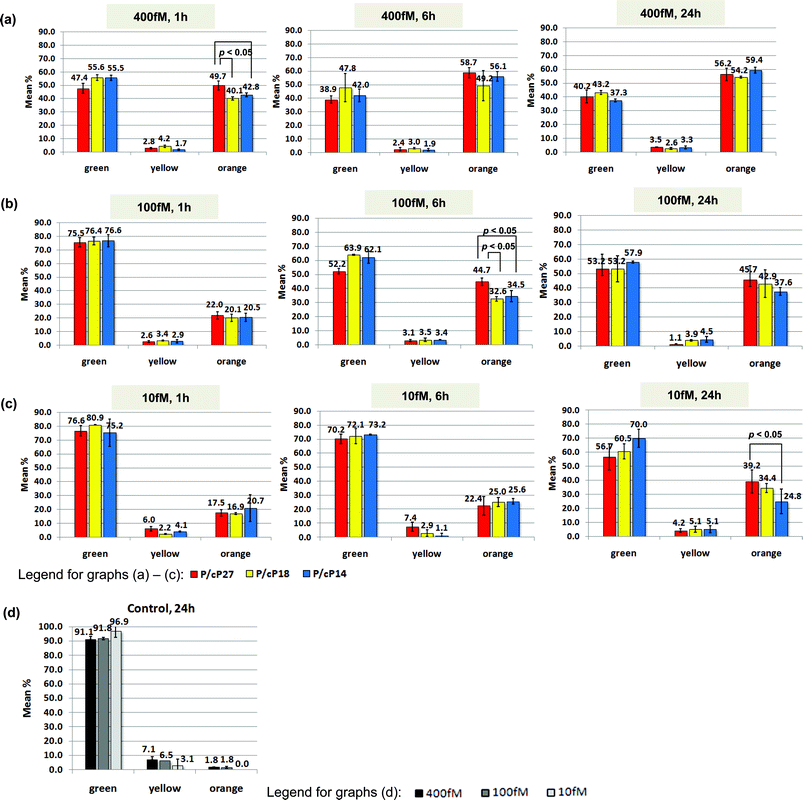 | ||
| Fig. 5 Probe concentration and hybridized duplex length study (Scheme 1). The charts summarize the hybridization percentage between complementary probes nAu-P/nAu-cP27 (red bars), nAu-P/nAu-cP18 (yellow bars), and nAu-P/nAu-cP14 (blue bars), at decreasing concentration of (a) 400 fM, (b) 100 fM, and (c) 10 fM while the ratio is kept at 1/1. The analysis is performed by counting the hybridized-clusters (categorized as ‘yellow’ and ‘orange’ due to their distinct LSPR plasmon shift) vs. the individual probes (categorized as ‘green’). The corresponding controls are illustrated in (d) for 400 fM (black bars), 100 fM (grey bars), and 10 fM (light-grey bars) where only a single type of probe exists, thus no hybridization take place. Each set of data in (a)–(d) consists of three repeats. Buffer: 140 mM Tris, 140 mM NaCl, 2 mM MgCl2. | ||
For the 400 fM probe concentration (Fig. 5a), P/cP27 (red bar), with complementary sequences 9 bp more than P/cP18 (yellow bar) and 13 bp more than P/cP14 (blue bar), appeared to have a higher degree of hybridization than the other two did after 1 h (p-value < 0.05). These affirmed that the hybridization efficiency increases with longer complementary sequences,55,56 although the distinction between P/cP18 and P/cP14 was not clear at this probe concentration. The degree of hybridization for P/cP27, P/cP18, and P/cP14 peaked at 6 h and did not exhibit any increase from 6 h to 24 h, indicating that hybridization may have achieved equilibrium within the first 6 h.
The effect of complementary duplex length on hybridization percentage was not obvious at 400 fM, but it was so at 100 fM (Fig. 5b). After 6 h, P/cP27 (red bar) showed the highest percentage (p-value < 0.05), compared with P/cP18 (yellow bar) and P/cP14 (blue bar), both of which exhibited similar percentages. At 24 h, however, all three pairs exhibited statistical equivalence in their hybridization percentages.
For the lowest nAu concentration of 10 fM (Fig. 5c), the hybridization percentages were low even after 24 h incubation. As a result, the effect of duplex lengths on nanoassembly formation shown in Scheme 1 was only obvious between P/cP27 and P/cP14 at 24 h (p-value < 0.05). Out of the three duplex structures, P/cP27 showed faster hybridization kinetics than the other two owing to its more stable duplex linker, which enhance co-operative linking. From the dark field micrographs, we can achieve a detection limit of about 6000 nAu.
Hybridization of free DNA in solution proceeds with relatively fast kinetics and reaches thermodynamic equilibrium in seconds or minutes. The stability of the hybridized DNA duplexes depends mainly on the complementary duplex length. However, the hybridization kinetics slows down substantially when the DNA are covalently bound57,58 on nAu,56 and parameters such as nAu concentration, hybridization time as well as complementary duplex length all affect the degree of hybridization and consequently the nanoassembly formation. As shown in Fig. 5, the degree of hybridization of DNA-modified nAu in bulk solution decreased with lower nAu concentrations and shorter duplex lengths, but it increased with the progression of time. In addition, the system with a longer duplex sequence reached equilibrium in a shorter time, and the equilibrium was only observed at the highest nAu concentration of 400 fM within the investigation period of 24 h (Fig. 5a). Although bulkiness of nAu can slow down hybridization kinetics, in our case of homogeneous hybridization (i.e. in bulk solution), appreciable hybridization was observed within 1 min after mixing of complementary probes in the 400 fM sample and, to lesser extent, in the 100 fM sample (Fig. S3†). This is substantially faster compared with the surface-bound hybridization reported in the literature, mostly likely due to additional interfacial restriction by the surface-bound particles.46
3.4 Size effect study (Scheme 1vs.Scheme 2)
To gain more information regarding the effect of nAu size on nanoassembly formation, we fixed one 50 nm nAu probe and varied the size of the complementary probe, in which 10 nm nAu was for its better size contrast against 50 nm nAu, and 20 nm nAu for its lower plasmon scattering contrast against 50 nm nAu. Thus, hybridization between complementary 50 nm/50 nm probes (homo-size system, Scheme 1), 50 nm/20 nm probes (hetero-size system, Scheme 2a), and 50 nm/10 nm probes (hetero-size system, Scheme 2b) were carried out at 1/1 molar ratio of 800 fM. All the nAu were fully saturated with ssDNA on their surfaces.The hybridization percentage for 50 nm/50 nm homo-size system were high, and the reaction almost reached equilibrium after 1 h (Fig. 6a); whereas the hybridization percentages for both 50 nm/20 nm and 50 nm/10 nm hetero-size system were surprisingly low (Fig. 6b and 6c respectively), and there was a continual increment in hybridization as time elapsed. In addition, the coupled-plasmon color change due to hybridization of 50 nm/10 nm probes (Fig. S4 IIIa†) was not as significant as that of 50 nm/20 nm probes (Fig. S4 IIa†), which in turn was not as significant as that of 50 nm/50 nm probes (Fig. S4 Ia†).
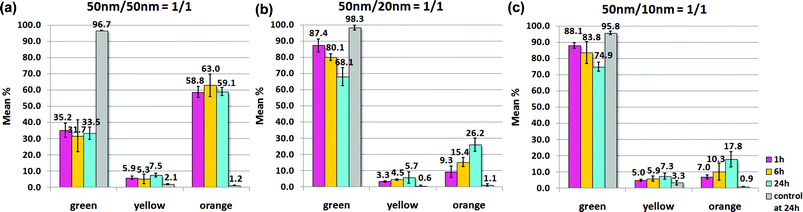 | ||
| Fig. 6 Size effect study (Scheme 1vs.Scheme 2) at 1 h, 6 h, and 24 h. Hybridization percentage between nAu-P/nAu-cP27 for (a) homo-size system with same probe size (50 nm/50 nm), (b) hetero-size system with different probe sizes (50 nm/20 nm), and (c) hetero-size system with different probe sizes (50 nm/10 nm). Ratio of complementary probes: 1/1; concentrations of all species: 800 fM. Data for homo-size system is the average of three repeats; for hetero-size system, each data reported consists of four repeats, two with 50 nm nAu-P/20 nm or 10 nm nAu-cP27, and the other two with 20 nm or 10 nm nAu-P/50 nm nAu-cP27. Images are quantified by counting the hybridized-clusters (‘yellow’ and ‘orange’) vs. the individual probes (‘green’). The corresponding controls for (a) homo-size system comprises single type of 50 nm probe at 1.6 pM, and for (b) hetero-size system , comprises 800 fM 50 nm probe and 800 fM 20 nm or 10 nm probe, both carrying the same type of ssDNA sequences. Buffer: 140 mM Tris, 140 mM NaCl, 2 mM MgCl2. | ||
It is well known that both the LSPR scattering cross-section and magnitude increase tremendously with the increase in nAu size. Here, 50 nm nAu possess sufficiently high light scattering yield44 and the strong plasmon scattering can be easily detected under the current dark field setup. Besides, the high and nearly saturated ‘orange’ hybridization percentage after 1 h (Fig. 6a) indicated that hybridization between complementary 50 nm probes was relatively easy. Hill et al.59 have reported that DNA coverage on 60 nm nAu and planar gold films were similar, and this shows that 60 nm nAu exhibit near planar surface curvature in terms of DNA loading. The quasi-planar surface curvature59 of 50 nm nAu can be expected to induce more than one hybridization between two 50 nm nAu to form stable nanoassemblies. This was shown by the low percentages of the ‘yellow’ category and high percentages of the ‘orange’ category in the 50 nm/50 nm system at all time points (Fig. 6a). Therefore, we deduce that stronger plasmon coupling between complementary 50 nm/50 nm homo-size probes and more stable links between neighboring nAu cause more nanoassemblies (‘orange’) to form than small nanoassemblies (‘yellow’), and leave relatively small amount of free ‘green’ particles, which is then reflected as the nearly-saturated high hybridization efficiency. Notice that for the fairly small 50 nm nAu and relatively large interparticle separation, we need not consider the multipole resonance contribution from 50 nm nAu.45,60
For the 50 nm/20 nm and 50 nm/10 nm hetero-size systems, hybridization was originally thought to be easier due to the higher surface curvature (hence higher deflection angle and less DNA crowding)59 and higher collision rate exhibited by the smaller 20 nm and 10 nm nAu. However, their observed hybridization percentages (Fig. 6b, c) were much lower than the 50 nm/50 nm homo-size system. Based on the size-dependent optical properties of metallic nanoparticles, the plasmon scatterings from a 20 nm and 10 nm nAu are reported to be 240 and 15000 times weaker than a 50 nm nAu.44 The inherent plasmon scattering from 10 nm nAu is so weak that by itself alone no visible scattering is detectable under the current conditions. As a result, the weak surface plasmon of 10 nm nAu induces relatively weak plasmon coupling between 50 nm and 10 nm nAu, and may cause indiscrete and unobservable plasmon shift even when the 50 nm nAu is hybridized a few 10 nm nAu. We were unable to determine the minimal number of 10 nm and 20 nm nAu required to attach on a 50 nm to bring upon an observable change in the plasmon color due to the instrumental limitations in our electron microscopy.46 Secondly, a 50 nm nAu with relatively larger surface area is able to hybridize to more 10 nm nAu, and reduces the available free 10 nm nAu61 and may leave many 50 nm nAu unhybridized. It is expected that, most small nAu have already hybridized to large nAu within the first hour owing to the high ssDNA density on the nAu surfaces. However, the weak coupled-plasmon shift results in the low observable hybridization percentage as a whole. This is further supported by the stronger plasmon shift of 50 nm/20 nm (Fig. S4 IIa†) compared to 50 nm/10 nm (Fig. S4 IIIa†). We believe that given a time longer than 24 h, hybridization percentage of the ‘orange’ category for both hetero-size systems would be elevated even higher.
With hybridization between complementary 50 nm/10 nm probes in 1/3 ratio (Fig. S5b†), more nanoassemblies with multiple 50 nm probes were observed at the same time frame than that of 1/1 ratio (Fig. S5a†). From the structure of nanoassemblies (Fig. S5†), most 10 nm nAu were trapped in between 50 nm nAu, making the 10 nm nAu not accessible to bridge with other 50 nm nAu. When the molar ratio increases, additional 10 nm nAu allow more bridging between the 50 nm nAu and hence results in the observed larger nanoassembly size. Similar binary nAu networks was reported62 using 31 nm/8 nm probes in 1/120 ratio, and core-satellite structures began to form.
It is important to highlight that all the data tabulated in this work are reproducible. For example, statistics based on 10 frames (Fig. S6a†) or 30 frames (Fig. S6b†) of dark field images show negligible difference between them, hence proving the validity of our quantification method.
3.5 Surface-ssDNA density study (Scheme 2)
In this study, the hetero-size systems were compared with the nAu bearing different surface-ssDNA densities. In the first set, all the nAu in both 50 nm/20 nm and 50 nm/10 nm hetero-size systems were fully saturated with ssDNA (Fig. 7 Ia and IIa, denoted as 50 nm/20 nm and 50 nm/10 nm respectively). The percentages of nanoassemblies continued to increase from 1 h to 24 h, and the fractions of ‘yellow’ relative to ‘orange’ category at all time frames were small and always less than unity. In the second set, 50 nm nAu were fully saturated with ssDNA but the 20 nm and 10 nm nAu were each conjugated with only 5 ssDNA followed by a monolayer of short oligonucleotides (dT5) for surface passivation (Fig. 7 Ib and IIb, denoted as 50 nm/20 nm(low) and 50 nm/10 nm(low) respectively). Consequently, the percentages of 50 nm/20 nm(low) nanoassemblies (Fig. 7 Ib) reduced substantially from 1 h to 24 h compared to 50 nm/20 nm (Fig. 7 Ia), with the most apparent reduction in the ‘orange’ nanoassemblies. The same trend was observed for 50 nm/10 nm(low) nanoassemblies (Fig. 7 IIb) compared to 50 nm/10 nm (Fig. 7 Ia). The percentages of 50 nm/10 nm(low) nanoassemblies also showed negligible change from 1 h to 24 h, and the fraction of ‘yellow’ relative to ‘orange’ category were always high and greater than unity. The low hybridization percentages were most likely due to the ineffective collision between the 50 nm and 20 nm or 10 nm nAu, the latter conjugated with low density of ssDNA. | ||
| Fig. 7 Surface-ssDNA density study (Scheme 2) at 1 h, 6 h, and 24 h. Hybridization percentage between nAu-P/nAu-cP27 for (I) 50 nm/20 nm hetero-size system with the 20 nm nAu (a) fully saturated with ssDNA, or (b) bearing only 5 ssDNA, and for (II) 50 nm/10 nm hetero-size system with the 10 nm nAu (a) fully saturated with ssDNA, or (b) bearing only 5 ssDNA. Ratio of complementary probes: 1/1; concentrations of all species: 800 fM. Each set of data reported consists of four repeats, two with 50 nm nAu-P/20 nm or 10 nm nAu-cP27, and the other two with 20 nm or 10 nm nAu-P/50 nm nAu-cP27. Images are quantified by counting the hybridized-clusters (‘yellow’ and ‘orange’) vs. the individual probes (‘green’). The corresponding controls comprise 800 fM 50 nm probe and 800 fM 20 nm or 10 nm probe, both carrying the same type of ssDNA sequences, for (a) fully saturated and (b) 5-ssDNA-bearing 20 nm or 10 nm nAu. Buffer: 140 mM Tris, 140 mM NaCl, 2 mM MgCl2. | ||
Apparently, reducing the surface-ssDNA density of 10 nm nAu resulted in the observed extremely low hybridization percentage of 50 nm/10 nm(low) system compared to 50 nm/10 nm system. This is not unexpected because the low surface-ssDNA density on 10 nm(low) nAu certainly reduces the number of effective collisions and imposes more steric constraint to form multi-50 nm nanoassemblies. The already weak plasmon coupling between 50 nm and 10 nm nAu (size effect study, vide supra) are now aggravated by the even lower hybridization kinetics brought by 10 nm(low) nAu. On the other hand, 10 nm nAu which are fully saturated with ssDNA enable multiple hybridization events to construct multi-50 nm nanoassemblies. This explains why the 50 nm/10 nm nAu system exhibited faster hybridization than 50 nm/10 nm(low) system. Finally, 50 nm/20 nm(low) exhibited the higher degree of nanoassembly formation (Fig. 7 Ib) compared with 50 nm/10 nm(low) (Fig. 7 IIb), most likely due to the stronger scattering yield of 20 nm. This shows that small nanoparticles are not as good as their larger counterparts in dark field detection.
4. Conclusions
Using Rayleigh scattering as a signal readout platform, we are able to detect/visualize the progress of nanoassembly formation of complementary nAu-DNA probes at as low as 10 fM concentration. Three studies were conducted to gain knowledge on the hybridization process of the nAu-DNA probes. For the first study, we found that hybridization of the complementary 50 nm nAu at equal amount increased with increasing nAu concentration, longer hybridization time, and longer complementary duplex DNA length. In the second study, due to the size-dependent optical scattering of nAu, higher hybridization percentages were observed in 50 nm/50 nm homo-size system compared to the 50 nm/20 nm hetero-size system, which in turn were higher than 50 nm/10 nm hetero-size system. For the third study, hybridization efficiency of the 50 nm/20 nm and 50 nm/10 nm hetero-size systems was significantly reduced by the low surface-DNA density on 20 nm and 10 nm nAu. Added together, our findings showed that the nanoassembly formation can be controlled by various parameters, namely the concentration and molar ratio of complementary nAu-DNA, hybridization length of duplex DNA, and surface-DNA density of the nanoparticles. Besides, this work highlights the importance of using nanoparticles with sufficient light scattering yield to facilitate the detection of nanoassembly formation by optical scattering. The information carried here is thus useful for the development of a number of assays designing to study conveniently the biomolecule interaction by the aid of specifically chosen nanoparticle system.Acknowledgements
This work was supported by research funding from the Singapore Ministry of Education Academic Research Fund Tier 1 via grant R279000282112.References
- C. F. Bohren and D. R. Huffman, in Absorption and Scattering of Light by Small Particles, Wiley, New York, 1983 Search PubMed.
- U. Kreibig and M. Vollmer, in Optical Properties of Metal Clusters, Springer, New York, 1995 Search PubMed.
- K. H. Su, Q. H. Wei, X. Zhang, J. J. Mock, D. R. Smith and S. Schultz, Nano Lett., 2003, 3, 1087–1090 CrossRef CAS.
- P. Nordlander, C. Oubre, E. Prodan, K. Li and M. I. Stockman, Nano Lett., 2004, 4, 899–903 CrossRef CAS.
- A. M. Funston, C. Novo, T. J. Davis and P. Mulvaney, Nano Lett., 2009, 9, 1651–1658 CrossRef CAS.
- P. K. Jain and M. A. El-Sayed, Chem. Phys. Lett., 2010, 487, 153–164 CrossRef CAS.
- H. Wang and B. M. Reinhard, J. Phys. Chem. C, 2009, 113, 11215–11222 CAS.
- B. M. Reinhard, S. Sheikholeslami, A. Mastroianni, A. P. Alivisatos and J. Liphardt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2667–2672 CrossRef CAS.
- B. M. Reinhard, M. Siu, H. Agarwal, A. P. Alivisatos and J. Liphardt, Nano Lett., 2005, 5, 2246–2252 CrossRef CAS.
- D. S. Sebba, J. J. Mock, D. R. Smith, T. H. LaBean and A. A. Lazarides, Nano Lett., 2008, 8, 1803–1808 CrossRef CAS.
- P. K. Jain and M. A. El-Sayed, Nano Lett., 2008, 8, 4347–4352 CrossRef CAS.
- J. Ling, Y. F. Li and C. Z. Huang, Anal. Chem., 2009, 81, 1707–1714 CrossRef CAS.
- C. Novo, A. M. Funston, I. Pastoriza-Santos, L. M. Liz-Marzán and P. Mulvaney, J. Phys. Chem. C, 2008, 112, 3–7 CAS.
- C. Sönnichsen, B. M. Reinhard, J. Liphardt and A. P. Alivisatos, Nat. Biotechnol., 2005, 23, 741–745 CrossRef.
- T. J. Yim, Y. Wang and X. Zhang, Nanotechnology, 2008, 19 Search PubMed.
- M. Ringler, T. A. Klar, A. Schwemer, A. S. Susha, J. Stehr, G. Raschke, S. Funk, M. Borowski, A. Nichtl, K. Kürzinger, R. T. Phillips and J. Feldmann, Nano Lett., 2007, 7, 2753–2757 CrossRef CAS.
- F. Svedberg, Z. Li, H. Xu and M. Käll, Nano Lett., 2006, 6, 2639–2641 CrossRef CAS.
- H. Xu and M. Käll, Phys. Rev. Lett., 2002, 89, 2468021–2468024 CrossRef.
- J. T. Bradshaw, S. B. Mendes and S. S. Saavedra, Anal. Chem., 2005, 77 Search PubMed.
- R. De Waele, A. F. Koenderink and A. Polman, Nano Lett., 2007, 7, 2004–2008 CrossRef CAS.
- A. F. Koenderink, R. De Waele, J. C. Prangsma and A. Polman, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76 Search PubMed.
- Y. Sato, K. Sato, K. Hosokawa and M. Maeda, Anal. Biochem., 2006, 355, 125–131 CrossRef CAS.
- A. Charrier, N. Candoni, N. Liachenko and F. Thibaudau, Biosens. Bioelectron., 2007, 22, 1881–1886 CrossRef CAS.
- S. Tokonami, H. Shiigi and T. Nagaoka, Anal. Chem., 2008, 80, 8071–8075 CrossRef CAS.
- W. J. Qin, O. S. Yim, P. S. Lai and L. Y. L. Yung, Biosens. Bioelectron., 2010, 25, 2021–2025 CrossRef CAS.
- L. Wang and P. C. H. Li, Biomicrofluidics, 2010, 4, 1–9 CrossRef.
- B. A. Du, Z. P. Li and C. H. Liu, Angew. Chem., Int. Ed., 2006, 45, 8022–8025 CrossRef CAS.
- J. Zhang, S. Song, L. Zhang, L. Wang, H. Wu, D. Pan and C. Fan, J. Am. Chem. Soc., 2006, 128, 8575–8580 CrossRef CAS.
- J. Li, J. H. Jiang, X. M. Xu, X. Chu, C. Jiang, G. Shen and R. Q. Yu, Analyst, 2008, 133, 939–945 RSC.
- S. J. Hurst, S. H. Min, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2007, 79, 7201–7205 CrossRef CAS.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 3468–3470 CrossRef CAS.
- M. S. Han, A. K. R. Lytton-Jean, B. K. Oh, J. Heo and C. A. Mirkin, Angew. Chem., Int. Ed., 2006, 45, 1807–1810 CrossRef CAS.
- H. D. Hill, S. J. Hurst and C. A. Mirkin, Nano Lett., 2009, 9, 317–321 CrossRef CAS.
- S. J. Hurst, H. D. Hill, R. J. Macfarlane, J. Wu, V. P. Dravid and C. A. Mirkin, Small, 2009, 5, 2156–2161 CrossRef CAS.
- W. J. Qin and L. Y. L. Yung, Bioconjugate Chem., 2008, 19, 385–390 CrossRef CAS.
- W. Zhao, L. Lin and I. M. Hsing, Bioconjugate Chem., 2009, 20, 1218–1222 CrossRef CAS.
- J. S. Lee, A. K. R. Lytton-Jean, S. J. Hurst and C. A. Mirkin, Nano Lett., 2007, 7, 2112–2115 CrossRef CAS.
- W. Zhao and I. M. Hsing, Chem. Commun., 2010, 46, 1314–1316 RSC.
- S. A. Claridge, H. W. Liang, S. R. Basu, J. M. J. Fréchet and A. P. Alivisatos, Nano Lett., 2008, 8, 1202–1206 CrossRef CAS.
- C. J. Loweth, W. Brett Caldwell, X. Peng, A. P. Alivisatos and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 1808–1812 CrossRef CAS.
- K. Suzuki, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2009, 131, 7518–7519 CrossRef CAS.
- H. N. Wang and T. Vo-Dinh, Small, 2011, 7, 3067–3074 CrossRef CAS.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS.
- J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 137–156 CrossRef CAS.
- S. Sheikholeslami, Y. W. Jun, P. K. Jain and A. P. Alivisatos, Nano Lett., 2010, 10, 2655–2660 CrossRef CAS.
- T. Sannomiya, C. Hafner and J. Voros, Nano Lett., 2008, 8, 3450–3455 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- H. Xia, S. Bai, J. Hartmann and D. Wang, Langmuir, 2010, 26, 3585–3589 CrossRef CAS.
- T. K. Sau, A. Pal, N. R. Jana, Z. L. Wang and T. Pal, J. Nanopart. Res., 2001, 3, 257–261 CrossRef CAS.
- P. Qiu and C. Mao, J. Nanopart. Res., 2009, 11, 885–894 CrossRef CAS.
- K. R. Brown and M. J. Natan, Langmuir, 1998, 14, 726–728 CrossRef CAS.
- K. R. Brown, D. G. Walter and M. J. Natan, Chem. Mater., 2000, 12, 306–313 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS.
- A. K. R. Lytton-Jean and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 12754–12755 CrossRef CAS.
- J. Xu and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 13227–13231 CrossRef CAS.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 29, 5163–5168 CrossRef CAS.
- A. W. Peterson, L. K. Wolf and R. M. Georgiadis, J. Am. Chem. Soc., 2002, 124, 14601–14607 CrossRef CAS.
- H. D. Hill, J. E. Millstone, M. J. Banholzer and C. A. Mirkin, ACS Nano, 2009, 3, 418–424 CrossRef CAS.
- A. Tcherniak, J. W. Ha, S. Dominguez-Medina, L. S. Slaughter and S. Link, Nano Lett., 2010, 10, 1398–1404 CrossRef CAS.
- S. J. Park, A. A. Lazarides, J. J. Storhoff, L. Pesce and C. A. Mirkin, J. Phys. Chem. B, 2004, 108, 12375–12380 CrossRef CAS.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Dark field micrographs, TEM images, and comparison of the statistical distributions as noted in the text. See DOI: 10.1039/c2ra20330h |
| This journal is © The Royal Society of Chemistry 2012 |